

Abstract
Cerebellar circuitry is important to controlling and modifying motor activity. It conducts the coordination and correction of errors in muscle contractions during active movements. Therefore, cerebrovascular lesions of the cerebellum or its pathways can cause diverse movement disorders, such as action tremor, Holmes’ tremor, palatal tremor, asterixis, and dystonia. The pathophysiology of abnormal movements after stroke remains poorly understood. However, due to the current advances in functional neuroimaging, it has recently been described as changes in functional brain networks. This review describes the clinical features and pathophysiological mechanisms in different types of movement disorders following cerebrovascular lesions in the cerebellar circuits.
Keywords: Movement disorder, Cerebellum, Cerebrovascular disorder, Strokes
INTRODUCTION
Cerebrovascular diseases and strokes are major causes of secondary movement disorders, although abnormal movements occur infrequently in association with stroke. It is important for clinicians to detect abnormal movements in stroke patients in lesion localizations and to determine basic pathophysiology. Strokes accompanied by abnormal movements are not limited to a specific vascular territory or lesion sites [1]. Movement disorders occur frequently when strokes involve the basal ganglia or their connections. However, they can occur with cerebrovascular lesions at diverse locations in the motor circuits. The cerebellar circuitry, like the basal ganglia circuitry, is also important for controlling and modifying motor activity. Therefore, cerebrovascular lesions of the cerebellum or its pathways can cause diverse movement disorders. These lesions typically result in the deterioration of coordination (ataxia, asynergia), misjudgment of distance (dysmetria), and intention tremors [2]. In addition to these typical symptoms of cerebellar dysfunction, various abnormal movements can occur in patients with cerebrovascular lesions of the cerebellum or its pathways. Common movement disorders in patients with stroke in the cerebellar circuitry are action tremor, Holmes’ tremor, palatal tremor, asterixis, and dystonia, but other movement disorders, such as stereotypy, can occur rarely (Table 1).
Table 1.
Common movement disorders after stroke in the cerebellar circuits
| Onset | Common phenotype | Involved circuit | Frequent location of stroke (except cerebellum) | |
|---|---|---|---|---|
| Cerebellar outflow tremor | Acute | Intention tremor, w/o postural tremor | Dentato-rubro-thalamic or dentato-rubro-olivary | Posterior thalamus |
| Holmes' tremor | Delayed (1-24 months) | Rest and intention tremor, w/o postural tremor | Both nigrostriatal dopaminergic and cerebello-thalamic/cerebello-olivary | Brainstem or thalamus |
| Palatal tremor | Delayed (1 week-49 months) | - | Dentato-rubro-olivary (Guillain-Mollaret tringle) | Brainstem |
| Asterixis | Acute | Unilateral or occasionally bilateral | Dentato-rubro-thalamic | Thalamus, basal ganglia, midbrain, or frontoparietal cortex |
| Dystonia | Delayed (1 month-15 years) | Focal or segmental | Cerebello-thalamic | Basal ganglia, thalamus, or brainstem |
| Stereotypy | Delayed (1 month) | - | - | Basal ganglia |
w/o: with or without.
Abnormal movements, including both acute and delayed phase complications, can appear in 1–4% of stroke patients [3]. However, the prevalence of movement disorders in patients with stroke in the cerebellar circuitry is uncertain. Post-stroke movement disorders are mostly reported as unusual cases or in series of case reports obtained retrospectively from stroke registries. In addition, stroke in the cerebellar circuitry comprises only a small proportion of these post-stroke movement disorders. Among 2,900 patients with acute stroke in the Lausanne Stroke Registry, only 29 cases of hyperkinetic movement disorders were found (1% prevalence), only 3 of which were related to cerebellar stroke [4]. Another stroke registry identified 56 acute or delayed (up to 1 year after the stroke) movement disorders from 1,500 patients with stroke (3.7% prevalence), and 5 of these cases were related to stroke in the cerebellar circuitry [5]. Therefore, it is no surprise that the natural history and pathophysiology of movement disorders in patients with stroke in the cerebellar circuitry are not well understood.
In this article, we summarize the current knowledge about the clinical features and pathophysiological mechanisms of different types of movement disorders following cerebrovascular lesions in the cerebellar circuits.
FUNCTIONAL ANATOMY AND PATHOPHYSIOLOGY
Cerebellar motor circuits
The motor system, which controls the entire range of human activity, encompasses a broad range of nervous system structures and pathways. Two parallel pathways, the cerebellar and basal ganglia circuits, control and modify motor activity. The basal ganglia are mainly concerned with learned, automatic behavior and with maintaining the background support or posture needed for voluntary motor activity, whereas the cerebellum conducts the coordination and correction of errors in muscle contractions during active movements [6]. The cerebellum plays a particularly important role in muscle activation at the correct time. Therefore, pathologic lesions in the cerebellum or its pathways can cause a loss of coordination and errors in the timing of muscle activations on the same side of the body. Typical neurologic symptoms in patients with lesions in the cerebellar circuitry are intention tremor, dysmetria, dysdiadochokinesia, gait ataxia, ataxic dysarthria, and nystagmus.
The anatomy of the cerebellar pathways is complicated, but there are simplified models [2]. In the current model, there are two circuits that are clinically significant. One is the main cortico-cerebellar-cortical circuit, and the other is the modulatory dentato-rubro-olivary circuit [the Guillain-Mollaret triangle (GMT)] (Figure 1).
Figure 1.
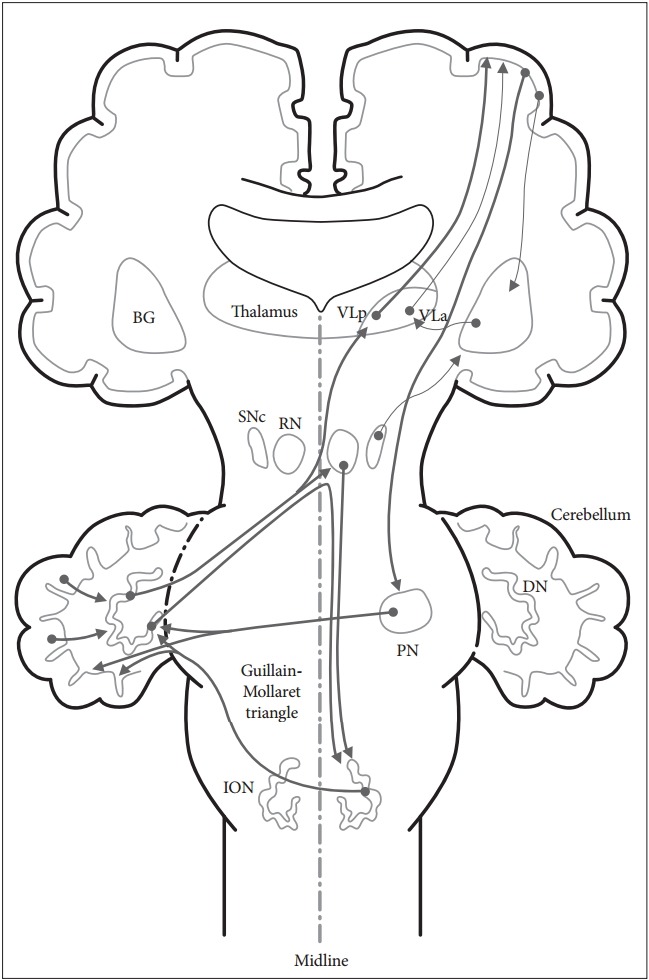
Schematic diagram of the cerebellar circuits involved in movement disorders.There are two cerebellar circuits that are clinically significant in movement disorders following cerebrovascular lesions. One is the cortico-cerebellar-cortical circuit and the other is the dentato-rubro-olivary circuit (the Guillain-Mollaret triangle). BG: basal ganglia, DN: dentate nucleus, ION: inferior olivary nucleus, PN: pontine nuclei, RN: red nucleus, SNc: substantia nigra pars compacta, VLa: ventral lateral anterior nucleus of the thalamus, VLp: ventral lateral posterior nucleus of the thalamus.
The cortico-cerebellar-cortical circuit
The main afferent fiber of the cerebellum is the cortico-ponto-cerebellar tract, which starts from the frontal lobe, creates synapses on the pontine nuclei, and finally arrives at the cerebellar cortex via the middle cerebellar peduncle. The main efferent fiber of the cerebellum is the dentato-rubro-thalamo-cortical tract, which starts from the dentate nucleus, passes through the superior cerebellar peduncle and the contralateral red nucleus, creates synapses with the ventrolateral thalamus, and finally arrives at the motor cortex.
The dentato-rubro-olivary circuit (the Guillain-Mollaret triangle)
This triangle is composed of a few fibers that connect the dentate nucleus with the contralateral red nucleus and inferior olivary nucleus (ION). Efferent fibers from the dentate nucleus pass through the superior cerebellar peduncle and create synapses with the contralateral red nucleus. Efferent fibers from the red nucleus traverse through the central tegmental tract and create synapses with the ipsilateral ION. Thereafter, efferent fibers from the ION pass through the inferior cerebellar peduncle and create synapses with the contralateral cerebellum to complete the triangular circuit. This triangle forms a feedback loop between the cerebellum and brainstem, and it serves to control spinal cord motor activity [7].
Both the basal ganglia circuit and the cerebellar circuit are subcortical loops that largely receive information from the cortex and provide information back to the cortex via the thalamus. Although both loops utilize the thalamus, their relay nuclei are separate, and the loops remain separate [2]. The ventral lateral posterior nucleus of the thalamus is a relay nucleus for the cerebellum, whereas the ventral lateral anterior nucleus of the thalamus is a relay nucleus for the basal ganglia.
Pathomechanism of movement disorders after the lesion of cerebellar circuit
Recent reports have suggested that most movement disorders occur from neural circuit dysfunctions and abnormalities in functional connectivity, rather than from a single lesion [8]. The pathophysiology of abnormal movements have been described as changes in functional brain networks due to the current advances in functional neuroimaging [8]. The destruction of the functional circuits to connect the neural network is critically involved in the expression of movement disorders [8].
The onset of abnormal movements after a stroke is very diverse, ranging from the day of stroke outbreak to a few years later. These relationships between the occurrence of stroke and the onset of abnormal movements depend on the type of movement disorder. Cerebellar tremor and asterixis seem to present in acute stages, but Holmes’ tremor, palatal tremor, and dystonia seem to occur after a time delay after the stroke in the cerebellar circuits. The duration from stroke onset to the appearance of delayed abnormal movements was several weeks to months in Holmes’ tremor [9], 2–49 months in palatal tremor [10], and 1 day to 5 years in dystonia [11]. However, the delay in the onset of movement disorders varies widely within each movement disorder. There was a negative correlation between age at the initial insult of static brain lesions and latency until subsequent movement disorders [12]. The latency from initial brain insult to the onset of abnormal movements was longer when the lesion occurred at a younger age [13]. This relationship might partly be explained by differences in brain metabolism with age and neuroplasticity [3,12].
The pathophysiology of movement disorders with acute onset after a stroke in the cerebellar circuity is probably related to acute disruption of cerebellar motor circuits. However, the pathophysiology of movement disorders with delayed onset after a stroke in the cerebellar circuitry remains poorly understood. One hypothesis for the delayed onset is that the time lag might indicate the time required for successful but unbalanced motor recovery and subsequent development of pathological pathways reflecting neuroplasticity [14-16]. Additionally, it could indicate the time required for plastic changes in the activities of neuronal synapses [14]. Other possible hypotheses are described in review articles published previously [3,13,17].
Recent advances in functional neuroimaging and neurophysiologic techniques have greatly enhanced our pathophysiological understanding of delayed movement disorders after stroke. Hypertrophic olivary degeneration has been associated with the development of delayed-onset palatal or Holmes’ tremor [9,18]. This degeneration is caused by a remote lesion occurring in the dentato-rubro-olivary pathway [9,18,19], and it usually appears at approximately 3 weeks following a brain lesion that blocks inhibitory pathways from the deep cerebellar nuclei to the inferior olive [20]. The inferior olive becomes hypertrophic, and its neurons enlarge gradually after a stroke. Three-dimensional binocular eye movement recordings recently demonstrated that oscillations in oculopalatal tremor arise from the hypertrophied inferior olive, and slow cerebellar learning plays a role in amplifying these oscillations [21]. Other examples include alterations in movement execution and the mental representation of movements in patients with unilateral dystonia secondary to a subcortical stroke, as shown on functional MRI [22]. Compared with control subjects, movements of the dystonic hand caused an increase in the activity of the bilateral cerebellum, as well as of the motor, premotor, and prefrontal cortex [22]. Alterations in the cerebellar functional networks in dystonia are in agreement with a previous imaging report using H2(15)O positron emission tomography (PET) in patients with post-stroke hemidystonia [23] and with a report using 18F fluorodeoxyglucose PET in patients with DYT1 dystonia [24].
CLINICAL FEATURES (PHENOMENOLOGY)
Common abnormal movements
Cerebellar outflow tremor
Tremor is characterized by an oscillatory rhythmical movement produced by involuntary contractions of agonist and antagonist muscles synchronously or alternatingly [25]. Tremor is classified as resting or action, and action tremor is divided into postural, isometric, or kinetic [26]. Intention tremor is a type of kinetic tremor, and it usually increases in amplitude as a moving object reaches the target. Cerebellar tremor is a dominant intention tremor, with a frequency mainly less than 5 Hz [26]. Postural tremor can appear, but rest tremor is commonly not identified in patients with cerebellar lesions [26]. Cerebellar tremor is usually present in the arms or the legs. The affected body part with cerebellar tremor is usually unilateral, segmental or multifocal, rather than focal or generalized [3]. Another form of cerebellar tremor is titubation, which normally appears in the trunk or head [27].
Different types of tremor are observed after stroke, and post-stroke tremor generally occurs on action [3]. Typical intention tremor and postural tremor are presumably generated within the dentato-rubrothalamo-cortical circuit or the GMT [28]. Common lesion locations of cerebellar tremors were the dentate nucleus, interpositus nucleus, and brachium conjunctivum [2]. Tremor appears to be rare due to lesions of the cerebellar cortex alone [2]. Lesions involving the posterior thalamus are also related to cerebellar circuits, as evidenced by successful alleviation of the tremor with a stereotaxic lesion of the nucleus ventralis intermedius of the thalamus, which is a relay nucleus for the cerebellum [29]. Cerebellar outflow tremor usually occurs in the acute stroke period [4].
The pathophysiology of cerebellar tremor is not yet thoroughly understood, but it is believed to be related to dysfunction of the cerebellar efferent pathways [27]. These pathways include the dentato-rubrothalamo-cortical and dentato-rubro-olivary circuits. MRI-based lesion-symptom mapping demonstrated that limb kinetic tremor might be correlated with cerebellar atrophy in intermediate and lateral zones [30]. Three possible mechanisms of cerebellar tremor are 1) serial voluntary corrections of positioning errors (serial dysmetria); 2) abnormality of transcortical and segmental proprioceptive feedback loops; and 3) the action of the central oscillators [2]. Each of these mechanisms seems to be important to some component of body oscillations in patients with cerebellar dysfunction under various circumstances [2].
Holmes’ tremor
Holmes’ tremor is an unusual tremor syndrome characterized by a combination of rest and intention tremor, involving the proximal and distal parts of the upper extremities with large amplitudes [26,31]. Postural tremor is also present in many cases [26]. The terms midbrain and Holmes’ tremor have been used to express a rest tremor that is more severe on postural maintenance and most severe at intention [16]. The frequency is usually approximately 4.5 Hz or less, and it can sometimes be irregular [31]. It generally appears in one upper limb, accompanied by dysmetria and dysdiadochokinesia on the ipsilateral side [31].
Holmes’ tremor is a rare symptomatic tremor; therefore, it has been reported in small case series. Stroke, as well as trauma in the brainstem, is a frequent cause of Holmes’ tremor. In one report of 3 cases of Holmes’ tremor following stroke, the lesion sites were the superior cerebellar peduncle, midbrain tegmentum, and posterior thalamus [32]. Most patients with Holmes’ tremor after brainstem stroke have accompanied hypertrophy of the ION, suggesting that interruption of the dentato-rubro-olivary circuit might play an important role in the development of Holmes’ tremor [9,19]. Holmes’ tremor was also reported in patients with hemi-cerebellar infarction [33]. Therefore, the human central nervous system might be able to produce Holmes’ tremor in the absence of unilateral cerebellar input. There is usually a various delay (1–24 months) from the onset of stroke to the first occurrence of the tremor [26].
Holmes’ tremor occurs when a lesion involves both the dopaminergic (nigrostriatal fibers) and the cerebello-thalamic/cerebello-olivary pathways [15,31], as demonstrated by pathoanatomic and PET data [26,34-36]. Damage to the cerebello-thalamic pathways might be related to kinetic/postural tremor, and damage to the nigrostriatal pathways might be related to tremor at rest.
Palatal tremor
Palatral tremor consists of slow, rhythmic movements of the soft palate and sometimes of other muscles in the pharynx, larynx, lower face, and trunk [18]. The frequency of palatal tremor is usually 1–3 Hz. There are two forms of palatal tremor: essential and symptomatic. No cause could be identified in essential palatal tremor, which constituted one-fourth of patients with palatal tremor [37]. Symptomatic palatal tremor usually occurs with a lesion in the brainstem or cerebellum [37].
The common causes of symptomatic palatal tremor include stroke, trauma, tumor, multiple sclerosis, Behçet’s disease, and encephalitis [10]. Stroke in the brainstem or cerebellum is the most common cause of symptomatic palatal tremor [10], and brainstem strokes represent 60–70% of cases [38]. Stroke in the frontoparietal cortex can rarely cause palatal tremor, likely related to epileptiform activity in the motor cortex [39,40]. Palatal tremor usually develops some time (1 week–49 months) after lesion onset in the brain [22].
Symptomatic palatal tremor is frequently caused by brainstem stroke disrupting dentato-rubro-olivary fibers or central tegmental tracts [18]. It is usually associated with hypertrophic degeneration of the ION, which can be seen on MR images as a high signal on T2- or proton density-weighted images with ION enlargement [18]. Hypertrophic olivary degeneration is a trans-synaptic neuronal degeneration in which degeneration is accompanied by hypertrophy [41]. Increased signals from the ION are present within one month of disease onset, and the high signal continues for several years [18]. Olivary hypertrophy typically appears 10–18 months following the lesion, but can occur as early as 6 months, and it vanishes by 4 years [18]. The pathophysiology of palatal tremor is believed to be denervation supersensitivity of the inferior olive due to lesions of the dentato-rubro-olivary pathways, and the inferior olive is considered to perform the role as pacemaker [39]. Changes in the ION appeared on MR images from patients with lesions in the GMT, and they were well correlated with chronological and morphometric changes on histopathologic analysis [18,42,43].
Asterixis
Asterixis, which is a negative myoclonus, is an involuntary movement of the hand characterized by brief flapping of the outstretched limb due to intermittent failure to maintain sustained muscle contraction [44,45]. It is commonly bilateral, and in these cases, it is observed in metabolic encephalopathy. Unilateral asterixis is more uncommon, and it usually occurs in patients with focal structural brain lesions [46]. Stroke is the most common cause of unilateral asterixis, occurring on the side contralateral to the lesion [46]. Cerebellar lesions sometimes cause ipsilateral asterixis, which can be explained by the decussation of dentato-rubro-thalamo-cortical fibers before they pass through or create synapses with the red nucleus [44]. Bilateral asterixis occasionally occurs in patients with unilateral lesions, indicating that posture maintenance is not absolutely controlled unilaterally [44].
A neuroimaging study showed that the lesion locations causing post-stroke asterixis were the thalamus, frontal lobe, midbrain, cerebellum, and lenticulocapsular area in order of frequency [44]. The incidence of post-stroke asterixis remains unknown, and cerebellar stroke constituted only 2 of 30 cases of post-stroke asterixis [44]. However, the cerebellar circuitry might be involved in many patients with post-stroke asterixis because ataxia was present in all patients with thalamic and posterior fossa stroke. Therefore, lesions in the cerebellar circuits were responsible for post-stroke asterixis in 23 of 30 cases of post-stroke asterixis [44]. Asterixis usually occurred in the acute phase of stroke [4,45].
Asterixis can be caused by the abnormal control of arm posture maintenance owing to dysfunctional regulation of the brainstem-spinal pathways from the cerebello-brainstem-thalamo-cortical system [44]. Tonic control of the limbs is associated with various brainstem-spinal tracts which are regulated by supratentoral structures [45]. The ventral lateral thalamic nucleus is the area where the cerebello-rubral or vestibulo-cerebellar fibers merge, and it is also connected to the prefrontal area [45]. Therefore, the development of asterixis in patients with lesions of the cerebellum or thalamus can be interpreted as the failure of supraspinal control over brainstem-spinal pathways.
Dystonia
Dystonia is a sustained muscle contraction causing repetitive, patterned and twisting movements or abnormal postures [3,7]. Dystonia was the second most frequent form of hyperkinetic movement disorder that occurred during and after stroke in the Lausanne Stroke Registry [4]. The form of post-stroke dystonia is typically hemidystonia on the contralateral side of the lesion. However, it can be focal, appearing in one hand or foot or in the cranial area, and can be segmental or even generalized [3]. The basal ganglia was the most frequent lesion location in patients with post-stroke dystonia [5], usually resulting in hemidystonia [47,48]. Cerebellar strokes were only associated with focal hand dystonia and segmental dystonia involving the upper extremity in the Lausanne Stroke Registry [4]. However, cerebellar stroke could cause various forms of dystonia, such as cervical dystonia [49,50], oromandibular dystonia [51,52], and blepharospasm [53].
Common lesion sites of post-stroke dystonia were the putamen, caudate, pallidum, internal capsule, thalamus, midbrain, and cortex [4,11]. Additionally, cerebellar strokes have been reported to cause dystonia [4,49-53]. Cerebellar stroke accounted for 2 of 8 cases of post-stroke dystonia in one report [4], but no cerebellar stroke cases were found among 16 patients with post-stroke dystonia in another report [5]. Post-stroke dystonia is frequently delayed (1 month–15 years) in onset [3].
Primary dystonia is usually associated with basal ganglia dysfunction and impairment in sensorimotor integration [8]. Therefore, it is natural that dystonia could occur due to damage to the basal ganglia loop [54]. However, a recent report suggested that the cerebellum might also be involved in the pathogenesis of dystonia [8]. A torsion dystonia-related pattern identified in the analysis of rest-state metabolic imaging in patients with primary dystonia, and the metabolic activity in these patients was increased in the cerebellum, as well as in the basal ganglia and the supplementary motor area [55]. Furthermore, nonmanifesting dystonia mutation carriers showed diminished integrity of the cerebello-thalamo-cortical tracts on magnetic resonance diffusion tensor imaging studies [56]. These imaging findings suggest a possible mechanism in which abnormal connectivity of the cerebello-thalamo-cortical tracts plays an important role in the clinical symptoms of dystonia.
Complex movement disorders
Movement disorders after stroke in the cerebellar circuits can have a complex form and can encompass several components, resulting in difficulty in classifying them. For example, post-stroke dystonia can be accompanied by other abnormal movements, such as tremor, myoclonus, or athetoid movements, and the resulting movements can have a complicated form, such as dystonic tremor or dystonic myoclonus [16]. Cerebrovascular lesions in the cerebellum and thalamus have been reported to cause “jerk dystonic unsteady hand”, which is a form of post-stroke dystonic myoclonus [3,4].
Miscellaneous abnormal movements
Stereotypy
Stereotypy is repetitive, non-goal-directed movement with the same form that continues for a certain period of time [57]. Stereotypy occurs commonly in patients with tardive dyskinesia, schizophrenia, intellectual disability, and autism [25]. One of the rare causes of stereotypy is stroke [57]. The frontal lobe and basal ganglia system seem to play important roles in stereotypy [58], and there have been a few reports of stereotypic behaviors occurring after a putaminal infarction [57,59]. The cerebellum has also been implicated in stereotypy [60], and there is a report of stereotypy after cerebellar infarction [61].
Restless legs syndrome
Restless legs syndrome (RLS) is a common neurologic disease characterized by a desire to move the legs due to unpleasant or uncomfortable feelings in them [12]. RLS was found in patients with stroke when the lesions involved diverse structures, such as the basal ganglia and/or corona radiata, pons, thalamus, internal capsule, and rarely, the cerebral cortex [62]. RLS did not occur in patients with cerebellar strokes [62]. There was a report that patients with pre-stroke RLS developed cerebellar infarctions, but post-stroke RLS was not related to a lesion in the cerebellum [63].
CONCLUSIONS
Movement disorders infrequently occur in association with cerebrovascular lesions in the cerebellar circuits. Nonetheless, recognition of abnormal movements in patients with cerebellar stroke could be important to localizing lesions, in determining the basic etiology, and in understanding the underlying pathophysiology. Cerebellar outflow tremor and asterixis can occur immediately after acute stroke in cerebellar circuits. In contrast, Holmes’ tremor, palatal tremor, and dystonia are more often delayed sequelae of cerebellar stroke. Movement disorders with acute onset after cerebellar stroke are likely related to acute disruption of cerebellar motor circuits. However, the pathophysiology of movement disorders with delayed onset after cerebellar stroke remains to be clarified.
Considerable evidence has indicated that these abnormal movements in patients with stroke represent circuit disorders resulting from neural circuit dysfunctions and abnormalities in functional connectivity [8]. However, the predisposing factors to developing abnormal movements after a stroke in the cerebellar circuitry have been poorly reported. Future studies should attempt to identify the factors that predispose cerebellar stroke patients to developing abnormal movements. Because involvement of the cerebellar circuits by a stroke rarely causes the development of abnormal movement, except for cerebellar outflow tremor, multicenter studies using advanced neuroimaging and neurophysiological techniques will be necessary to study the disease progress, pathophysiology, and management of these movement disorders.
REFERENCES
- 1.Edlow JA, Selim MH. Atypical presentations of acute cerebrovascular syndromes. Lancet Neurol. 2011;10:550–560. doi: 10.1016/S1474-4422(11)70069-2. [DOI] [PubMed] [Google Scholar]
- 2.Fahn S, Jankovic J, Hallett M. Motor control: physiology of voluntary and involuntary movements. In: Fahn S, Jankovic J, Hallett M, editors. Principles and practice of movement disorders. 2nd ed. Philadelphia, PA: Elsevier Saunders; 2011. pp. 36–54. [Google Scholar]
- 3.Mehanna R, Jankovic J. Movement disorders in cerebrovascular disease. Lancet Neurol. 2013;12:597–608. doi: 10.1016/S1474-4422(13)70057-7. [DOI] [PubMed] [Google Scholar]
- 4.Ghika-Schmid F, Ghika J, Regli F, Bogousslavsky J. Hyperkinetic movement disorders during and after acute stroke: the Lausanne Stroke Registry. J Neurol Sci. 1997;146:109–116. doi: 10.1016/s0022-510x(96)00290-0. [DOI] [PubMed] [Google Scholar]
- 5.Alarcón F, Zijlmans JC, Dueñas G, Cevallos N. Post-stroke movement disorders: report of 56 patients. J Neurol Neurosurg Psychiatry. 2004;75:1568–1574. doi: 10.1136/jnnp.2003.011874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Westmoreland BF, Benarroch EE, Daube JR, Reagan TJ, Sandok BA. The motor system. In: Westmoreland BF, Benarroch EE, Daube JR, Reagan TJ, Sandok BA, editors. Medical neuroscience: an approach to anatomy, pathology, and physiology by systems and levels. 3rd ed. Boston: Little, Brown and Co; 1994. pp. 167–207. [Google Scholar]
- 7.Bansil S, Prakash N, Kaye J, Wrigley S, Manata C, Stevens-Haas C, et al. Movement disorders after stroke in adults: a review. Tremor Other Hyperkinet Mov (N Y) 2012 Mar 20; doi: 10.7916/D86W98TB. [Epub]. http://dx.doi.org/10.7916/D86W98TB. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holtbernd F, Eidelberg D. Functional brain networks in movement disorders: recent advances. Curr Opin Neurol. 2012;25:392–401. doi: 10.1097/WCO.0b013e328355aa94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang YW, Chang FC, Tsai CH, Wu JC, Lu CS, Kuo CC, et al. Clinical and magnetic resonance imaging manifestations of Holmes tremor. Acta Neurol Taiwan. 2005;14:9–15. [PubMed] [Google Scholar]
- 10.Deuschl G, Toro C, Valls-Solé J, Zeffiro T, Zee DS, Hallett M. Symptomatic and essential palatal tremor. 1. Clinical, physiological and MRI analysis. Brain. 1994;117(Pt 4):775–788. doi: 10.1093/brain/117.4.775. [DOI] [PubMed] [Google Scholar]
- 11.Choi YC, Lee MS, Choi IS. Delayed-onset focal dystonia after stroke. Yonsei Med J. 1993;34:391–396. doi: 10.3349/ymj.1993.34.4.391. [DOI] [PubMed] [Google Scholar]
- 12.Allen RP, Picchietti D, Hening WA, Trenkwalder C, Walters AS, Montplaisi J, et al. Restless legs syndrome: diagnostic criteria, special considerations, and epidemiology. A report from the restless legs syndrome diagnosis and epidemiology workshop at the National Institutes of Health. Sleep Med. 2003;4:101–119. doi: 10.1016/s1389-9457(03)00010-8. [DOI] [PubMed] [Google Scholar]
- 13.Scott BL, Jankovic J. Delayed-onset progressive movement disorders after static brain lesions. Neurology. 1996;46:68–74. doi: 10.1212/wnl.46.1.68. [DOI] [PubMed] [Google Scholar]
- 14.Kim JS. Delayed onset mixed involuntary movements after thalamic stroke: clinical, radiological and pathophysiological findings. Brain. 2001;124(Pt 2):299–309. doi: 10.1093/brain/124.2.299. [DOI] [PubMed] [Google Scholar]
- 15.Béjot Y, Giroud M, Moreau T, Benatru I. Clinical spectrum of movement disorders after stroke in childhood and adulthood. Eur Neurol. 2012;68:59–64. doi: 10.1159/000336740. [DOI] [PubMed] [Google Scholar]
- 16.Handley A, Medcalf P, Hellier K, Dutta D. Movement disorders after stroke. Age Ageing. 2009;38:260–266. doi: 10.1093/ageing/afp020. [DOI] [PubMed] [Google Scholar]
- 17.Louis ED, Lynch T, Ford B, Greene P, Bressman SB, Fahn S. Delayed-onset cerebellar syndrome. Arch Neurol. 1996;53:450–454. doi: 10.1001/archneur.1996.00550050080027. [DOI] [PubMed] [Google Scholar]
- 18.Goyal M, Versnick E, Tuite P, Cyr JS, Kucharczyk W, Montanera W, et al. Hypertrophic olivary degeneration: meta-analysis of the temporal evolution of MR findings. AJNR Am J Neuroradiol. 2000;21:1073–1077. [PMC free article] [PubMed] [Google Scholar]
- 19.Kipfer S, Frigerio SB. Post-ischemic stroke Holmes’ tremor of the upper limb. Mov Disord. 2013;28:1347. doi: 10.1002/mds.25621. [DOI] [PubMed] [Google Scholar]
- 20.Sanverdi SE, Oguz KK, Haliloglu G. Hypertrophic olivary degeneration in children: four new cases and a review of the literature with an emphasis on the MRI findings. Br J Radiol. 2012;85:511–516. doi: 10.1259/bjr/60727602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shaikh AG, Hong S, Liao K, Tian J, Solomon D, Zee DS, et al. Oculopalatal tremor explained by a model of inferior olivary hypertrophy and cerebellar plasticity. Brain. 2010;133(Pt 3):923–940. doi: 10.1093/brain/awp323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lehéricy S, Gerardin E, Poline JB, Meunier S, Van de Moortele PF, Le Bihan D, et al. Motor execution and imagination networks in post-stroke dystonia. Neuroreport. 2004;15:1887–1890. doi: 10.1097/00001756-200408260-00010. [DOI] [PubMed] [Google Scholar]
- 23.Ceballos-Baumann AO, Passingham RE, Marsden CD, Brooks DJ. Motor reorganization in acquired hemidystonia. Ann Neurol. 1995;37:746–757. doi: 10.1002/ana.410370608. [DOI] [PubMed] [Google Scholar]
- 24.Eidelberg D, Moeller JR, Antonini A, Kazumata K, Nakamura T, Dhawan V, et al. Functional brain networks in DYT1 dystonia. Ann Neurol. 1998;44:303–312. doi: 10.1002/ana.410440304. [DOI] [PubMed] [Google Scholar]
- 25.Fahn S, Jankovic J, Hallett M. Clinical overview and phenomenology of movement disorders. In: Fahn S, Jankovic J, Hallett M, editors. Principles and practice of movement disorders. 2nd ed. Philadelphia, PA: Elsevier Saunders; 2011. pp. 1–35. [Google Scholar]
- 26.Deuschl G, Bain P, Brin M. Consensus statement of the movement disorder society on tremor. Ad Hoc Scientific Committee. Mov Disord. 1998;13 Suppl 3:2–23. doi: 10.1002/mds.870131303. [DOI] [PubMed] [Google Scholar]
- 27.Javalkar V, Khan M, Davis DE. Clinical manifestations of cerebellar disease. Neurol Clin. 2014;32:871–879. doi: 10.1016/j.ncl.2014.07.012. [DOI] [PubMed] [Google Scholar]
- 28.Elble R, Deuschl G. Milestones in tremor research. Mov Disord. 2011;26:1096–1105. doi: 10.1002/mds.23579. [DOI] [PubMed] [Google Scholar]
- 29.Lyons KE, Pahwa R. Deep brain stimulation and tremor. Neurotherapeutics. 2008;5:331–338. doi: 10.1016/j.nurt.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Timmann D, Brandauer B, Hermsdörfer J, Ilg W, Konczak J, Gerwig M, et al. Lesion-symptom mapping of the human cerebellum. Cerebellum. 2008;7:602–606. doi: 10.1007/s12311-008-0066-4. [DOI] [PubMed] [Google Scholar]
- 31.Puschmann A, Wszolek ZK. Diagnosis and treatment of common forms of tremor. Semin Neurol. 2011;31:65–77. doi: 10.1055/s-0031-1271312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berkovic SF, Bladin PF. Rubral tremor: clinical features and treatment of three cases. Clin Exp Neurol. 1984;20:119–128. [PubMed] [Google Scholar]
- 33.Brittain JS, Jenkinson N, Holland P, Joundi RA, Green AL, Aziz TZ. Development of Holmes’ tremor following hemicerebellar infarction. Mov Disord. 2011;26:1957–1959. doi: 10.1002/mds.23704. [DOI] [PubMed] [Google Scholar]
- 34.Krack P, Deuschl G, Kaps M, Warnke P, Schneider S, Traupe H. Delayed onset of “rubral tremor” 23 years after brainstem trauma. Mov Disord. 1994;9:240–242. doi: 10.1002/mds.870090225. [DOI] [PubMed] [Google Scholar]
- 35.Defer GL, Remy P, Malapert D, Ricolfi F, Samson Y, Degos JD. Rest tremor and extrapyramidal symptoms after midbrain haemorrhage: clinical and 18F-dopa PET evaluation. J Neurol Neurosurg Psychiatry. 1994;57:987–989. doi: 10.1136/jnnp.57.8.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Remy P, de Recondo A, Defer G, Loc’h C, Amarenco P, Planté-Bordeneuve V, et al. Peduncular ‘rubral’ tremor and dopaminergic denervation: a PET study. Neurology. 1995;45(3 Pt 1):472–477. doi: 10.1212/wnl.45.3.472. [DOI] [PubMed] [Google Scholar]
- 37.Deuschl G, Wilms H. Clinical spectrum and physiology of palatal tremor. Mov Disord. 2002;17 Suppl 2:S63–S66. doi: 10.1002/mds.10062. [DOI] [PubMed] [Google Scholar]
- 38.Borruat FX. Oculopalatal tremor: current concepts and new observations. Curr Opin Neurol. 2013;26:67–73. doi: 10.1097/WCO.0b013e32835c60e6. [DOI] [PubMed] [Google Scholar]
- 39.Salazar R, Miller D. Symptomatic palatal tremor of cortical origin due to stroke. J Clin Neurosci. 2013;20:757–759. doi: 10.1016/j.jocn.2012.05.049. [DOI] [PubMed] [Google Scholar]
- 40.Jung HJ, Choi SM, Lee JK, Lee SH, Kim BC. Palatal tremor as a manifestation of epilepsia partialis continua caused by acute precentral gyral infarction. J Clin Neurosci. 2013;20:1460–1461. doi: 10.1016/j.jocn.2012.07.024. [DOI] [PubMed] [Google Scholar]
- 41.Sen D, Gulati YS, Malik V, Mohimen A, Sibi E, Reddy DC. MRI and MR tractography in bilateral hypertrophic olivary degeneration. Indian J Radiol Imaging. 2014;24:401–405. doi: 10.4103/0971-3026.143902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goto N, Kaneko M. Olivary enlargement: chronological and morphometric analyses. Acta Neuropathol. 1981;54:275–282. doi: 10.1007/BF00697000. [DOI] [PubMed] [Google Scholar]
- 43.Goto N, Kakimi S, Kaneko M. Olivary enlargement: stage of initial astrocytic changes. Clin Neuropathol. 1988;7:39–43. [PubMed] [Google Scholar]
- 44.Kim JS. Asterixis after unilateral stroke: lesion location of 30 patients. Neurology. 2001;56:533–536. doi: 10.1212/wnl.56.4.533. [DOI] [PubMed] [Google Scholar]
- 45.Siniscalchi A, Gallelli L, Di Benedetto O, De Sarro G. Asterixis as a presentation of cerebellar ischemic stroke. West J Emerg Med. 2012;13:507–508. doi: 10.5811/westjem.2012.1.6900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tatu L, Moulin T, Martin V, Chavot D, Rousselot JP, Monnier G, et al. [Unilateral asterixis and focal brain lesions. 12 cases] Rev Neurol (Paris) 1996;152:121–127. [PubMed] [Google Scholar]
- 47.Pettigrew LC, Jankovic J. Hemidystonia: a report of 22 patients and a review of the literature. J Neurol Neurosurg Psychiatry. 1985;48:650–657. doi: 10.1136/jnnp.48.7.650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marsden CD, Obeso JA, Zarranz JJ, Lang AE. The anatomical basis of symptomatic hemidystonia. Brain. 1985;108(Pt 2):463–483. doi: 10.1093/brain/108.2.463. [DOI] [PubMed] [Google Scholar]
- 49.Zadro I, Brinar VV, Barun B, Ozretić D, Habek M. Cervical dystonia due to cerebellar stroke. Mov Disord. 2008;23:919–920. doi: 10.1002/mds.21981. [DOI] [PubMed] [Google Scholar]
- 50.Usmani N, Bedi GS, Sengun C, Pandey A, Singer C. Late onset of cervical dystonia in a 39-year-old patient following cerebellar hemorrhage. J Neurol. 2011;258:149–151. doi: 10.1007/s00415-010-5685-2. [DOI] [PubMed] [Google Scholar]
- 51.Waln O, LeDoux MS. Delayed-onset oromandibular dystonia after a cerebellar hemorrhagic stroke. Parkinsonism Relat Disord. 2010;16:623–625. doi: 10.1016/j.parkreldis.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 52.Akin A, Yilmaz R, Selcuk F, Akbostancı MC. Sudden onset of oromandibular dystonia after cerebellar stroke. Tremor Other Hyperkinet Mov (N Y) 2014;4:262. doi: 10.7916/D8C24TN3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.O’Rourke K, O’Riordan S, Gallagher J, Hutchinson M. Paroxysmal torticollis and blepharospasm following bilateral cerebellar infarction. J Neurol. 2006;253:1644–1645. doi: 10.1007/s00415-006-0202-3. [DOI] [PubMed] [Google Scholar]
- 54.Krystkowiak P, Martinat P, Defebvre L, Pruvo JP, Leys D, Destée A. Dystonia after striatopallidal and thalamic stroke: clinicoradiological correlations and pathophysiological mechanisms. J Neurol Neurosurg Psychiatry. 1998;65:703–708. doi: 10.1136/jnnp.65.5.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Poston KL, Eidelberg D. Functional brain networks and abnormal connectivity in the movement disorders. Neuroimage. 2012;62:2261–2270. doi: 10.1016/j.neuroimage.2011.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Argyelan M, Carbon M, Niethammer M, Ulug AM, Voss HU, Bressman SB, et al. Cerebellothalamocortical connectivity regulates penetrance in dystonia. J Neurosci. 2009;29:9740–9747. doi: 10.1523/JNEUROSCI.2300-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Edwards MJ, Lang AE, Bhatia KP. Stereotypies: a critical appraisal and suggestion of a clinically useful definition. Mov Disord. 2012;27:179–185. doi: 10.1002/mds.23994. [DOI] [PubMed] [Google Scholar]
- 58.Stern E, Silbersweig DA, Chee KY, Holmes A, Robertson MM, Trimble M, et al. A functional neuroanatomy of tics in Tourette syndrome. Arch Gen Psychiatry. 2000;57:741–748. doi: 10.1001/archpsyc.57.8.741. [DOI] [PubMed] [Google Scholar]
- 59.Maraganore DM, Lees AJ, Marsden CD. Complex stereotypies after right putaminal infarction: a case report. Mov Disord. 1991;6:358–361. doi: 10.1002/mds.870060418. [DOI] [PubMed] [Google Scholar]
- 60.Grossman R, Verobyev L. The neurobiology of stereotypic behaviors and stereotypic movement disorders. Psychiatr Ann. 1998;28:317–326. [Google Scholar]
- 61.Lee D, Lee D, Ahn TB. Stereotypy after cerebellar infarction. J Neurol Sci. 2014;344:227–228. doi: 10.1016/j.jns.2014.06.019. [DOI] [PubMed] [Google Scholar]
- 62.Lee SJ, Kim JS, Song IU, An JY, Kim YI, Lee KS. Poststroke restless legs syndrome and lesion location: anatomical considerations. Mov Disord. 2009;24:77–84. doi: 10.1002/mds.22303. [DOI] [PubMed] [Google Scholar]
- 63.Gupta A, Shukla G, Mohammed A, Goyal V, Behari M. Restless legs syndrome, a predictor of subcortical stroke: a prospective study in 346 stroke patients. Sleep Med. 2015 Jul 20; doi: 10.1016/j.sleep.2015.05.025. [Epub]. http://dx.doi.org/10.1016/j.sleep.2015.05.025. [DOI] [PubMed] [Google Scholar]