

Abstract
Movement disorders are primarily associated with the basal ganglia and the thalamus; therefore, movement disorders are more frequently manifest after stroke compared with neurological injuries associated with other structures of the brain. Overall clinical features, such as types of movement disorder, the time of onset and prognosis, are similar with movement disorders after stroke in other structures. Dystonia and chorea are commonly occurring post-stroke movement disorders in basal ganglia circuit, and these disorders rarely present with tremor. Rarer movement disorders, including tic, restless leg syndrome, and blepharospasm, can also develop following a stroke. Although the precise mechanisms underlying the pathogenesis of these conditions have not been fully characterized, disruptions in the crosstalk between the inhibitory and excitatory circuits resulting from vascular insult are proposed to be the underlying cause. The GABA (gamma-aminobutyric acid)ergic and dopaminergic systems play key roles in post-stroke movement disorders. This review summarizes movement disorders induced by basal ganglia and thalamic stroke according to the anatomical regions in which they manifest.
Keywords: Basal ganglia, Thalamus, Movement disorder, Cerebrovascular disease
INTRODUCTION
Movement disorders that manifest following a stroke are most frequently associated with lesions in the basal ganglia (44%) and the thalamus (37%) [1]. The probability of developing a movement disorder after an infarction of deep nuclei infarction, such as one affecting the basal ganglia and thalamus, is three times greater compared with a surface infarction [2]. The characteristic clinical features of movement disorders induced by lesions in the basal ganglia circuit are similar to those observed in post-stroke movement disorders associated with other regions of the brain. Movement disorders resulting from basal ganglia infarction typically present unilaterally in the contralateral side of the ischemic lesion, and there is a delay in the onset of these conditions after a stroke occurs. Movement disorders present with various symptoms, including dystonia, chorea/ballism, and tremor, that spontaneously resolve within several years. The frequencies at which various movement disorders appear after stoke are shown in Table 1 [1,3]. Small vessel infarctions in the basal ganglia and thalamus are the most common types of stroke implicated in movement disorders. Movement disorders that present after cardioembolism, large artery infarction, and intracerebral hemorrhage have also been reported. Strokes confined to the basal ganglia circuit can induce hyperkinetic movement disorders, including dystonia, chorea, asterixis, and hypokinetic movement disorders such as vascular Parkinsonism. Similar to movement disorders mediated by other regions of the brain, the precise mechanisms underlying movement disorders associated with stroke in basal ganglia circuit are not fully understood.
Table 1.
The frequency of post-stroke movement disorders
| Lausanne stroke registry (%) | Ecuador stroke registry (%) | |
|---|---|---|
| Chorea | 38 | 36 |
| Dystonia | 17 | 29 |
| Limb shaking | 10 | |
| Myoclonus-dystonia | 10 | |
| Stereotypic | 7 | |
| Asterixis | 7 | |
| Tremor | 3 | 25 |
| Hemi-akathisia | 3 | |
| Dysarthria, dyskinetic hand | 2 | |
| Parkinsonism | 10 |
FUNCTIONAL ANATOMY
Basal ganglia-thalamocortical circuit
The basal ganglia comprise the principal subcortical component of the circuits that link the cerebral cortex and the thalamus. The basal ganglia comprise four deep nuclei: 1) The putamen, which is the source of input to the basal ganglia, receives input from fibers emanating from the motor cortex. 2) The internal segment of the globus pallidus, also referred to as the globus pallidus interna (GPi), and the substantia nigra pars reticulata (SNr) are the major sources of output from the basal ganglia, and these structures receive projections emanating from the putamen. 3) A portion of the external globus pallidus and subthalamic nucleus (STN) function as the relay nuclei in the indirect pathway. 4) The ventrolateral (VL) thalamus receives projections from the output structures (GPi and SNr) and projects them back to the motor cortex [4]. The basal ganglia reach the cerebral cortex via the ventral anterior nucleus of thalamus, thereby contributing to the pathway that mediates the preparatory phase of movements (Figure 1).
Figure 1.
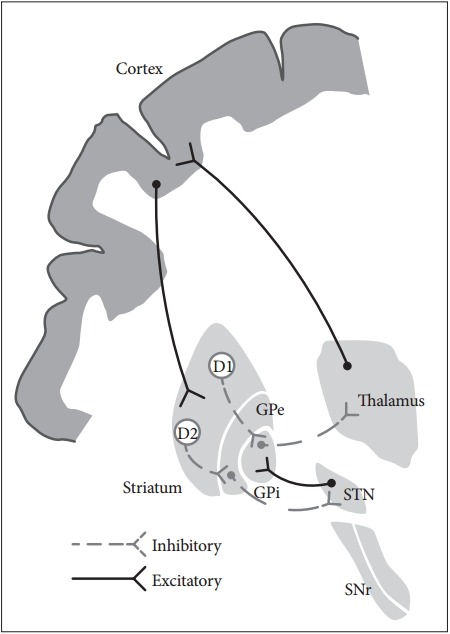
The anatomic connection of the basal ganglia circuit. Two types of pathways (direct and indirect pathway) are originated from striatum to basal ganglia output nuclei. Hyperdirect pathway relays direct cortical projection to the STN. GPe: globus pallidus externa, GPi: globus pallidus interna, SNr: substantia nigra pars reticulata, STN: subthalamic nucleus.
The nuclei of the basal ganglia receive input from the cerebral cortex and send output to the cerebellum and spinal cord via the thalamus. The intrinsic basal ganglia connections are categorized into two connecting pathways: the direct pathway and the indirect pathway. The direct pathway, which uses gamma-aminobutyric acid (GABA) to perform its functions, emanates from the putamen and projects directly into the output regions (GPi/SNr). When phasic excitatory input activates the direct pathway from the striatum to the pallidum, the tonically active neurons in the pallidum are suppressed, and the thalamocortical drive is ultimately activated. In contrast, the indirect pathway, which originates from the putamen, projects to the basal ganglia output region through a network that directly involves the globus pallidus externa (GPe) and the STN. The indirect pathway comprises several connections between the striatum and the GPe, the GPe and the interna, and the GPi and the STN. These circuits enhance the inhibition of the thalamocortical drive. Thus, the direct pathway provides positive feedback, and the indirect pathway provides negative feedback in the circuit between basal ganglia and thalamus [5].
A growing body of recent evidence suggests that a novel hyperdirect pathway relays direct cortical projections to the STN [6]. This dynamic cortico-STN-pallidal hyperdirect pathway has been referred to as the center-surround model of basal ganglia function [7]. When voluntary movement is initiated, the cortico-STN-pallidal hyperdirect pathway activates GPi neurons, which ultimately results in the inhibition of the thalamocortical tract. The direct pathway subsequently conveys a signal to the GPi and inhibits pallidal neurons in the central region. Inhibition of the pallidal neurons relieves the inhibition of select thalamocortical neurons. The indirect pathway ultimately reaches the GPi, thereby activating neurons and reestablishing inhibition of the thalamocortical tract. As a result of this sequence of events, a specific motor program can be initiated, executed, and terminated. This novel mechanism mediated by the basal ganglia emphasizes the functional significance of the STN in motor control.
The temporal relationship between stroke onset and movement disorders (acute or delayed onset)
The latency of movement disorders after basal ganglia infarction varies according to the type of movement disorder (Table 2). Ballism, chorea, myoclonus, and asterixis primarily develop as acute manifestations, whereas tremor and dystonia tend to manifest as delayed onset movement disorders after basal ganglia infarction. Ballism and chorea exhibit the shortest delay in onset and present with within a few days after stroke (mean: 4.3 days) [8]. Tremor is associated with the longest delay in onset, although a few cases of acute onset tremor have been reported (from 1 month to 4 years) [9]. Dystonia is another movement disorder with a delayed onset, but it tends to manifest earlier than tremor (mean: 9.5 months) [10]. The development of features resembling Parkinson’s, a disorder referred to as vascular Parkinsonism, occurs within the first year after basal ganglia infarction [11].
Table 2.
Commonly reported movement disorders occurring after stroke in the basal ganglia circuit
| Onset | Common clinical phenotype | Location of stroke | |
|---|---|---|---|
| Chorea/ballism | Acute | Hemichorea/hemiballism (m/c) | Putamen |
| STN | |||
| Caudate nucleus | |||
| Dystonia | Delayed (9.5 months) | Hemidystonia | Putamen |
| Focal dystonia | Thalamus (posterolateral nucleus) | ||
| Tremor | Delayed (1 month-4 years) | Resting, postural and kinetic | Thalamus (posterior nucleus) |
| Slow frequency (1-3 Hz) | |||
| Asterixis | Acute | Thalamus (ventrolateral nucleus, ventroposterolateral nucleus) | |
| Vascular Parkinsonism | Acute | Bradykinesia (predominantly lower limb) | GPe |
| Lack of tremor | Putamen |
STN: subthalamic nucleus, GPe: globus pallidus externa.
HYPERKINETIC MOVEMENT DISORDERS INDUCED BY LESIONS IN THE BASAL GANGLIA CIRCUIT
Pathophysiology
Hyperkinetic movement disorders have been described on the basis of the basal ganglia model. Typically, lesions of the STN decrease the excitation of the basal ganglia output, and consequently reduce thalamic inhibition. The striatum is commonly involved in hyperkinetic movement disorders. According to the box theory, lesions in the striatum interrupt the transport of GABA to the GPe and, as a result, inhibit the STN. This change can lead to the loss of control of the GPi and consequently relieve the inhibition of thalamic output. In turn, the decrease in striatal inhibition of neurons in the globus pallidus enhances STN inhibition, thereby relieving the inhibition of basal ganglia output to the thalamus.
The GABAergic and dopaminergic system might also contribute to post-stroke movement disorders. Animal studies demonstrated that ischemic insult alters dopaminergic receptors and that this change leads to the degeneration of the GABAergic system in the SNr [12,13]. Notably, GABA plays a major role in the modulation of inhibitory synaptic transmission in the brain.
However, hyperkinetic movement disorders are relatively rare even when the basal ganglia are markedly damaged. This observation might be explained by individual susceptibility and brain plasticity. The basal ganglia network might be composed of parallel processing and have a compensatory mechanism that protects against the loss of motor control [14].
Involuntary movements that arise after basal ganglia infarction typically result from clearly defined lesions of the nuclei or from imbalances in the neurotransmitter system in basal ganglia [15]. Recent evidence suggests that most movement disorders induced basal ganglia lesions result from defects in functional connectivity rather than from a single lesion [9].
A reduction in the activity of the indirect pathway results in hyperkinetic movement disorders, including chorea and ballism. Studies in animal models strongly support the hypothesis that STN lesions induce hyperkinetic movement. An experimental monkey model demonstrated that STN lesions can induce hyperkinetic movements such as hemiballism and chorea [16]. STN lesions in monkeys decreased the tonic discharge of GPi neurons and consequently increased the response of thalamocortical neurons to cortical input. Together, these events culminate in involuntary movement. Lesions in the putamen can also reduce GPi discharge and enhance the response of thalamocortical neurons.
Several mechanisms by which post-stroke delayed onset movement disorders such as tremor and dystonia develop have been proposed. For example, delayed onset movement disorders that arise after basal ganglia ischemia might be a consequence of dendritic plasticity and changes in synaptic activity patterns. Changes in brain metabolism in response to ischemic insults vary with the age at which the injury occurs [17]. A negative correlation between the age at which ischemic insult occurs and the time between the insult and the onset of movement disorders has been previously reported [10]. It is also possible that the cumulative effect of multiple variables, including location, the severity of the injury, individual susceptibility, and age of occurrence, may play a role in determining movement disorder after basal ganglia infarction.
Types of movement disorder
Ballism/chorea
Neuroimaging studies have demonstrated that hemichorea or ballism are most commonly associated with the STN, caudate, and putamen [18]. In an animal model, chorea developed as a result of STN inactivation and a consequential reduction in basal ganglia output [19]. Ballism and chorea are known to share a common pathophysiology [20].
Ballism and chorea are the most frequently occurring movement disorders associated with basal ganglia infarction. Chorea presents with irregular, abrupt, rapid, and transient movements that can affect the entire body, and chorea typically manifests in the distal region of the body. Ballism is a severe form of chorea that presents with proximal limb involvement [21]. Although, hemiballism and hemichorea are more frequently reported compared with ballism in chorea, these disorders can occur in combination with ballism and chorea of the limbs [22]. Chorea and ballism affect similar bodily regions, indicating that the two movement disorders are strongly associated. In contrast with dystonia, approximately 80% of cases of ballism or chorea develop immediately after a stroke [23]. In total, 54% of cases of chorea associated with infarctions in the basal ganglia circuit completely resolve; however, chorea associated with STN lesions do not resolve.
While the STN is considered the primary region of the brain associated with hemichorea, lesions to the striatum are the most frequent cause hemichorea [18,24]. However, rare cases of ipsilateral hemiballism and contralateral hemiparesis have been reported [25]. Very few cases of chorea or ballism associated with isolated thalamic infarction have been reported. One study demonstrated that hypoperfusion of the STN and globus pallidus using a (99m) Tc-ethyl cysteinate dimer-single photon emission computed tomography associated with the development of chorea after stroke [26].
Dystonia
A previous study demonstrated that lesions in the posterior putamen and GPe, which might affect the direct and indirect pathway, induce dystonia [27]. Lesions of the thalamus, especially those in the ventroanterior (VA) and VL nuclei, and cerebellar lesions can cause dystonia. Neuroimaging studies demonstrated that lesions in the putamen and pallidum can also cause dystonia [28]. A review of the literature indicates that dystonia is frequently associated with lesions in the basal ganglia, especially in the lentiform nucleus and that dystonia is the most commonly occurring movement disorder [2,29]. Lesions in the lentiform nucleus are most frequently implicated in dystonia, especially those in the putamen [9,30,31]. Dystonia is defined as sustained muscle contractions or abnormal posture. Dystonia commonly presents in combination with hemidystonia on the contralateral side. However, focal and segmental dystonia can also occur [1,3,8]. Rare cases of paroxysmal dystonia have also been reported [32]. In most patients, dystonia completely or partially resolves. The average delay in the onset of dystonia is 9.5 months, which is a relatively long period of time compared with other movement disorders. Most patients that develop dystonia following basal ganglia and thalamic infarctions initially present with hemiparesis and develop dystonia once the hemiparesis resolves [33]. The length of time between the acute infarction and the onset of dystonia indicates that the recovery process plays a role in the development of dystonia [34].
MRI studies revealed that the postcommissural putamen, which is the lateral and ventral part of the head in the caudate nucleus that mediates the sensorimotor functions of the striatum, is associated with dystonia [3,31]. Pure thalamic stroke can also induce dystonia [35]. The posterolateral, paramedian, and centromedian nuclei are typically involved in dystonia, despite the fact that the VL and VA nucleus receive input from the basal ganglia. Lesions in the centromedian nucleus have also been implicated in dystonia [32]. The precise mechanisms by which lesions in these regions of the brain induce dystonia have not been fully characterized. Dystonia might be a consequence of the hypertonicity caused by the suppression of motor system inhibition at several levels.
Myoclonus and asterixis
Myoclonus can arise as a result of damage to the cerebral cortex, brainstem, spinal cord or peripheral nerve [36]. Pure cortical myoclonus can also be associated with the cortical myoclonus [37]. There have been few reports of myoclonus induced by striatal lesions [38]. Consistent with this observation, myoclonus is a rarely reported observed in strokes other than those affecting the basal ganglia. The development of general myoclonus following basal ganglia infarction has not been reported [39]. Asterixis, which is also referred to as negative myoclonus, is characterized by the failure to maintain muscle contraction in a fixed position. Asterixis has been observed following the occurrence of thalamic infarctions involving the brainstem and cerebellum [40]. Unilateral asterixis is more commonly observed, but some cases of bilateral asterixis have been reported.
Asterixis may be attributed with the loss of proprioception due to an interruption of the cerebellum/brainstem/thalamus/frontal lobe system, which has also been implicated in asterixis [41]. Lesions in the ventral lateral and ventroposterolateral nuclei in the thalamus can also induce the development of unilateral asterixis.
Tremor
Tremor after stroke often exhibits a heterogeneous presentation of that includes resting, action, and intention tremors [9]. Unilateral, multifocal, and segmental tremor are more commonly observed compared with generalized and focal tremor [3]. Tremor in the absence of other involuntary movement is extremely rare. The frequency of post-stroke tremor is relatively low (1–3 Hz) and is similar to the frequency of rubral tremors [42]. The delayed onset of post-stroke tremor can range from several months to several years after the event.
There have been reports of tremor after basal ganglia infarction; however, tremor induced by lesions to the thalamus is more frequently observed. Lesions to the posterior nucleus in the thalamus frequently induce tremor [43]. Although less common, tremor induced by strokes affecting the intermediate and anterior nuclei of the thalamus have also been reported [44,45]. Tremor arising from thalamic infarctions more frequently present in combination with other movement disorders, rather than occurring in isolation. Clinicopathological studies suggest that the dentatorubrocerebellar tract might also play a role in the development of tremor following thalamic infarction [43].
Complex movement disorders
It is not uncommon for patients to present with a combination of symptoms associated with multiple involuntary movement disorders following a thalamic stroke. Complex movement disorders can present with symptoms of dystonia, athetosis, chorea, and action tremor, and these symptoms can vary in their degree of severity [34]. The development of tremor in combination with dystonia following thalamic stroke has been reported [46]. Myoclonic dystonia in the ventral intermediate and posterior nuclei following thalamic infarction has also been reported [47]. The lesions associated with these conditions involve the posterolateral thalamus, and complex movement disorders can also be associated with sensory deficits and limb ataxia. Complex movement disorders frequently involve the hands and fingers, and typically exhibit a relatively long delay of 2 weeks to 24 months before involuntary movements develop.
Miscellaneous
Rarer involuntary movements that can manifest after basal ganglia infarction include tic, blepharospasm, restless leg syndrome (RLS), akathisia, and istereotypic movement disorder behaviors.
Tic
Tic is defined as sudden, repetitive, nonrhythmic motor movement or vocalization. Very few cases of tic following basal ganglia infarction have been reported. Although there have been reports of tics developing as a result of infarctions of the caudate and lentiform nuclei [48,49]. Tic and dystonia often present in combination. The cortico-striato-thalamo-cortical circuit might play a crucial role in the development of tics as ischemic lesions can disrupt this circuit [50].
Blepharospasm
Blepharospasm is rarely reported after basal ganglia infarction, although rare cases of blepharospasm after striatal infarction and bilateral basal ganglia infarction have been reported [51,52]. Functional MRI studies demonstrated that activation of the putamen is required for the development of blepharospasm, suggesting that the striatum is involved in the initiation and execution of eyelid spasms [53].
Restless leg syndrome
RLS, which is characterized by unpleasant and uncomfortable sensations in the legs when lying down, can also develop following acute ischemic or hemorrhagic stroke in various regions of the brain [54]. Among RLS after cerebral infarction, approximately 30.4% of cases of RLS following cerebral infarction are associated with basal ganglia lesions, and 14.3% are associated with thalamic lesions [55]. RLS typically exhibits an acute onset and develops within 1 week of an infarction. The classic motor and sensory symptoms of stroke are not associated with RLS. A study using positron emission tomography (PET) demonstrated that RLS is associated with dopaminergic hypofunction of the caudate nucleus and the putamen [56]. The basal ganglia-brainstem circuit might also contribute to the development of RLS.
HYPOKINETIC MOVEMENT DISORDERS IN LESION OF BASAL GANGLIA CIRCUIT
Pathophysiology
The mechanism underlying aberrant hypokinetic movements in Parkinsonian bradykinesia have been well established. A reduction in dopamine levels stimulates the indirect pathway and results in a strong inhibition of neurons in the GPe. These events lead to the tonic inhibition of the STN, and exert a strong excitatory effect on basal ganglia output neurons. Thus, hypokinetic movements in Parkinson’s disease are attributed to the strong inhibition of basal ganglia output. However, ischemic vascular insult is not associated with dopaminergic loss at the level of the substantia nigra. Although the pathophysiology of vascular Parkinsonism after stroke is not entirely clear, ischemic lesion might interrupt the pallido-/nigro-thalamic circuit, thereby disrupting basal ganglia output to the thalamic VL and VA nuclei [57].
Vascular Parkinsonism
Vascular Parkinsonism, which was initially referred to as atherosclerotic Parkinsonism by Critchley, is condition in which features of Parkinson’s disease appear after a stroke. Vascular Parkinsonism is a poorly defined disease, and shares clinical and pathological features of other diseases. Therefore, if vascular Parkinsonism represents a distinct disorder has been debated. There are no established diagnostic clinical criteria to diagnose vascular Parkinsonism, although some groups have proposed guidelines for diagnosis and appropriate management. Vascular Parkinsonism has been reported in 2% to 3% of all cases of Parkinsonism; however, the true incidence is not known [58]. Vascular Parkinsonism can be unilateral or bilateral involvement, can be caused by lesions to the lentiform nucleus and striatum, as well as pontine lesions and extensive subcortical white matter lesions [58]. Two distinct subtypes of vascular Parkinsonism have been proposed. The first type is described as Parkinsonism after acute basal ganglia infarction and the second type is described as chronic diffuse white matter degeneration involving the lentiform nucleus and the striatum. Despite continuous debate, vascular Parkinsonism shares several clinical features with Parkinson’s disease, including symmetrical presentation, the absence of tremor, predominantly lower body involvement, and poor response to L-dopa [59]. Infarctions affecting basal ganglia lacunae, including the thalamus, GPe and putamen, that extend into the caudate and internal capsule, can induce features of Parkinson’s disease [60]. There have been no reports of vascular Parkinsonism associated with STN infarction. In cases of vascular Parkinsonism after acute basal ganglia infarction, ischemic lesions affecting the pallido-/nigro-thalamic pathway might interrupt basal ganglia output to the thalamic VL and VA nuclei [57]. Unilateral lesions can induce features of Parkinson’s disease on the side of the body opposite of the legion, suggesting the existence of a causal relationship between basal ganglia infarction and vascular Parkinsonism.
CONCLUSIONS
The characteristics and anatomical regions associated with the various movement disorders that result from lesions in the basal ganglia circuit are described in Table 2 and Figure 2. Disorders involving both hyperkinetic and hypokinetic movements can occur following a stroke. Chorea/ballism and dystonia are the most common types of hyperkinetic movement disorders that arise following basal ganglia and thalamic infarctions. Tremor is often develops following thalamic infarction and exhibits extensive cerebellar tract involvement. Although myoclonus is rarely observed following strokes in the basal ganglia circuit, asterixisis often reported in this context. Chorea/ballism developed at the acute stage post-stroke, whereas dystonia and tremor exhibit a relatively long delay in their onset. Vascular Parkinsonism can also exhibit an acute onset after basal ganglia stroke, but it is rarely manifests as a result of thalamic or STN infarction.
Figure 2.
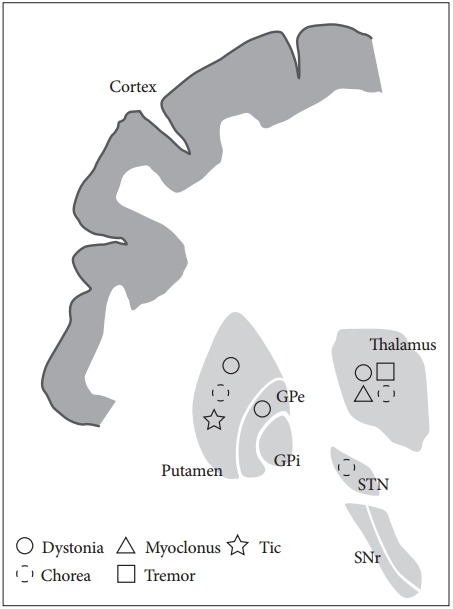
Anatomical correlation with movement disorders after basal ganglia infarction. GPe: globus pallidus externa, GPi: globus pallidus interna, SNr: substantia nigra pars reticulata, STN: subthalamic nucleus.
To date, few studies describe the pathophysiology of movement disorders associated with strokes in the basal ganglia circuit have been reported. Recently, new neuroimaging techniques, including diffusion tensor MRI, functional MRI, and PET, have provided new insights into various brain functions. Future studies employing these new approaches are expected to provide novel insights into the mechanisms underlying post-stroke movement disorders.
Footnotes
Conflicts of Interest
The author has no financial conflicts of interest.
REFERENCES
- 1.Ghika-Schmid F, Ghika J, Regli F, Bogousslavsky J. Hyperkinetic movement disorders during and after acute stroke: the Lausanne Stroke Registry. J Neurol Sci. 1997;146:109–116. doi: 10.1016/s0022-510x(96)00290-0. [DOI] [PubMed] [Google Scholar]
- 2.Siniscalchi A, Gallelli L, Labate A, Malferrari G, Palleria C, Sarro GD. Post-stroke movement disorders: clinical manifestations and pharmacological management. Curr Neuropharmacol. 2012;10:254–262. doi: 10.2174/157015912803217341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alarcón F, Zijlmans JC, Dueñas G, Cevallos N. Post-stroke movement disorders: report of 56 patients. J Neurol Neurosurg Psychiatry. 2004;75:1568–1574. doi: 10.1136/jnnp.2003.011874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wolters EC, Van Laar T, Berendse HW. Parkinsonism and related disorders. 3rd ed. Amsterdam: VU University Press; 2010. [Google Scholar]
- 5.Fahn S, Jankovic J, Hallett M. Principles and practice of movement disorders. Philadelphia: Elsevier Health Sciences; 2011. [Google Scholar]
- 6.Nougaret S, Meffre J, Duclos Y, Breysse E, Pelloux Y. First evidence of a hyperdirect prefrontal pathway in the primate: precise organization for new insights on subthalamic nucleus functions. Front Comput Neurosci. 2013;7:135. doi: 10.3389/fncom.2013.00135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nambu A, Tokuno H, Takada M. Functional significance of the cortico-subthalamo-pallidal ‘hyperdirect’ pathway. Neurosci Res. 2002;43:111–117. doi: 10.1016/s0168-0102(02)00027-5. [DOI] [PubMed] [Google Scholar]
- 8.Handley A, Medcalf P, Hellier K, Dutta D. Movement disorders after stroke. Age Ageing. 2009;38:260–266. doi: 10.1093/ageing/afp020. [DOI] [PubMed] [Google Scholar]
- 9.Mehanna R, Jankovic J. Movement disorders in cerebrovascular disease. Lancet Neurol. 2013;12:597–608. doi: 10.1016/S1474-4422(13)70057-7. [DOI] [PubMed] [Google Scholar]
- 10.Scott BL, Jankovic J. Delayed-onset progressive movement disorders after static brain lesions. Neurology. 1996;46:68–74. doi: 10.1212/wnl.46.1.68. [DOI] [PubMed] [Google Scholar]
- 11.Zijlmans JC, Daniel SE, Hughes AJ, Révész T, Lees AJ. Clinicopathological investigation of vascular parkinsonism, including clinical criteria for diagnosis. Mov Disord. 2004;19:630–640. doi: 10.1002/mds.20083. [DOI] [PubMed] [Google Scholar]
- 12.Lin B, Levy S, Raval AP, Perez-Pinzon MA, Defazio RA. Forebrain ischemia triggers GABAergic system degeneration in substantia nigra at chronic stages in rats. Cardiovasc Psychiatry Neurol. 2010;2010:506952. doi: 10.1155/2010/506952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nagasawa H, Araki T, Kogure K. Alteration of dopamine D1 receptor in the strionigral system of the postischemic rat brain. Neurosci Lett. 1992;134:271–274. doi: 10.1016/0304-3940(92)90533-d. [DOI] [PubMed] [Google Scholar]
- 14.Bhatia KP, Marsden CD. The behavioural and motor consequences of focal lesions of the basal ganglia in man. Brain. 1994;117(Pt 4):859–876. doi: 10.1093/brain/117.4.859. [DOI] [PubMed] [Google Scholar]
- 15.Holtbernd F, Eidelberg D. Functional brain networks in movement disorders: recent advances. Curr Opin Neurol. 2012;25:392–401. doi: 10.1097/WCO.0b013e328355aa94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ohye C, Le Gayader C, Feger J. Responses of subthalamic and pallidal neurons to striatal stimulation: an extracellular study on awake monkeys. Brain Res. 1976;111:241–252. doi: 10.1016/0006-8993(76)90769-1. [DOI] [PubMed] [Google Scholar]
- 17.Vannucci RC. Mechanisms of perinatal hypoxic-ischemic brain damage. Semin Perinatol. 1993;17:330–337. [PubMed] [Google Scholar]
- 18.Chung SJ, Im JH, Lee MC, Kim JS. Hemichorea after stroke: clinical-radiological correlation. J Neurol. 2004;251:725–729. doi: 10.1007/s00415-004-0412-5. [DOI] [PubMed] [Google Scholar]
- 19.Crossman AR, Sambrook MA, Jackson A. Experimental hemichorea/hemiballismus in the monkey. Studies on the intracerebral site of action in a drug-induced dyskinesia. Brain. 1984;107(Pt 2):579–596. doi: 10.1093/brain/107.2.579. [DOI] [PubMed] [Google Scholar]
- 20.Albin RL. The pathophysiology of chorea/ballism and Parkinsonism. Parkinsonism Relat Disord. 1995;1:3–11. doi: 10.1016/1353-8020(95)00011-t. [DOI] [PubMed] [Google Scholar]
- 21.Bhidayasiri R, Truong DD. Chorea and related disorders. Postgrad Med J. 2004;80:527–534. doi: 10.1136/pgmj.2004.019356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vidaković A, Dragasević N, Kostić VS. Hemiballism: report of 25 cases. J Neurol Neurosurg Psychiatry. 1994;57:945–949. doi: 10.1136/jnnp.57.8.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bansil S, Prakash N, Kaye J, Wrigley S, Manata C, Stevens-Haas C, et al. Movement disorders after stroke in adults: a review. Tremor Other Hyperkinet Mov (N Y) 2012 Mar 20; doi: 10.7916/D86W98TB. [Epub]. http://dx.doi.org/10.7916/D86W98TB. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Whittier JR. Ballism and the subthalamic nucleus hypothalamicus; corpus luysi) review of the literature and study of 30 cases. Arch Neurol Psychiatry. 1947;58:672–692. doi: 10.1001/archneurpsyc.1947.02300350022002. [DOI] [PubMed] [Google Scholar]
- 25.Krauss JK, Pohle T, Borremans JJ. Hemichorea and hemiballism associated with contralateral hemiparesis and ipsilateral basal ganglia lesions. Mov Disord. 1999;14:497–501. doi: 10.1002/1531-8257(199905)14:3<497::aid-mds1019>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 26.Takahashi T, Kanamori H, Shigehara R, Takahashi SN, Tamura M, Takasu T, et al. Pure hemi-chorea resulting from an acute phase of contralateral thalamic lacunar infarction: a case report. Case Rep Neurol. 2012;4:194–201. doi: 10.1159/000345227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vidailhet M, Grabli D, Roze E. Pathophysiology of dystonia. Curr Opin Neurol. 2009;22:406–413. doi: 10.1097/WCO.0b013e32832d9ef3. [DOI] [PubMed] [Google Scholar]
- 28.Fross RD, Martin WR, Li D, Stoessl AJ, Adam MJ, Ruth TJ, et al. Lesions of the putamen: their relevance to dystonia. Neurology. 1987;37:1125–1129. doi: 10.1212/wnl.37.7.1125. [DOI] [PubMed] [Google Scholar]
- 29.Netravathi M, Pal PK, Indira Devi B. A clinical profile of 103 patients with secondary movement disorders: correlation of etiology with phenomenology. Eur J Neurol. 2012;19:226–233. doi: 10.1111/j.1468-1331.2011.03469.x. [DOI] [PubMed] [Google Scholar]
- 30.Pettigrew LC, Jankovic J. Hemidystonia: a report of 22 patients and a review of the literature. J Neurol Neurosurg Psychiatry. 1985;48:650–657. doi: 10.1136/jnnp.48.7.650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chuang C, Fahn S, Frucht SJ. The natural history and treatment of acquired hemidystonia: report of 33 cases and review of the literature. J Neurol Neurosurg Psychiatry. 2002;72:59–67. doi: 10.1136/jnnp.72.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee MS, Marsden CD. Movement disorders following lesions of the thalamus or subthalamic region. Mov Disord. 1994;9:493–507. doi: 10.1002/mds.870090502. [DOI] [PubMed] [Google Scholar]
- 33.Baik JS, Lee MS. Movement disorders associated with moyamoya disease: a report of 4 new cases and a review of literatures. Mov Disord. 2010;25:1482–1486. doi: 10.1002/mds.23130. [DOI] [PubMed] [Google Scholar]
- 34.Kim JS. Delayed onset mixed involuntary movements after thalamic stroke: clinical, radiological and pathophysiological findings. Brain. 2001;124(Pt 2):299–309. doi: 10.1093/brain/124.2.299. [DOI] [PubMed] [Google Scholar]
- 35.Krystkowiak P, Martinat P, Defebvre L, Pruvo JP, Leys D, Destée A. Dystonia after striatopallidal and thalamic stroke: clinicoradiological correlations and pathophysiological mechanisms. J Neurol Neurosurg Psychiatry. 1998;65:703–708. doi: 10.1136/jnnp.65.5.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kojovic M, Cordivari C, Bhatia K. Myoclonic disorders: a practical approach for diagnosis and treatment. Ther Adv Neurol Disord. 2011;4:47–62. doi: 10.1177/1756285610395653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Espay AJ, Chen R. Myoclonus. Continuum (Minneap Minn) 2013;19(5 Movement Disorders):1264–1286. doi: 10.1212/01.CON.0000436156.54532.1a. [DOI] [PubMed] [Google Scholar]
- 38.Defebvre L. Myoclonus and extrapyramidal diseases. Neurophysiol Clin. 2006;36:319–325. doi: 10.1016/j.neucli.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 39.Bogousslavsky J, Caplan LR. Stroke syndromes. 2nd ed. Cambridge: Cambridge University Press; 2001. [Google Scholar]
- 40.Kim JS. Asterixis after unilateral stroke: lesion location of 30 patients. Neurology. 2001;56:533–536. doi: 10.1212/wnl.56.4.533. [DOI] [PubMed] [Google Scholar]
- 41.Massey EW, Goodman JC, Stewart C, Brannon WL. Unilateral asterixis: motor integrative dysfunction in focal vascular disease. Neurology. 1979;29:1180–1182. doi: 10.1212/wnl.29.8.1180. [DOI] [PubMed] [Google Scholar]
- 42.Miwa H, Hatori K, Kondo T, Imai H, Mizuno Y. Thalamic tremor: case reports and implications of the tremor-generating mechanism. Neurology. 1996;46:75–79. doi: 10.1212/wnl.46.1.75. [DOI] [PubMed] [Google Scholar]
- 43.Nowak DA, Seidel B, Reiner B. Tremor following ischemic stroke of the posterior thalamus. J Neurol. 2010;257:1934–1936. doi: 10.1007/s00415-010-5635-z. [DOI] [PubMed] [Google Scholar]
- 44.Cho C, Samkoff LM. A lesion of the anterior thalamus producing dystonic tremor of the hand. Arch Neurol. 2000;57:1353–1355. doi: 10.1001/archneur.57.9.1353. [DOI] [PubMed] [Google Scholar]
- 45.Krystkowiak P, Martinat P, Cassim F, Pruvo JP, Leys D, Guieu JD, et al. Thalamic tremor: correlations with three-dimensional magnetic resonance imaging data and pathophysiological mechanisms. Mov Disord. 2000;15:911–918. doi: 10.1002/1531-8257(200009)15:5<911::aid-mds1023>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 46.Kim JS. Delayed onset hand tremor caused by cerebral infarction. Stroke. 1992;23:292–294. doi: 10.1161/01.str.23.2.292. [DOI] [PubMed] [Google Scholar]
- 47.Lehéricy S, Vidailhet M, Dormont D, Piérot L, Chiras J, Mazetti P, et al. Striatopallidal and thalamic dystonia. A magnetic resonance imaging anatomoclinical study. Arch Neurol. 1996;53:241–250. doi: 10.1001/archneur.1996.00550030051022. [DOI] [PubMed] [Google Scholar]
- 48.Kwak CH, Jankovic J. Tourettism and dystonia after subcortical stroke. Mov Disord. 2002;17:821–825. doi: 10.1002/mds.10207. [DOI] [PubMed] [Google Scholar]
- 49.Gomis M, Puente V, Pont-Sunyer C, Oliveras C, Roquer J. Adult onset simple phonic tic after caudate stroke. Mov Disord. 2008;23:765–766. doi: 10.1002/mds.21955. [DOI] [PubMed] [Google Scholar]
- 50.Peterson BS. Neuroimaging studies of Tourette syndrome: a decade of progress. Adv Neurol. 2001;85:179–196. [PubMed] [Google Scholar]
- 51.Keane JR, Young JA. Blepharospasm with bilateral basal ganglia infarction. Arch Neurol. 1985;42:1206–1208. doi: 10.1001/archneur.1985.04060110088025. [DOI] [PubMed] [Google Scholar]
- 52.Grandas F, López-Manzanares L, Traba A. Transient blepharospasm secondary to unilateral striatal infarction. Mov Disord. 2004;19:1100–1102. doi: 10.1002/mds.20109. [DOI] [PubMed] [Google Scholar]
- 53.Schmidt KE, Linden DE, Goebel R, Zanella FE, Lanfermann H, Zubcov AA. Striatal activation during blepharospasm revealed by fMRI. Neurology. 2003;60:1738–1743. doi: 10.1212/01.wnl.0000063306.67984.8c. [DOI] [PubMed] [Google Scholar]
- 54.Sechi G, Agnetti V, Galistu P, Murgia B, Marrosu F, Puligheddu M, et al. Restless legs syndrome and periodic limb movements after ischemic stroke in the right lenticulostriate region. Parkinsonism Relat Disord. 2008;14:157–160. doi: 10.1016/j.parkreldis.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 55.Lee SJ, Kim JS, Song IU, An JY, Kim YI, Lee KS. Poststroke restless legs syndrome and lesion location: anatomical considerations. Mov Disord. 2009;24:77–84. doi: 10.1002/mds.22303. [DOI] [PubMed] [Google Scholar]
- 56.Wetter TC, Eisensehr I, Trenkwalder C. Functional neuroimaging studies in restless legs syndrome. Sleep Med. 2004;5:401–406. doi: 10.1016/j.sleep.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 57.Peters S, Eising EG, Przuntek H, Müller T. Vascular parkinsonism: a case report and review of the literature. J Clin Neurosci. 2001;8:268–271. doi: 10.1054/jocn.2000.0807. [DOI] [PubMed] [Google Scholar]
- 58.Winikates J, Jankovic J. Clinical correlates of vascular parkinsonism. Arch Neurol. 1999;56:98–102. doi: 10.1001/archneur.56.1.98. [DOI] [PubMed] [Google Scholar]
- 59.Gupta D, Kuruvilla A. Vascular parkinsonism: what makes it different? Postgrad Med J. 2011;87:829–836. doi: 10.1136/postgradmedj-2011-130051. [DOI] [PubMed] [Google Scholar]
- 60.Zijlmans JC, Thijssen HO, Vogels OJ, Kremer HP, Poels PJ, Schoonderwaldt HC, et al. MRI in patients with suspected vascular parkinsonism. Neurology. 1995;45:2183–2188. doi: 10.1212/wnl.45.12.2183. [DOI] [PubMed] [Google Scholar]