

Abstract
Molecular chaperones such as heat shock protein 90 (Hsp90) have been shown to form complexes with tumor antigens and can be used to prepare anticancer vaccines largely due to this property. Earlier studies had suggested that, mice immunized with a molecular chaperone based vaccine derived from tumors became immune to further vaccination and that both CD8+ and CD4+ T cells were activated by the chaperone vaccine in a manner dependent on scavenger receptor SREC-I. Here we have investigated mechanisms whereby SREC-I might facilitate uptake of Hsp90 conjugated peptides by APC into the MHC class II pathway for presentation to CD4+ T cells. Our studies showed that antigenic peptides associated with Hsp90 were taken up into the Class II pathway by a mechanism dependent on SREC-I binding and internalization and presented to CD4+ T cells. In addition our studies showed that SREC-I could associate with MHC class II molecules on the cell surface and in intracellular endosomes, suggesting a mechanism involving facilitated uptake of peptides into the MHC class II pathway. These studies in addition to our earlier findings showed SREC-I to play a primary role in chaperone-associated antigen uptake both through cross priming of MHC class I molecules and entry into the class II pathway.
Keywords: Hsp90, SREC-I, Ova, Antigen Presentation, Dendritic cell
Introduction
Molecular chaperones such as heat shock Hsp90 have been shown to form complexes with tumor antigens and have thus been used in preparing anticancer vaccines, largely due to this property (Kunisawa and Shastri, 2006; Murshid et al., 2011). HSPs therefore could function as antigen carriers and it has been suggested that they might use this property to mediate antigen presentation in target APC by facilitating endocytosis of bound polypeptides (Murshid et al., 2010; Srivastava 2010 and Murshid et al., 2012). APCs such as dendritic cells (DC) were shown to take up external antigens and process such polypeptides for presentation to T cells through two distinct pathways including: (1) the classical MHC class II pathway and (2) cross presentation through the MHC class I pathway. Soluble antigens have been shown previously to be partitioned between these two alternative pathways by interacting with different cell surface receptors in DC such as CLEC9, DC-SIGN, DEC205 and the mannose receptor 1 (Murshid et al., 2012; Bonifaz et al., 2002; Burgdorf and Kurts, 2008). Triage between the two pathways thus occurs at the cell surface through the dedicated antigen binding receptors. It was however, not clear whether HSP chaperoned antigens adhered to these mechanisms for selective entry into pathways of antigen presentation. Previous studies showed that HSP-peptide complexes were internalized by a distinct group of receptors compared to free polypeptides (Theriault et al., 2006). These receptors were from the scavenger receptor families and included LOX-1, SREC-I/SCARF-I, and FEEL1/Stabilin-1. Both SREC-I and LOX-1 have been shown to mediate the cross presentation of molecular chaperone bound antigens and lead to activation of CD8+ T lymphocytes (Theriault et al., 2006; Delneste et al., 2002; Murshid et al., 2011). In addition, earlier studies showed that mice immunized with a molecular chaperone based vaccine derived from tumors became immune to further tumor inoculation and that both CD8+ and CD4+ T cells were activated in a SREC-I dependent manner by the chaperone vaccines (Gong et al., 2010; Gong et al., 2009).
We have set out here to determine whether SREC-I can directly mediate entry of Hsp90-bound antigens into the MHC class II pathway and stimulation of CD4+ T cells.
Materials and Methods
Mice
Mice, B6C3F1 (H-2bxk) used in this study were obtained from The Jackson Laboratory (Bar Harbor, ME). All mice were kept in a specific pathogen-free mouse facility. Studies were done according to institutional guidelines for animal use and care.
Reagents and Abs
OVA was purchased from Sigma-Aldrich (St. Louis, MO). PL19 peptide was custom synthesized (AnaSpec, Fremont, CA) and purified to 95% by HPLC. Rabbit polyclonal human anti–SREC-I Ab was rabbit monoclonal was custom synthesized by GenScript (Piscataway, NJ) against specific peptide sequence (TQGTQGSTLDPAGQC). Commercially available anti-SREC-I ab was purchased from Atlas Antibodies. Anti–MycII ab was from Sigma. Rat anti-mouse IFN-γ was from BD Pharmingen (San Diego, CA). GM-CSF was purchased from PeproTech (Rocky Hill, NJ). Cy3-transferrin and all other secondary fluorescent Abs were from Jackson Immunoresearch Laboratories. Alexa 555 labeling kit was from Life Sciences. Clostridium deficile ToxinB was purchased from Calbiochem. Anti-pMHC-II mAb L243 was purchased from BD Biosciences. CIITA plasmid was from Addgene.
Cells and culture conditions
BMDCs were generated from the femur and tibiae of, B6C3F1 mice. The bone marrow was flushed out and cultured in RPMI-1640 supplemented with 10% heat-inactivated FBS and 40 ng/ml GM-CSF for 6 d. On day 3, one third of the medium was replaced by fresh growth medium. On day 6, the immature BMDCs were used for experiment. Wild-type COS7 and Hek293 cells were transfected with human full length SREC-I in pcDNA3 for stable expression of SREC-I. Both cell lines were maintained in Ham’s F12K medium supplemented with 10% heat inactivated FBS. For generation of stable SREC-I–expressing cell lines, cells were selected and maintained in the same medium plus 400 mg/ml G418. Hek293 and Raw 264.7 cell lines were maintained in DMEM supplemented with 10% heat-inactivated FBS. KG-1 cells were maintained in RPMI-1640 supplemented with 10% heat-inactivated FBS.
Plasmids
The pcDNA3.1-SREC-I (human) was a generous gift from Dr. H. Adachi. FLAG–SREC-I construct was made in 3xFLAG-CMV vector. MHCII molecules were gifts from Dr. R. Germain of NIH.
Hsp90–peptide/protein complex and anti–SREC-I Ab–OVA complex formation
OVA was dialyzed for 36 h at 4°C with several changes of the buffer (PBS) to remove degradation products as well as possible contaminating peptides from the solution. Small peptides, PL19 (PDEVSGLEQLESIINFEKL) were loaded onto Hsp90 in solution at a molar ratio of 50:1 in PBS, incubated for 10 min at 45–50°C, and cooled at room temperature for 30–40 min. For Hsp90–OVA conjugate preparation, soluble Hsp90 and excess OVA (1:2 ratio) were mixed and incubated for 10 min at 45°C. The solution mixtures were then incubated at room temperature for 30 min. Free OVA was removed using Microcon YM-100 (Millipore, Bedford, MA) with a 100-kDa cutoff. Alexa 488- and Alexa 555-labeled Hsp90-peptide/OVA (Hsp90.PC) conjugates were made according to the manufacturer’s protocol (Invitrogen). For anti–SREC-I Ab-OVA preparation, OVA was coupled to anti–SREC-I Ab using bis-(sulfosuccinimidyl) suberate as described by the manufacturer (Thermo Fisher Scientific, Waltham, MA) and labeled with Alexa fluorochromes as described for Hsp90 and purified using Microcon YM-100 ultrafiltration (Millipore, Billerica, MA).
Immunofluorescence and microscopic analysis of Hsp90 internalization
COS7 cells were labeled with Alexa Fluor-conjugated Hsp90 complexes for 20–30 min on ice. The ice-cold medium was then replaced by warm medium and incubated at 37°C for different periods. Cells were later washed with ice-cold stripping buffer (50 mM sodium citrate and 280 mM sucrose [pH 4.6]) to remove unbound Hsp90.PC. Later, the cells were fixed with 4% para-formaldehyde and permeabilized with 0.1% Triton X-100. Cells were stained with different Abs and later analyzed using a Zeiss 510 confocal microscope (Carl Zeiss, Jena, Germany). For analysis of binding to BMDCs, FcRs were pre-blocked. For blocking FcR-mediated nonspecific binding, immature BMDCs were treated with anti-FcγR Ab (CD16/32 specific for FcγRIII, FcγRII) for 10 min at 4°C at a concentration of 1 mg/ml/million cells. To prevent complication of experiments by Hsp90 interaction with HSP receptor LOX1, BMDCs were also treated with goat anti-mouse LOX1 Ab (10 mg/ml) for 10–15 min on ice. Fluorophores were visualized using the following filter sets: 488 nm excitation and band pass 505–530 emission filter for Alexa 488; 543 nm excitation and band pass 560–615 for Cy3; and 633 excitation and long pass 650 for Cy5. Images were taken using a 633 numerical aperture 1.4 oil immersion objective lens (Carl Zeiss, Jena, Germany). Figures were made using Adobe Photoshop 7.0 (Adobe Systems, San Jose, CA) with little or no contrast adjustments without altering original images.
In vitro cross-presentation assay
Immature BMDCs from mice were plated in 96-well plates post purification. Cells were then pulsed with 10 mg/ml Hsp90 and its OVA conjugates (Hsp90–PL19/OVA), 10mg/ml free OVA, and also with 10 mg/ml anti–SREC-I–OVA complexes for 5 min to 2 h. In all Ag cross-presentation experiments, a preparation of the BMDC was pulsed with 100 ng/ml PL19 as positive control. For inhibitors and drug treatment, cells were incubated with the drugs before they were pulsed with different Ag preparations. The ligands were then removed and cells fixed with para-formaldehyde for 10 min at room temperature. Later, peptide-specific T cell hybridoma (KZO, a generous gift from Dr. N. Shastri, Department of Molecular and Cellular Biology, University of California, Berklely, CA) was added to the fixed cells at 37°C for 20 h.
Western Blotting and Coimmuoprecipitation assay
HeLa cells were transfected with FLAG-SREC-I and CIITA-Myc for 22 hours. Cells were then incubated with or without Hsp90.Ova for 30 mins. Cells were then washed with iced-cold Dulbecco’s phosphate buffered saline (PBS) and lysed with NP-40 lysis buffer (containing 1% Nonidet P-40, 150mM NaCl, 1mM EDTA, 1mM PMSF, 1× HALT protease and phosphatase inhibitor cocktail, Thermo Scientific). For immunoprecipitation, 1 mg of cell extract was incubated with 5 μg of anti FLAG antibody (M2) for 2 hours at 4°C followed by incubation with 20 μl of protein A (50% slurry, GE healthcare) plus-sepharose beads for overnight at 4°C. The beads were then washed with NP40-lysis buffer and complexes were eluted by boiling in Laemmle sample buffer. For Western blotting, immunoprecipitated samples were resolved by 4–15% gradient SDS-PAGE and transferred to PVDF (polyvinylidene fluoride) membranes. Membranes were immunoblotted with anti-HLA-DR antibody and later secondary antibody that are HRP-conjugated. The membrane reactions were visualized by Perkin Elmer enhanced chemiluminescence reagents.
Cdc42 activation assay
Cdc42 activation assay was performed in Hek293 cells with or without Hsp90.PL19 according to the manufacturer’s protocol (Active Rac1/Cdc42 pull down and detection kit, Thermo Scientific).
Results
We first examined the potential role of purified Hsp90 and cell surface SREC-I in uptake of antigens into the MHC class II pathway. Hsp90 was purified from Sf9 insect cells that expressed the human Hsp90α gene (after infection by baculovirus encoding the protein) as described previously (Murshid et al., 2010). Recombinant Hsp90 was then bound to a peptide PL19 (as described below) derived from ovalbumin and complexes isolated as described previously and labeled with a fluorescent dye Alexa-555 fluorophore (Murshid et al., 2010). We ensured that the Hsp90 preparations could interact with SREC-I on the cell surface as determined by fluorescence microscopy. Indeed, such Alexa 555-Hsp90-Ova complexes were localized on the cell surface with SREC-I in Raw 264.7 cells and became co-internalized with the receptor on incubation at 37°C (Fig. 1A and B). Repeating a similar experiment in the dendritic cell line KG1 we found again that Hsp90.PL19 complexes became bound to FLAG-SREC-on the surface and appeared to become co-internalized in intracellular vesicles (Fig. 1C). The binding and internalization of Hsp90-peptide to SREC-I was thus seen both in macrophage and dendritic cells.
Figure 1. Hsp90-Ova can interact with SREC-I at the cell surface and in intracellular compartments.

A and B, Hsp90-PL19 can interact with SREC-I in Raw264.7 cells. Raw 264.7 cells were transfected with a construct expressing FLAG-SREC-I for 18 hours. Cells were then incubated with Alexa 555-Hsp90-PL19 peptide (10ug/ml) complexes on ice for 20 minutes followed by incubation at 37°C for 10 minutes. The cells were then permeabilized for 5 minutes and then fixed, stained for FLAG with anti-FLAG ab (green). C, KG1 cells (DC cell line) were transfected with FLAG-SREC-I and then incubated with Alexa 555-Hsp90.Ova for 5–10 mins. Cells were then fixed and either permeabilized (D) or not (C) with 4% PFA. Scale bar 5μm.
To examine the role of SREC-I in class II presentation, we incubated bone marrow-derived mouse dendritic cells (BMDC) from B6C3F1 mice with KZO T cell hybridoma cells that recognize the peptide (PDEVSGIEQLESIINFEKL or PL19) derived from ovalbumin (Ova) in the context of MHC class II species I-Ak. For the antigen presentation experiments Hsp90 was complexed to either PL19 or to purified full length Ova. To test a causal role for SREC-I in Ova presentation through the Class II pathway, we prepared anti-SREC-I antibodies cross-linked to full length Ova. Antigen presentation was assayed by measuring interferon gamma (IFNγ) release by the KZO T cells. Exposure to uncomplexed, antigen free Hsp90 had minimal effect on IFNγ release from the KZO target cells (Fig 2A). However, when Hsp90 was complexed to either purified Ova or the synthetic peptide (PL19), a large increase in IFNγ release from the KZO cells was observed which exceeded the effects of free, uncomplexed Ova (Fig. 2A). Hsp90 thus appeared to stimulate presentation of Ova via the Class II pathway. In addition, a similarly powerful activation of IFN γ release was seen when cells were exposed to anti-SREC-I antibody-Ova complexes further suggesting a receptor role for SREC-I in processing of Hsp90-Ova complexes through the MHC class II pathway.
Figure 2. Hsp90-OVA polypeptide complexes bind to SREC-I and mediate OVA Ag presented to KZO cells after uptake.

A, Immature BMDCs from B6C3F1 mice were incubated with Hsp90, free OVA, Hsp90-OVA peptide (247–265, PL19), Hsp90-OVA, and anti–SREC-I-Ab–OVA complexes for 2h at 37°C. Cells were then fixed, and cultured with KZO T cell hybridoma cells overnight before assaying secreted IFN-γ by ELISA. B, COS7 cells were transfected with SREC-I and constructs expressing murine MHCII molecules (Aαk, Aβk and murine Invariant chain, mIi/p31) for 22 h and then incubated with Hsp90, free OVA, OVA peptide, and Hsp90-PC for 2h. Cells were then fixed, washed, and cultured with KZO cells overnight. The IFN-γ secreted by KZO T cells was assayed by ELISA. C, D In a control experiment for B, COS7 cells were either transfected (D) or not (C) with MHCII molecules alone for 22 h and incubated with free or Hsp90-conjugated OVA Ags as in B for 2 h and assayed for Ag presentation. Data shown in A–C are the mean + SD of triplicate wells. The results shown (A–C) are representative of at least three independent experiments.
To confirm an additional role of SREC-I in directing Hsp90 bound antigens to MHC class II, we employed non-APC cells (COS-7 cells) that do not express SREC-I and MHCII engineered to transiently express MHC class II alpha and beta chains along with Invariant chain (Aαk, Aβk and murine Invariant chain, mIi/p31). As these cells do not express either SREC-I or surface MHC class II complexes under basal conditions, we were then able to compare the dependence on SREC-I for responding to Hsp90-chaperoned Ova peptides as in Fig. 2A. In COS7 cells expressing both MHCII and SREC-I, Hsp90 was able to chaperone polypeptides through the MHC class II pathway, lead to enhanced presentation of Ova and Ova peptides as evidenced by enhanced IFNγ release from the KZO cells (Fig. 2B). As in the BMDC, Ova covalently associated with anti-SREC-I antibody was preferentially presented by COS7 cells expressing SREC-I to KZO cells compared with free Ova (Fig. 2B). In the absence of SREC-I, presentation of Hsp90 chaperoned peptides by the COS7 cells was almost completely absent, confirming the role of this scavenger receptor in efficient presentation of peptides via the MHC class II pathway (Fig. 2C). On the other hand COS7-MHCII cells were somewhat able to present Ova peptide (PL19) and Ova via MHCII alone, despite the absence of SREC-I but were incapable of internalizing and engulfing Hsp90 bound Ova or PL19 (Fig. 2D)
Our previous studies have shown that cross presentation of antigens bound to Hsp90 by APC involves internalization of SREC-I through a clathrin-independent endocytic pathway. Hsp90-SREC-I complexes were instead taken up through the GPI-anchored-protein-enriched endosomal compartment (GEEC) pathway (Murshid et al., 2010). The GEEC pathway was shown to be regulated by a specific subgroup of small (Rho) GTPases including Cdc42 but excluding Rac1 for endocytosis. Rho family GTPases play a central role in F actin structure modulation.
It was also demonstrated that activated Cdc42 could induce filopodia formation, a process involving cytoskeleton remodeling in the neuritic phenotype (Chen et al., 1999, Yamaguchi et al., 2001). We observed that SREC-I expressing cells showed a morphology resembling neuritic cells after transfection of SREC-I expression vectors in CHO-K1, HeLa and Hek293 cells (data not shown). On the basis of this observation we proposed that SREC-I might modulate signal transduction effects of Cdc42 involved in the MHC class II pathway. Filopodia formation is likely to be due to interaction of the downstream adaptor protein WASP family members with Arp2/3 (actin nucleating complex) and thus cytoskeleton remodeling. Indeed, we found that SREC-I co-localized with Arp3 on the cell surface especially in those protrusions in the presence of its ligand, Hsp90-PL19, suggesting a role for ligand-bound SREC-I in actin cytoskeleton remodeling, receptor internalization (data not shown). We also observed co-localization of SREC-I and Cdc42 in peri-Golgi tubulovesicular structures, further suggesting the role of dynamic actin cytoskeleton in the formation of carries in the secretory pathway, Golgi to plasma membrane (Fig. 3B).
Figure 3. Hsp90-peptide complexes trigger Cdc42 activation.

A and B, CHO-SREC-I cells were either used as controls (A) or incubated (B) with Hsp90-PL19 (10ug/ml) for 30 mins. Cells were then fixed, permeabilized with 4% p-formaldehyde and stained for SREC-I (Alexa 488, green) and Cdc42 (Cy3, red). C, Hek293 cells expressing both SREC-I and MHCII were incubated with or without Hsp90-PL19, GTPγS or GDP. Cell lysates were then incubated with GST-PAK1-PBD (20ug) and a glutathione resin. The cell lysates were separated by 4–20% SDS-PAGE gradient gels, transferred to PVDF membrane and probed with anti-Cdc42 ab. The results shown here are representation of 2 independent experiments.
We next used a GST-PAK1-PBD pull down assay to measure intracellular GTPase activity in response to exposure to Hsp90 peptide complexes. Active Rho GTPases Cdc42 or Rac1 can bind specifically to the p21-binding domain (PBD) of p21 activated protein kinase 1 (PAK1) leading its activation. Therefore PBD of PAK1 was used as a probe to specifically isolate the active forms of Cdc42 or Rac1. We found that exposure to Hsp90-Ova complexes activated Cdc42 GTPase (Fig. 3C). Intracellular Cdc42 GTPase activity was high in cells exposed to Hsp90-PL19 complexes (Fig. 3C, 4th lane) and was comparable to the positive control incubated with the non-metabolizable NTP GTPγS (Fig 3C, 5th lane) Fig. 3C. Activity in cells exposed to Hsp90-PL19 complexes was inhibited by addition of GDP to the incubations, indicating specific, chaperone activated, GDP-inhibitable Cdc42 activity (Fig. 3C, 3rd lane). However, statistically insignificant activation of another GTPase Rac1 was observed with or without Hsp90 (Data not shown). These data indicated involvement of Cdc42 and the GEEC pathway in Hsp90-peptide-SREC-I interactions.
We next examined the potential role of SREC-I associated Cdc42 (and the GEEC pathway) in Hsp90-antigen complex presentation though the MHC class II pathway. We found as in Fig 2 that full length Ova or synthetic peptide PL19 presentation by BMDC to CD4+ KZO T cell hybridoma cells could be stimulated by association of the antigen with either Hsp90 or SREC-I (Fig. 4). However, when the incubations were carried out in the presence of Toxin B, an inhibitor of Cdc42, T cell stimulation by Ova associated with Hsp90 or SREC-I was abolished (Fig. 4). By contrast, presentation of free Ova was not markedly affected by exposure to Toxin B indicating utilization of an alternative uptake pathway not requiring SREC-I (Fig. 4).
Figure 4. Presentation of Hsp90-chaperoned antigens after binding SREC-I requires actin, Rho-GTPase activity.

Immature BMDCs were treated with Toxin B (2 ng/ml) and incubated at 37°C. Cells were then pulsed with OVA polypeptides, Hsp90-PC complexes, and anti–SREC-I-Ab-OVA for 2h at 37°C, fixed, washed, then cultured with KZO T cell hybridoma cells overnight. IFN-γ secreted by KZO cells was measured by sandwich ELISA using anti–IFN-γ Ab. Data shown are the means + SD of triplicate assays. The data shown are representative of at least three independent experiments. Scale bar 5μm.
Next we examined the localization of MHCII and SREC-I in cells with and without exposure to the chaperone complexes in CIITA overexpressing HeLa (Fig. 5). To examine the localization of the receptor, we overexpressed both CIITA and SREC-I in a non-DC/macrophage cell lines, HeLa cells. The expression of CIITA in this cell line induced endogenous expression of both MHCII and invariant chain (Ii). These cells therefore could act as antigen presenting cells and were capable of activating CD4+ T cells. Localization studies of MHCII and invariant chain were convenient in HeLa-CIITA cells (Walseng et al., 2008). In the absence of ligand, in cells incubated at 4°C, MHCII and SREC-I appeared to be independently distributed on the cell surface (Fig. 5A). However, in cells warmed to 37°C, SREC-I and MHC class II complexes appeared to become substantially co-localized in intracellular structures reminiscent of ER and Golgi, suggesting intracellular co-trafficking of these two receptors (Fig. 5B). Exposure of cells to Hsp90-Ova complexes caused SREC-I to redistribute and co-localize with MHC class II complexes on the surface of the HeLa-CIITA cells (compare Fig. 5A and C). Incubations were initially performed at 4°C to reduce endocytosis and to increase binding. Subsequent, warming of cells to 37°C led to close co-localization SREC-I and MHC class II into intracellular endosomal structures (Fig 5D). This distribution was quite different from the intracellular localization observed in control cells that had not been exposed to Hsp90 (comparison of B, D). We also observed biochemical association between HLA-DR (MHCII) and SREC-I in HeLa-CIITA cells (Fig. 2E). These data were consistent with a mechanism involving association of SREC-I/Hsp90-peptide complexes with MHC II at the plasma membrane and coordinate internalization to endosomes. The experiments therefore suggested that Hsp90 binding to SREC-I could potentially bring the internalized peptide into close proximity with MHC class II molecules on the cell surface and assist antigen loading in intracellular vesicles.
Figure 5. SREC-I co-localizes with MHCIIA on the cell surface and in intracellular organelles.

A, B) HeLa cells stably expressing Myc-CIITA were transfected with FLAG-SREC-I for 18 hours and then incubated at 4°C (A), or 37°C (B). Cells were then fixed and stained for SREC-I (FLAG-SREC-I, red) and MHCII ab (MHCII, green) (A) or permeabilized and stained as in (A). C, D) HeLa-CIITA cells were again transfected as in 5A and then incubated with Hsp90-Ova for 5 minutes. Cells were then stained for MHCII (MHCII, green) and FLAG (red) without permeabilization for cell surface localization assay. D) Transfected HeLa cells (as in A, B) were incubated with Hsp90-Ova for 15 minutes on ice and then incubated with warm growth media at 37°C. Cells were permeabilized and stained for MHCII (green) and FLAG (red). E) HeLa-FLAG-SREC-I and HeLa cells were transfected with CIITA-myc for 22 hours. Transfected and non-transfected HeLa cells were incubated with or without Hsp90.Ova (Hsp90) for 30 minutes. Cells were then incubated with anti-FLAG antibody beads for overnight at 4°C. Cell precipitates were collected, lysed and run for SDS-PAGE and later blotted for MHCII with anti-HLA-DR ab. Scale bar 5μm.
Discussion
Our experiments suggested that stimulation of CD4+ T cells by DC that had been activated by Hsp90-peptide complexes involved pathways that share many of the essentials mechanisms involved in stimulation of antigen cross presentation by similar chaperone/antigen complexes (Figs 1–5). These included binding of Hsp90-peptide complexes to SREC-I, internalization via the GEEC compartment and regulation by small GTPases, notably Cdc423 (Fig. 3). Thus the processes involved in triage of antigenic peptides and the routing of such peptides to these two pathways were different in essence from the mechanisms used by DC for other soluble antigens, which instead were shown to involve the use of distinct receptors to access the two routes for processing and presenting external antigens (Bonifaz et al., 2002; Calderwood et al., 2009). It appeared therefore that Hsp90 could trigger multiple pathways of CTL programming after binding the SREC-I receptor. These included CD8+ T lymphocyte activation by the antigen cross presentation pathway, innate immune activation through TLR4 and, as was shown here CD4+ T cell activation by triggering antigen uptake through the MHC class II route (Fig. 2) (Murshid et al., 2010; Calderwood et al., 2009). The significance of SREC-I association with MHCII on the cell surface was not entirely clear (Fig. 5). MHC class II complexes have been thought to cycle from the Golgi compartment to endosomes/lysosomes via the plasma membrane (Berger and Roche, 2009). Formation of SREC-I/Hsp90-peptide complexes at the cell surface could be involved in recruiting MHC class II, and these complexes would then also appear in endosomes (Fig 1). The role, if any of SREC-I/Hsp90 in antigen loading was unclear, although peptides could potentially be transferred from Hsp90 bound to SREC-I to MHC class II complexes in endosomes or lysosomes (Fig. 1). Loading of antigens to MHC class II complexes has been shown to involve chaperoning by HLA-DM molecules and Hsp90 could facilitate this process (Busch et al., 2005).
Our results also indicated that actin assembly and polymerization signaling pathways might be regulated by ligand dependent SREC-I interaction with Cdc42, leading to activation and bringing active Cdc42 to the region of filopodia. Activated Cdc42 could in turn recruit the N-WASP and Arp3 to trigger the formation of actin-based membrane protrusions. This might be the one of many downstream pathways in filopodium formation involving Cdc42. SREC-I might thus modulate motility in APC, an important aspect of localizing and scavenging for antigens within the immune response.
Our findings of roles for SREC-I in both the Class I and Class II mediated pathways of antigen cross presentation may be significant in that CTL programming was shown to require simultaneous or consecutive engagement of the APC by both CD4+ and CD8+ T lymphocytes, a process that resulted in sturdy activation of CTL that could evade apoptosis and engage the cellular target (Murshid et al., 2012; Kurts et al., 2010). A number of questions however remain to be answered with regard to the assignment of HSP-bound antigens to pathways of antigen presentation. These include in particular whether there are intracellular factors that determine if the internalized SREC-I-Hsp90-peptide complexes are transported to either the MHC class I or the class II pathway, or whether the process of assignment to one or other of the pathways is mostly stochastic. In addition other receptors for molecular chaperones have been observed, most notably LOX-1, and the existence of additional receptors in APC might also influence the route of antigen processing and presentation (Theriault et al., 2006).
In conclusion we have characterized a novel property of SREC-I in fostering presentation of molecular chaperone-bound antigens through association with MHC class II and facilitation of Class II presentation.
Figure 6. Schematic diagram of Hsp90-mediated Ag presentation. Endosomal pathway of Hsp90 chaperoned antigen presentation.
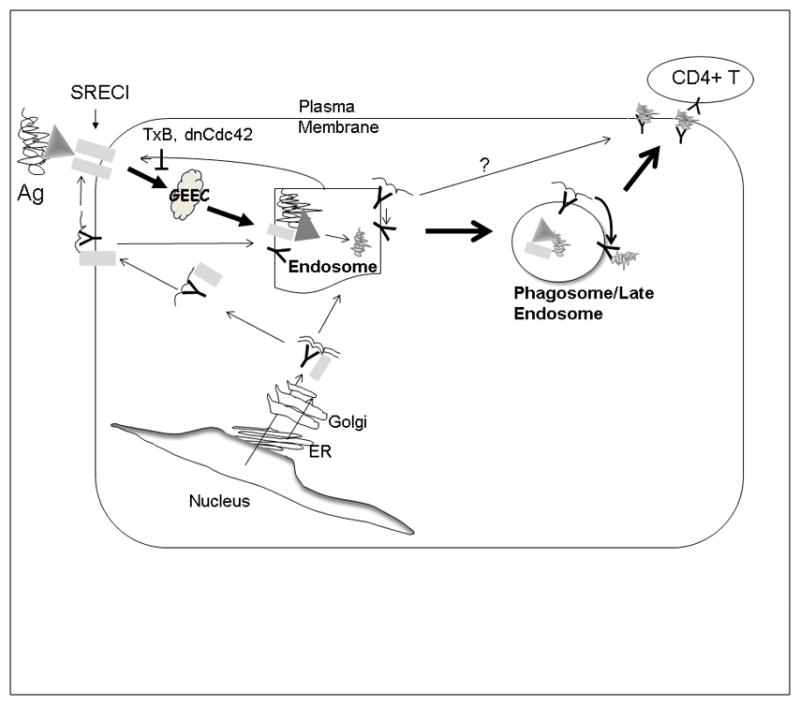
Hsp90-OVA peptide complexes are internalized by APC in an SREC-I receptor-mediated, Cdc42-regulated pinocytic pathway into the GEEC compartment, which is also regulated by the activity Src-tyrosine kinase. OVA peptides then traverse early endosomes complexed to Hsp90 and SREC-I and can be processed and loaded onto MHC-II molecules in recycling endosomes. MHC-II-OVA peptide complexes can then recycle to the plasma membrane and encounter CD4+ T cells. In an addition, MHCII and SREC-I appear to be closely associated in the cell. Both receptors are trafficked to plasma membrane from Golgi and endocytosed in the presence of Hsp90 complexed with Ova and this association may be involved in peptide loading onto MHCII.
Acknowledgments
This work was supported by NIH research grants RO-1CA047407, R01CA119045 and RO-1CA094397
Footnotes
Conflict of interest
The authors have no conflicts of interest that would interfere with the objectivity of the findings.
References
- 1.Kunisawa J, Shastri N. Hsp90alpha chaperones large C-terminally extended proteolytic intermediates in the MHC class I antigen processing pathway. Immunity. 2006;24:523–534. doi: 10.1016/j.immuni.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 2.Murshid A, Gong J, Stevenson MA, Calderwood SK. Heat shock proteins and cancer vaccines: developments in the past decade and chaperoning in the decade to come. Expert review of vaccines. 2011;10:1553–1568. doi: 10.1586/erv.11.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murshid A, Gong J, Calderwood SK. Heat shock protein 90 mediates efficient antigen cross presentation through the scavenger receptor expressed by endothelial cells-I. Journal of immunology. 2010;185:2903–2917. doi: 10.4049/jimmunol.0903635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Srivastava PK. Heat shock protein-based novel immunotherapies. Drug News Perspect. 2000;13:517–522. doi: 10.1358/dnp.2000.13.9.858479. [DOI] [PubMed] [Google Scholar]
- 5.Murshid A, Gong J, Calderwood SK. The role of heat shock proteins in antigen cross presentation. Frontiers in immunology. 2012;3:63. doi: 10.3389/fimmu.2012.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonifaz L, et al. Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J Exp Med. 2002;196:1627–1638. doi: 10.1084/jem.20021598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burgdorf S, Kurts C. Endocytosis mechanisms and the cell biology of antigen presentation. Curr Opin Immunol. 2008;20:89–95. doi: 10.1016/j.coi.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 8.Theriault JR, Adachi H, Calderwood SK. Role of scavenger receptors in the binding and internalization of heat shock protein 70. Journal of immunology. 2006;177:8604–8611. doi: 10.4049/jimmunol.177.12.8604. [DOI] [PubMed] [Google Scholar]
- 9.Delneste Y, et al. Involvement of LOX-1 in dendritic cell-mediated antigen cross-presentation. Immunity. 2002;17:353–362. doi: 10.1016/s1074-7613(02)00388-6. [DOI] [PubMed] [Google Scholar]
- 10.Murshid A, Theriault J, Gong J, Calderwood SK. Investigating receptors for extracellular heat shock proteins. Methods in molecular biology. 2011;787:289–302. doi: 10.1007/978-1-61779-295-3_22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gong J, et al. A heat shock protein 70-based vaccine with enhanced immunogenicity for clinical use. Journal of immunology. 2010;184:488–496. doi: 10.4049/jimmunol.0902255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gong J, et al. T cell activation by heat shock protein 70 vaccine requires TLR signaling and scavenger receptor expressed by endothelial cells-1. Journal of immunology. 2009;183:3092–3098. doi: 10.4049/jimmunol.0901235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rock KL. The ins and outs of cross-presentation. Nat Immunol. 2003;4:941–943. doi: 10.1038/ni1003-941. [DOI] [PubMed] [Google Scholar]
- 14.Calderwood SK, Murshid A, Prince T. The shock of aging: molecular chaperones and the heat shock response in longevity and aging--a mini-review. Gerontology. 2009;55:550–558. doi: 10.1159/000225957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berger AC, Roche PA. MHC class II transport at a glance. Journal of cell science. 2009;122:1–4. doi: 10.1242/jcs.035089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Busch R, et al. Achieving stability through editing and chaperoning: regulation of MHC class II peptide binding and expression. Immunological reviews. 2005;207:242–260. doi: 10.1111/j.0105-2896.2005.00306.x. [DOI] [PubMed] [Google Scholar]
- 17.Kurts C, Robinson BW, Knolle PA. Cross-priming in health and disease. Nat Rev Immunol. 2010;10:403–414. doi: 10.1038/nri2780. [DOI] [PubMed] [Google Scholar]
- 18.Chen XQ, Tan I, Leung T, Lim L. The myotonic dystrophy kinase-related Cdc42-binding kinase is involved in the regulation of neurite outgrowth in PC12 cells. J Biol Chem. 1999;274:19901–5. doi: 10.1074/jbc.274.28.19901. [DOI] [PubMed] [Google Scholar]
- 19.Yamaguchi Y, Katoh H, Yasui H, Mori K, Negishi M. RhoA inhibits the nerve growth factor-induced Rac1 activation through Rho-associated kinase-dependent pathway. J Biol Chem. 2001;276:18977–83. doi: 10.1074/jbc.M100254200. Epub 2001 Mar 15. [DOI] [PubMed] [Google Scholar]
- 20.Walseng E, Bakke O, Roche PA. Major histocompatibility complex class II-peptide complexes internalize using a clathrin- and dynamin-independent endocytosis pathway. J Biol Chem. 2008;283:14717–27. doi: 10.1074/jbc.M801070200. [DOI] [PMC free article] [PubMed] [Google Scholar]