

Abstract
Background. KDM6A (Lysine (K)-Specific Demethylase 6A) is the driver gene related to esophageal squamous cell carcinoma (ESCC). In order to provide more biological insights into KDM6A, in this paper, we treat PPI (protein-protein interaction) network derived from KDM6A as a conceptual framework and follow it to review its biological function. Method. We constructed a PPI network with Cytoscape software and performed clustering of network with Clust&See. Then, we evaluate the pathways, which are statistically involved in the network derived from KDM6A. Lastly, gene ontology analysis of clusters of genes in the network was conducted. Result. The network includes three clusters that consist of 74 nodes connected via 453 edges. Fifty-five pathways are statistically involved in the network and most of them are functionally related to the processes of cell cycle, gene expression, and carcinogenesis. The biology themes of clusters 1, 2, and 3 are chromatin modification, regulation of gene expression by transcription factor complex, and control of cell cycle, respectively. Conclusion. The PPI network presents a panoramic view which can facilitate for us to understand the function role of KDM6A. It is a helpful way by network approach to perform system review on a certain gene.
1. Introduction
Esophageal cancer is the eighth most common malignancy and the sixth cause of cancer deaths worldwide [1, 2]. Esophageal squamous cell carcinoma (ESCC) and adenocancer are the two histologic types that make up for greater than 90 percent of the diagnoses of esophageal cancers [3]. In East Asia, the majority of esophageal cancers are ESCC [2]. As the most of cancers, the pathomechanism of ESCC remains elusive. There is an agreement that cancers arise owing to mutations in a subset of genes that confer growth advantage. Not all mutations can contribute to the development of the cancers. The genes which harbor mutation conferring benefit to survival of tumor cell are called driver genes. The counterpart of driver gene is passenger gene, whose mutation does not contribute to oncogenesis [4]. Defining a driver gene in physiologic terms is easy but identifying which mutations are drivers and which are passengers is more difficult, so numerous statistical methods to identify driver genes have been described. Vogelstein and colleagues identified about 140 driver genes from Catalogue of Somatic Mutations in Cancer database by their unique ruler [5]. Of them, KDM6A is the driver gene related to ESCC.
In eukaryotic cells, DNA is packaged into chromatin whose functional unit is nucleosome. A nucleosome is an octameric structure composed of two histones each of H2A, H2B, H3, and H4 encircled by 147 bp of DNA [6]. Modifications of histones such as methylation, acetylation, phosphorylation, ubiquitination, and sumoylation regulate the structure of chromatin and determine the active or repressive chromatin states. Of them, histone methylation can function in gene activation or repression, depending on which residues are targeted. Methylation of histone H3 on Lysine 4 (H3K4me) is an active chromatin modification, while methylation of histone H3 on Lysine 27 (H3K27me) is associated with repression of gene activity [7]. The PRC2 (polycomb repressive complex 2) mediates transcriptional repression by methylation of histone on H3K27 [8–11]. KDM6A counteracts PRC2 and activates chromatin transcriptionally by demethylation of H3K27 [12]. This is the main biological function of KDM6A. However, KDM6A is vital in a wide array of functions including cell cycle regulation, cell differentiation, and stem cell specification [13–15]. The mutation of KDM6A is involved in various types of cancer across both solid and liquid tumors [16–18].
As we all know, proteins do not function alone while they orchestrate all biological processes by interacting with other proteins. Analysis of PPI network consisting of KDM6A and its interactors facilitates providing more biological insights into KDM6A for us. In this paper, we treat PPI network derived from KDM6A as a conceptual framework and follow it to review its biological function.
2. Method
2.1. Construction of PPI Network Derived from KDM6A
We obtained PPIs from STRING database, a precomputed database for the exploration of protein-protein interactions. The prediction methods in STRING include neighborhood gene fusion, cooccurrence, coexpression, experiments, databases, and text mining. A confidence score can be assigned to each prediction method and every interaction has a final aggregate score. The newest version of STRING 9.1 covers approximately 2.5 million proteins from 630 different organisms [19]. In this study, the interactions restricted to Homo sapiens were downloaded. In order to avoid false link in greatest extent, we set the score of inclusion criteria larger than 0.9. We constructed a network that consists of not only the direct PPI neighbors of KDM6A but also their secondary neighbors. The network was constructed using Cytoscape, a popular and highly versatile software platform for the analysis, operation, and visualization of large networks [20].
2.2. Construction of Cellular Pathways Database
All of the pathways with their m gene members were downloaded from an integrated pathway database, Molecular Signatures Database (MSigDB) [21], which is a large collection of annotated functional gene sets. There are 880 canonical pathways with 6804 proteins members in the database, including the metabolic and signaling pathways collected from BioCarta (http://www.biocarta.com/), KEGG [22], and Reactome [23].
2.3. Identification of Pathways Involved in PPI Network Derived from KDM6A
To examine the evidence of association of a given pathway with PPI network derived from KDM6A, Fisher's exact test based on the cumulative hypergeometric distribution was employed. The p value was calculated to evaluate statistical significance of a given pathway by the formula as follows:
| (1) |
In this formula, N represents the total number of proteins in the background population, n represents the number of proteins in the PPI network derived from KDM6A, and m denotes the number of proteins within the given pathways. The number of proteins that overlapped with both proteins in the PPI network and this pathway is denoted as k. In this study, a pathway is considered statistically associating with the PPI network derived from KDM6A under the condition of its p value being less than 0.05.
2.4. Ontology Analysis of Proteins in PPI Network Derived from KDM6A
Usually, gene ontology analysis is mainly conducted in individual gene. However, in this study, we perform ontology enrichment analysis based on a group of genes. As a web tool, NOA not only is able to perform analysis based on a group of genes but also takes interactions between genes into consideration [24]. Every group of genes corresponds to the proteins in the clusters of PPI network derived from KDM6A. There are many clustering methods that have been used to highlight groups of densely connected nodes [25]. Clusters in networks can offer mechanistic hypotheses of disease because they are highly interconnected molecular complexes or signaling pathways [26]. In this study, clusters were found with Cytoscape plugin called Clust&See dedicated to the identification, visualization, and analysis of clusters extracted from PPI network [27].
3. Result and Discussion
3.1. General Description of the PPI Network Derived from KDM6A
The network consists of 74 nodes connected via 453 edges. KDM6A has seven direct neighbors, which are RBBP5, WDR5, ASH2L, MLL2, PAXIP1, NCOA6, and RBL2, respectively (Figure 1). KDM6A contains a tetratricopeptide motif predicted to mediate protein-protein interactions [28] and is not only a member of a stable multiprotein complex that demethylates H3K27me3 but also a member of the MLL2 H3K4 methyltransferase complex which can facilitate gene expression [29, 30]. Its catalytic activity has been linked to regulation of homeobox (HOX) and RB transcriptional networks [31]. Different protein partners modulate the recruitment of KDM6A to specific chromatin regions to target specific genes [32, 33]. There are three clusters whose nodes are distinguished with different colors in the network (Figure 1). Every cluster has its biological theme detailed later and there are links among these clusters.
Figure 1.

PPI network derived from KDM6A. KDM6A has seven direct neighbors tagged with middle node size. There are three clusters, whose nodes are distinguished with different colors (cluster 1: red; cluster 2: green; cluster 3: blue).
3.2. Pathways Involved in PPI Network Derived from KDM6A
Of eight hundred and eighty (880) pathways, fifty-five (55) pathways are statistically involved in PPI network derived from KDM6A (p < 0.05, see Table 1 in Supplementary Material available online at http://dx.doi.org/10.1155/2016/1970904). These fifty-five pathways can be catalogue to eight classes including cell cycle, gene expression, lipid metabolism, cancer, apoptosis, signal transduction, development, and DNA repair. Most of pathways are related to cell cycle, gene expression, and cancer (Figure 2).
Figure 2.
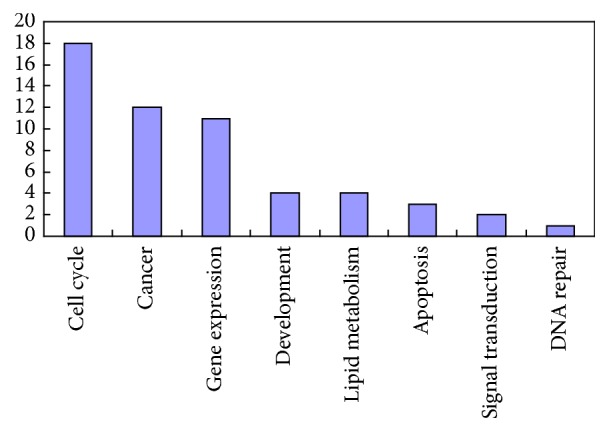
The frequency of fifty-five pathways. Fifty-five pathways can be catalogue to eight classes including cell cycle, gene expression, lipid metabolism, cancer, apoptosis, signal transduction, development, and DNA repair.
There are two points in current mainstream discipline about KDM6A: (i) as a general factor, KDM6A activates gene transcription during development. HOX genes encode transcription factors that regulate embryogenesis and guide tissue differentiation [34]. KDM6A removes H3K27me3 from HOX genes to restore their activity and control HOX gene expression [35]. (ii) KDM6A acts with methyltransferase complex to facilitate gene expression for regulating transcriptional networks of RB [36].
The evidence indicates that KDM6A evolved in pathways such as inflammation, apoptosis, cell cycle, and DNA repair related. KDM6A is implicated in IL-4 mediated transcriptional activation of the arachidonate 15-lipoxygenase-1 (ALOX15) gene. ALOX15 oxygenates polyunsaturated fatty acids and biomembranes, which generate multiple lipid signaling mediators involved in inflammation [37]. KDM6A is required for hormone-mediated transcriptional regulation of apoptosis and autophagy genes in Drosophila salivary glands [38]. DNA methylation affects the expression of genes involved in cell cycle checkpoint, apoptosis, and DNA repair [39].
3.3. Ontology Analysis of the Protein Interactions in the Three Clusters
We manually choose top three terms ranked by p value generated by analysis tool NOA (Table 1). The theme of cluster 1 is chromatin modification by methyl transfer of histone. It is the main function of KDM6A as previously mentioned. As part of an H3K4-methyltransferase complex containing MLL2, PTIP, ASC2, ASH2, RBQ3, and WDR5, KDM6A can promote H3K4 methylation [30]. It indicates that KDM6A and methyltransferase complex concert with active chromatin modification by demethylation of histone on H3K27 and methylation on K3K4. Clinically, KDM6A or MLL4 was associated with poor prognosis in patients with breast cancer. KDM6A interacts with a c-terminal region of MLL4 and coordinates regulation of cotarget gene expression [40].
Table 1.
The functional characterization of protein interaction networks in three clusters.
| Cluster | GO:term | Term name | p value | |
|---|---|---|---|---|
| Cluster 1 | Biological processes | GO:0016568 | Chromatin modification | 7.9E − 21 |
| GO:0006325 | Chromatin organization | 1.3E − 20 | ||
| GO:0051568 | Histone H3-K4 methylation | 9.2E − 20 | ||
| Cellular components | GO:0034708 | Methyltransferase complex | 5.6E − 38 | |
| GO:0035097 | Histone methyltransferase complex | 5.6E − 38 | ||
| GO:0044451 | Nucleoplasm part | 3.4E − 20 | ||
| Molecular functions | GO:0042054 | Histone methyltransferase activity | 2.5E − 20 | |
| GO:0008170 | N-Methyltransferase activity | 5.6E − 19 | ||
| GO:0008276 | Protein methyltransferase activity | 5.6E − 19 | ||
|
| ||||
| Cluster 2 | Biological processes | GO:0045944 | Positive regulation of transcription from RNA polymerase II promoter | 4.7E − 16 |
| GO:0006357 | Regulation of transcription from RNA polymerase II promoter | 4.6E − 15 | ||
| GO:0010628 | Positive regulation of gene expression | 9.5E − 15 | ||
| Cellular components | GO:0005634 | Nucleus | 3.4E − 9 | |
| GO:0044451 | Nucleoplasm part | 6.2E − 9 | ||
| GO:0005667 | Transcription factor complex | 4.3E − 7 | ||
| Molecular functions | GO:0008134 | Transcription factor binding | 6.2E − 23 | |
| GO:0016563 | Transcription activator activity | 1.6E − 18 | ||
| GO:0030528 | Transcription regulator activity | 9.6E − 16 | ||
|
| ||||
| Cluster 3 | Biological processes | GO:0000082 | G1/S transition of mitotic cell cycle | 1.8E − 11 |
| GO:0007049 | Cell cycle | 9.7E − 10 | ||
| GO:0022402 | Cell cycle process | 5.1E − 9 | ||
| Cellular components | GO:0000307 | Cyclin-dependent protein kinase holoenzyme complex | 8.9E − 14 | |
| GO:0005654 | Nucleoplasm | 4.3E − 10 | ||
| GO:0044428 | Nuclear part | 6.7E − 9 | ||
| Molecular functions | GO:0004693 | Cyclin-dependent protein kinase activity | 3.7E − 6 | |
| GO:0016538 | Cyclin-dependent protein kinase regulator activity | 9.5E − 5 | ||
| GO:0030332 | Cyclin binding | 3.0E − 3 | ||
The proteins in cluster 2 mainly compose the transcription factor complex. The direct neighbor of KDM6A, NCOA6 (nuclear receptor coactivator 6, NCOA6), binds nuclear receptors and stimulates the transcriptional activities in a hormone-dependent fashion. Mass spectrometry analysis demonstrated that PTIP associated with ASH2L, RBBP5, WDR5, NCOA6, and KDM6A [29]. It suggests that there exist interactions among NCOA6, KDM6A, and other methyltransferases.
The nodes in cluster 3 are RBL2 (retinoblastoma-like 2, RBL2) and its interactors which control cell cycle. RBL2 is a key regulator of entry into cell division, directly involved in heterochromatin formation by maintaining overall chromatin structure through stabilizing histone methylation. Even though there is no direct experimental evidence that KDM6A interacts with RBL2, a study found that ectopic expression of KDM6A enhanced the expression of retinoblastoma tumor suppressor gene RB and its related gene RBL2 [36].
4. Conclusion and Prospect
PPI network derived from KDM6A presents a panoramic view which can facilitate the understanding of the function role of KDM6A for us. The results from both sides of identification of pathway and ontology analysis of network confirm and complement one another. It is a helpful way by network approach to perform system review on a certain gene. Considering that these could be nonspecific interactions, the network can only be regarded as a guide map. Some links between the proteins in the network need to be validated by experiment to explore the biology role of KDM6A in the progression of carcinogenesis. For example, we need to investigate the expression pattern of these genes at mRNA or protein level and the possible regulatory relationships between these genes in clinical ESCC specimen or esophageal cancer cell line. Similar studies in other cancers were completed and reported [40, 41].
Supplementary Material
List of pathways involved in PPI network derived from KDM6A.
Acknowledgments
The present study was supported by a grant from the National Natural Science Foundation of China (Grant no. 81460359).
Competing Interests
The authors declare that they have no competing interests.
References
- 1.Kamangar F., Dores G. M., Anderson W. F. Patterns of cancer incidence, mortality, and prevalence across five continents: defining priorities to reduce cancer disparities in different geographic regions of the world. Journal of Clinical Oncology. 2006;24(14):2137–2150. doi: 10.1200/jco.2005.05.2308. [DOI] [PubMed] [Google Scholar]
- 2.Mathé E. A., Giang H. N., Bowman E. D., et al. MicroRNA expression in squamous cell carcinoma and adenocarcinoma of the esophagus: associations with survival. Clinical Cancer Research. 2009;15(19):6192–6200. doi: 10.1158/1078-0432.ccr-09-1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Daly J. M., Fry W. A., Little A. G., et al. Esophageal cancer: results of an American College of Surgeons patient care evaluation study. Journal of the American College of Surgeons. 2000;190(5):562–572. doi: 10.1016/s1072-7515(00)00238-6. [DOI] [PubMed] [Google Scholar]
- 4.Greenman C., Stephens P., Smith R., et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446(7132):153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vogelstein B., Papadopoulos N., Velculescu V. E., Zhou S., Diaz L. A., Jr., Kinzler K. W. Cancer genome landscapes. Science. 2013;339(6127):1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Margueron R., Reinberg D. Chromatin structure and the inheritance of epigenetic information. Nature Reviews Genetics. 2010;11(4):285–296. doi: 10.1038/nrg2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martin C., Zhang Y. The diverse functions of histone lysine methylation. Nature Reviews Molecular Cell Biology. 2005;6(11):838–849. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- 8.Czermin B., Melfi R., McCabe D., Seitz V., Imhof A., Pirrotta V. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell. 2002;111(2):185–196. doi: 10.1016/s0092-8674(02)00975-3. [DOI] [PubMed] [Google Scholar]
- 9.Cao R., Wang L., Wang H., et al. Role of histone H3 lysine 27 methylation in polycomb-group silencing. Science. 2002;298(5595):1039–1043. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 10.Muller J., Hart C. M., Francis N. J., et al. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell. 2002;111(2):197–208. doi: 10.1016/s0092-8674(02)00976-5. [DOI] [PubMed] [Google Scholar]
- 11.Kuzmichev A., Nishioka K., Erdjument-Bromage H., Tempst P., Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the enhancer of zeste protein. Genes and Development. 2002;16(22):2893–2905. doi: 10.1101/gad.1035902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schuettengruber B., Chourrout D., Vervoort M., Leblanc B., Cavalli G. Genome regulation by polycomb and trithorax proteins. Cell. 2007;128(4):735–745. doi: 10.1016/j.cell.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 13.Sen G. L., Webster D. E., Barragan D. I., Chang H. Y., Khavari P. A. Control of differentiation in a self-renewing mammalian tissue by the histone demethylase JMJD3. Genes and Development. 2008;22(14):1865–1870. doi: 10.1101/gad.1673508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang J. K., Tsai M.-C., Poulin G., et al. The histone demethylase UTX enables RB-dependent cell fate control. Genes and Development. 2010;24(4):327–332. doi: 10.1101/gad.1882610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Satoh T., Takeuchi O., Vandenbon A., et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nature Immunology. 2010;11(10):936–944. doi: 10.1038/ni.1920. [DOI] [PubMed] [Google Scholar]
- 16.Dalgliesh G. L., Furge K., Greenman C., et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010;463(7279):360–363. doi: 10.1038/nature08672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gui Y., Guo G., Huang Y., et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nature Genetics. 2011;43(9):875–878. doi: 10.1038/ng.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mar B. G., Bullinger L., Basu E., et al. Sequencing histone-modifying enzymes identifies UTX mutations in acute lymphoblastic leukemia. Leukemia. 2012;26(8):1881–1883. doi: 10.1038/leu.2012.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Franceschini A., Szklarczyk D., Frankild S., et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Research. 2013;41(1):D808–D815. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shannon P., Markiel A., Ozier O., et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Research. 2003;13(11):2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liberzon A., Subramanian A., Pinchback R., Thorvaldsdóttir H., Tamayo P., Mesirov J. P. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27(12):1739–1740. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kanehisa M., Goto S., Furumichi M., Tanabe M., Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Research. 2009;38(1):D355–D360. doi: 10.1093/nar/gkp896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Croft D., O'Kelly G., Wu G., et al. Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Research. 2011;39(1):D691–D697. doi: 10.1093/nar/gkq1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang J., Huang Q., Liu Z.-P., et al. NOA: a novel Network Ontology Analysis method. Nucleic Acids Research. 2011;39, article e87 doi: 10.1093/nar/gkr251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aittokallio T., Schwikowski B. Graph-based methods for analysing networks in cell biology. Briefings in Bioinformatics. 2006;7(3):243–255. doi: 10.1093/bib/bbl022. [DOI] [PubMed] [Google Scholar]
- 26.Ideker T., Sharan R. Protein networks in disease. Genome Research. 2008;18(4):644–652. doi: 10.1101/gr.071852.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spinelli L., Gambette P., Chapple C. E., et al. Clust&See: a Cytoscape plugin for the identification, visualization and manipulation of network clusters. BioSystems. 2013;113(2):91–95. doi: 10.1016/j.biosystems.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 28.Swigut T., Wysocka J. H3K27 Demethylases, at Long Last. Cell. 2007;131(1):29–32. doi: 10.1016/j.cell.2007.09.026. [DOI] [PubMed] [Google Scholar]
- 29.Cho Y.-W., Hong T., Hong S., et al. PTIP associates with MLL3- and MLL4-containing histone H3 lysine 4 methyltransferase complex. The Journal of Biological Chemistry. 2007;282(28):20395–20406. doi: 10.1074/jbc.m701574200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Issaeva I., Zonis Y., Rozovskaia T., et al. Knockdown of ALR (MLL2) reveals ALR target genes and leads to alterations in cell adhesion and growth. Molecular and Cellular Biology. 2007;27(5):1889–1903. doi: 10.1128/MCB.01506-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van der Meulen J., Speleman F., Van Vlierberghe P. The H3K27me3 demethylase UTX in normal development and disease. Epigenetics. 2014;9(5):658–668. doi: 10.4161/epi.28298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Agger K., Cloos P. A. C., Christensen J., et al. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature. 2007;449(7163):731–734. doi: 10.1038/nature06145. [DOI] [PubMed] [Google Scholar]
- 33.Welstead G. G., Creyghton M. P., Bilodeau S., et al. X-linked H3K27me3 demethylase Utx is required for embryonic development in a sex-specific manner. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(32):13004–13009. doi: 10.1073/pnas.1210787109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weatherbee S. D., Halder G., Kim J., Hudson A., Carroll S. Ultrabithorax regulates genes at several levels of the wing-patterning hierarchy to shape the development of the Drosophila haltere . Genes and Development. 1998;12(10):1474–1482. doi: 10.1101/gad.12.10.1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lan F., Bayliss P. E., Rinn J. L., et al. A histone H3 lysine 27 demethylase regulates animal posterior development. Nature. 2007;449(7163):689–694. doi: 10.1038/nature06192. [DOI] [PubMed] [Google Scholar]
- 36.Terashima M., Ishimura A., Yoshida M., Suzuki Y., Sugano S., Suzuki T. The tumor suppressor Rb and its related Rbl2 genes are regulated by Utx histone demethylase. Biochemical and Biophysical Research Communications. 2010;399(2):238–244. doi: 10.1016/j.bbrc.2010.07.061. [DOI] [PubMed] [Google Scholar]
- 37.Han H., Xu D., Liu C., Claesson H.-E., Björkholm M., Sjöberg J. Interleukin-4-mediated 15-lipoxygenase-1 trans-activation requires UTX recruitment and H3K27me3 demethylation at the promoter in A549 cells. PLoS ONE. 2014;9(1, article e85085) doi: 10.1371/journal.pone.0085085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Denton D., Aung-Htut M. T., Lorensuhewa N., et al. UTX coordinates steroid hormone-mediated autophagy and cell death. Nature Communications. 2013;4, article 2916 doi: 10.1038/ncomms3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Toyota M., Suzuki H., Nishizaka T., Sato A., Yamashita T. Molecular mechanisms involved in epigenetic alterations in cancer. Gan To Kagaku Ryoho. 2010;37(9):1650–1653. [PubMed] [Google Scholar]
- 40.Kim J.-H., Sharma A., Dhar S. S., et al. UTX and MLL4 coordinately regulate transcriptional programs for cell proliferation and invasiveness in breast cancer cells. Cancer Research. 2014;74(6):1705–1717. doi: 10.1158/0008-5472.CAN-13-1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang X. L., Li C. L., Yang L. H., Jiang Z. M., Gui Y. T., Cai Z. M. Clinical significance of high expression with UTX in renal cell carcinoma. Beijing Da Xue Xue Bao. 2014;46(6):926–930. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of pathways involved in PPI network derived from KDM6A.