

Abstract
Background
Microdeletions at 17q11.2 often encompass NF1 gene, is the cause for NF1 microdeletion syndrome. Microdeletion at 17q11.2 without the involvement of NF1 gene is rarely reported.
Case presentation
Here we reported a patient carrying a novel de novo deletion at 17q11.2 adjacent to NF1 gene, who presented with developmental delay, short stature, postnatal microcephaly, underweight and dysmorphic features including flat facial profile, dolicocephaly, hypertelorism, short philtrum, flat nasal bridge and posteriorly rotated and low set ears. Chromosomal microarray analysis revealed a 1.69 Mb de novo deletion at 17q11.2 adjacent to NF1 gene, which involves 43 RefSeq genes. We compared this with four overlapping deletions at this interval.
Conclusions
A rare de novo microdeletion at 17q11.2 not involving NF1 gene is associated with developmental delay and dysmorphic features. Seven genes, TAOK1, PHF12, NUFIP2, SLC26A4, SEZ6, GIT1 and TRAF4 are possible candidates for the clinical features of our patient. The delineation of this rare deletion and description of associated clinical phenotypes will help to understand the genotype-phenotype correlation of genomic imbalances at this locus.
Keywords: Developmental delay, Short stature, Microcephaly, Chromosomal microarray, SNP array, 17q11.2, Microdeletion
Background
Chromosomal microarray analysis (CMA) has been extensively used to investigate submicroscopic copy number variants (CNVs) that were not detectable by karyotyping [1]. It has been applied to examining patients with developmental delay/intellectual disability, autism spectrum disorders, and multiple congenital anomalies [2, 3].
Microdeletions at 17q11.2 region often involve a heterozygous 1.5 Mb deletion including NF1 gene, known as NF1 microdeletion syndrome, which is responsible for 5–20 % of all patients with neurofibromatosis type 1 (NF1) [4]. The deletion was mediated by non-allelic homologous recombination between the NF1 repetitive sequence (REP) [5]. Patients with the typical 1.5 Mb deletion are also known to have more severe phenotype than NF1 patients caused by point mutations, which mean that more than one gene in the 17q11.2 region may be involved with clinical phenotypes [6]. Microdeletion at 17q11.2 without the involvement of NF1 gene has not been reported. Here, we described a novel microdeletion at 17q11.2 adjacent to NF1 gene in a patient with developmental delay and dysmorphic facial features.
Case presentation
The patient was born to a healthy unrelated 25-year-old mother and a 27-year-old father without family history of multiple congenital anomalies, intellectual disability, recurrent pregnancy loss, or infertility. The girl was born by natural delivery at 37 weeks of gestation. Her birth weight was 2.520 kg (approximately, -2SD), length 48 cm (approximately, -1SD), and head circumference 33.5 cm (approximately, -1SD). At birth, her metabolic and neonatal hearing screening were all normal.
Her develop milestones were delayed. Head holding was acquired at 6 months of age, she started to sit up at 10 months and walked with support at 21 months. At age of 21 months, her weight was 7.6 kg (approximately, -3SD), her height was 76 cm (approximately, -2SD), and her head circumference was 43.2 cm (approximately, -3SD). She presented with mild dysmorphic features including flat facial profile, dolicocephaly, hypertelorism, short philtrum, flat nasal bridge and posteriorly rotated and low set ears. She also had a short fifth finger on both hand (Fig. 1). Developmental Screening Test (DST) showed that the DQ and MI scores was 58 and 55, respectively.
Fig. 1.
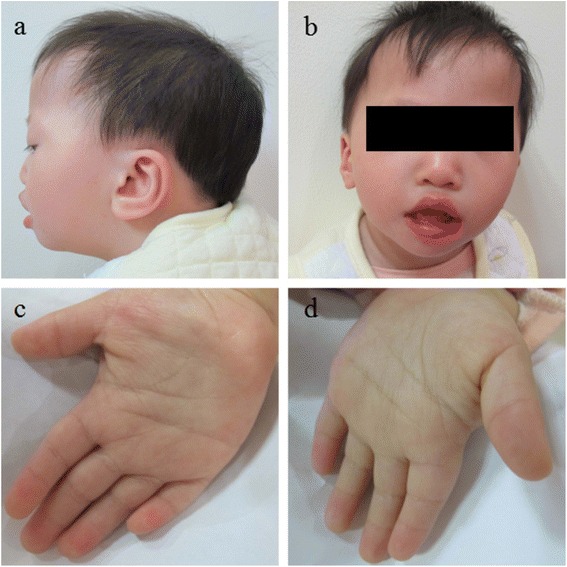
Clinical features of the patients. Note the facial profile, dolicocephaly and low-set posteriorly rotated ear (a); hypertelorism, low nasal bridge and short philtrum (b); short fifth fingers (c and d)
Methods and results
DNA samples were extracted from peripheral blood of the trios using Lab-Aid DNA kit (Zeesan Biotech Co, Ltd, China), DNA concentration was determined with NanoDrop ND-2000 spectrophotometer and soft-ware (NanoDrop Technologies, Berlin, Germany). Genomic wide single nucleotide polymorphism (SNP) array analysis was performed using the Illumina infinium-cytosnp-850 k, which includes over 850 k SNPs in the human genome. Hybridization and array scanning were performed according to the manufacturer’s instruction. Data were analyzed with Illumina Genome Studio and KaryoStudio software. CNVs identified in the samples were visualized by using the UCSC Genome Browser website (http://genome.ucsc.edu) and compared to the Database of Genomic Variants (http://projects.tcag.ca/variation) to exclude CNVs considered as benign variants. The Decipher database and CNV Morbidity Map of Developmental Delay were consulted as resources to aid genotype-phenotype correlation.
A deletion of 1,697,561 bp on chromosome 17 (Fig. 2a) was detected (chr17:27,064,286-28,761,847) (hg19). This position correspond to cytogenetic bands 17q11.2. The chromosomal constitution of the patient was reported as following: arr17q11.2(27064286-28761847) × 1. The parents did not carry the 1.69 Mb copy number variant (CNV), indicating a de novo origin of the rearrangement. The deletion involves 43 genes.
Fig. 2.
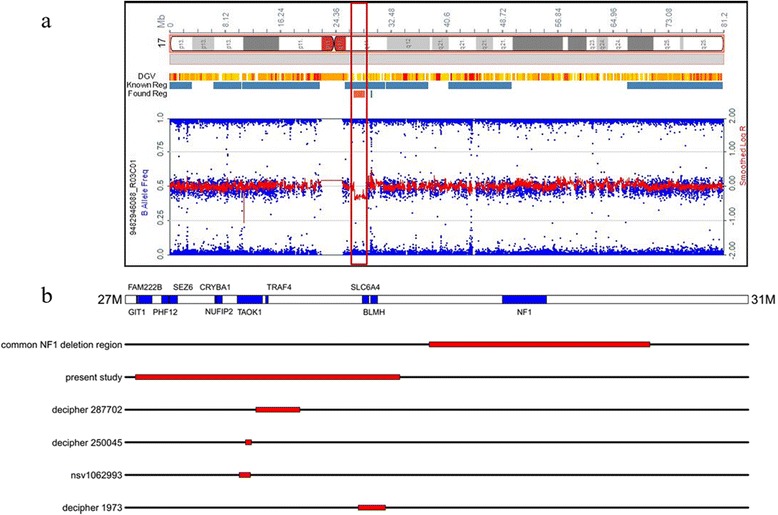
SNP array profile showing a deletion on 17q11.2 (a), and schematic representation of 27 Mbp to 31Mbp in 17q11.2 region (b). Blue bars represent genes discussed in this paper. Also shown, the extent of the deletions (in red) observed in reported cases are compared with the presented case
Discussion
Here we presented a 17q11.2 de novo deletion characterized by SNP array in a patient with developmental delay and mild dysmorphic features. This is a novel deletion, relative large in size and not listed among the reported CNVs in phenotypically normal individuals in the Database of Genomic Variants, therefore, it is considered as a likely pathogenic CNV.
We searched literature and databases for overlapping deletions at this interval without the involvement of NF1 gene. There are only four deletion cases currently reported within this interval, all are much smaller in size (Fig. 2b). One (nsv1062993) is reported in the morbidity map of developmental delay, and the other three (#250045, #287702, #1973) are reported in the Decipher database. The case #250045, presented with microcephaly and seizures, carrying a de novo deletion of 38 kb (chr17:27771342-27809321) involving part of TAOK1 gene. The case with 72 kb deletion (chr17:27730573-27802767) from the morbidity map of developmental delay (nsv1062993) also intercept with TAOK1 gene but the inheritance status and detailed clinical phenotypes are not known, except the patient is presumed to have developmental delay. The deletion nsv1062993 completely overlaps the CNV of #250045, and the two partial overlap TAOK1 gene, we assume that TAOK1 gene plays an important role in the phenotype of patient. The patient #287702 carried a de novo deletion of 17q11.2 (chr17:27837697-28120076) and a maternally inherited duplication of 15q13.3, and showed dysphasia, poor motor coordination and specific learning disability. The patient #1973 carried a deletion of 173 kb (chr17:28496019-28669139) in size, involving genes including SLC6A4, BLMH and part of NSRP1. He showed depression, intellectual disability and psychosis. The breakpoints of this four cases are different, however, their phenotype have some common ground, mainly defined on the basis of the central nervous system.
There are a total of 43 RefSeq genes involved in the deletion in our patient. Seven genes (FAM222B, BLMH, PHF12, CRYBA1, NUFIP2, TAOK1 and SLC6A4) were predicted to have a haploinsufficiency score less than 10, suggestive of possible clinical consequences with one copy deletion. Little clue is available about the potential relationship of FAM222B and BLMH with neurodevelopment. PHF12 is a type of PHD (plant homeodomain) finger protein, acts as a transcriptional repressor. Several proteins with PHD finger are known to epigenetically regulate gene expression and loss of function mutations are associated with intellectual disability phenotypes in human [7, 8]. CRYBA1 is one of the β-crystallins families, encoding both the beta-A3- and beta-A1-crystallins. Different beta-crystallin proteins can interact with each other and other lens proteins, which play a key role in maintaining the transparency of the lens. The mutations of CRYBA1 may be destroy the structure and function of crystallins leading to abnormalities in the development and maturation of the retinal vasculature, and have been identified to be causative for congenital cataracts [9, 10]. Up to now, the patient did not present typical phenotype of congenital cataracts, it may be associated with the phenotypic heterogeneity of congenital cataracts.
NUFIP2 (nuclear fragile X mental retardation protein interacting protein 2) gene encodes an 82-kD protein (called 82-FIP), distributed in various regions of the brain. It was demonstrated that NUFIP2 interacts with FMRP, whose absence causes the fragile-X syndrome [11]. 82-FIP might have a role in the development of the nervous system and in cognitive function. The deregulation of NUFIP2 was reported to be associated with mental retardation or cognitive impairment [12].
The gene of TAOK1 has the lowest predicted haploinsufficiency score. It encodes hTAOK1, which is a member of the Ste20 group of kinases with the kinase domain located at the N-terminus. hTAOK1 was initially cloned from human fetal brain [13], and highly expressed in human brain, as shown by Northern analysis (http://www.kazusa.or.jp/huge/gfpage/KIAA1361/). TAOK1 may play a role in the developing human brain by inducing neuronal apoptosis and regulating microtubule dynamics and checkpoint signaling [14, 15]. Of significance, the case nsv1062993 and #250045 both partially overlapped TAOK1 gene, presenting developmental delay and microcephaly, respectively. Accordingly, we hypothesize that TAOK1 might be involved in the developmental delay and microcephaly in our patient. In addition, a closely related family member TAOK2 is regarded as a autism spectrum disorder susceptibility gene, are shown to regulated basal dendrite development in cortical neurons [16].
The solute carrier family 6 (serotonin neurotransmitter transporter) member 4 gene (SLC6A4) encodes an integral membrane protein that transports the neurotransmitter serotonin from synaptic spaces into presynaptic neurons. Therefore, SLC6A4 gene may play a role in terminating the synaptic actions of serotonin and recycles it into the neurotransmitter pool [17]. Allelic heterogeneity at this gene have been implicated in speech delay, atypical autism, anxiety and obsessive compulsive disorder [18].
In addition, several other genes are known to be functionally important for the development and function of the central nervous system.
SEZ6 (seizure related 6 homolog (mouse)) specific expresses in the brain, especially in the developing forebrain [19]. SEZ6 may have the function on cell adhesion or recognition and protein-protein interaction [20]. The mutations of SEZ6 were associated with febrile seizures and epilepsy [21].
GIT1 (G protein-coupled receptor kinase interacting ArfGAP 1) is a multifunctional signaling adaptor protein. GIT1 interacts with various proteins and forms signaling complex to modulate the development of dendritic spines and neuronal synapses [22]. Git1–/– mice and dGitex21C Drosophila mutant were studied and displayed a microcephaly-like brain size reduction decreased neuronal cell body size, and behavioral deficits such as impaired motor coordination and learning [23].
TRAF4 gene encodes a member of the TNF receptor associated factor (TRAF) family. TRAF proteins are associated with, and mediate signal transduction from members of the TNF receptor superfamily. TRAF4 protein has been shown to interact with neurotrophin receptor, p75 (NTR/NTSR1), and negatively regulate NTR induced cell death and NF-kappa B activation [24]. TRAF4-deficient mice exhibited a high incidence of spina bifida, a defect likened to neural tube defects (NTDs), which revealed that TRAF4 participates in neurulation in vivo [25].
Thus, multiple genes at this interval are likely contributing to the clinical presentations of our patient. Further study is warranted to understand the underlying pathological mechanism.
Conclusions
We described a patient with developmental delay, short stature, postnatal microcephaly, underweight and dysmorphic features. A novel deletion adjacent to the NF1 locus was detected. Several genes are functionally important for neurodevelopment and candidate for this novel microdeletion disorder. Additional overlapping cases will help to better understand the clinical presentation of this disorder and critical genes involved.
Abbreviations
CMA, chromosomal microarray analysis; CNV, copy number variant; DQ, developmental quotient; MI, mental index; RefSeq, reference sequence; SNP, single nucleotide polymorphism
Acknowledgements
We are grateful to the family for participating in this study.
Funding
This work is supported by the project of science and technology of Guangxi Zhuang Autonomous Region (gui-ke-gong 14124004-1-8), and there is no role for funding agent in this work.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article. More details are available on request.
Authors’ contribution
SKC and YPS conceived and designed the experiments. XF and JSL diagnosed and followed up the patient. YQL, SJZ and QY prepared reagents, materials and analysis tools, RYC and JW performed the experiments. CYF and SY analyzed the data. BBX followed up the patient and wrote the first draft of the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent
Written informed consent was obtained from patient’s parents for the publication of this report and any accompanying images.
Contributor Information
Shaoke Chen, Phone: 86-771-2860792, FAX: 86-771-2860799, Email: chenshaoke123@163.com.
Yiping Shen, Phone: 617-553-5832, FAX: 617-730-0338, Email: yiping.shen@childrens.harvard.edu.
References
- 1.Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86(5):749–64. doi: 10.1016/j.ajhg.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sagoo GS, Butterworth AS, Sanderson S, Shaw-Smith C, Higgins JP, Burton H. Array CGH in patients with learning disability (mental retardation) and congenital anomalies: updated systematic review and meta-analysis of 19 studies and 13,926 subjects. Genet Med. 2009;11(3):139–46. doi: 10.1097/GIM.0b013e318194ee8f. [DOI] [PubMed] [Google Scholar]
- 3.Hochstenbach R, van Binsbergen E, Engelen J, Nieuwint A, Polstra A, Poddighe P, et al. Array analysis and karyotyping: workflow consequences based on a retrospective study of 36,325 patients with idiopathic developmental delay in the Netherlands. Eur J Med Genet. 2009;52(4):161–9. doi: 10.1016/j.ejmg.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 4.Kehrer-Sawatzki H, Kluwe L, Sandig C, Kohn M, Wimmer K, Krammer U, et al. High frequency of mosaicism among patients with neurofibromatosis type 1 (NF1) with microdeletions caused by somatic recombination of the JJAZ1 gene. Am J Hum Genet. 2004;75(3):410–23. doi: 10.1086/423624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dorschner MO, Sybert VP, Weaver M, Pletcher BA, Stephens K. NF1 microdeletion breakpoints are clustered at flanking repetitive sequences. Hum Mol Genet. 2000;9(1):35–46. doi: 10.1093/hmg/9.1.35. [DOI] [PubMed] [Google Scholar]
- 6.Venturin M, Guarnieri P, Natacci F, Stabile M, Tenconi R, Clementi M, et al. Mental retardation and cardiovascular malformations in NF1 microdeleted patients point to candidate genes in 17q11.2. J Med Genet. 2004;41(1):35–41. doi: 10.1136/jmg.2003.014761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim HG, Kim HT, Leach NT, Lan F, Ullmann R, Silahtaroglu A, et al. Translocations disrupting PHF21A in the Potocki-Shaffer-syndrome region are associated with intellectual disability and craniofacial anomalies. Am J Hum Genet. 2012;91(1):56–72. doi: 10.1016/j.ajhg.2012.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jensen LR, Amende M, Gurok U, Moser B, Gimmel V, Tzschach A, et al. Mutations in the JARID1C gene, which is involved in transcriptional regulation and chromatin remodeling, cause X-linked mental retardation. Am J Hum Genet. 2005;76(2):227–36. doi: 10.1086/427563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qi Y, Jia H, Huang S, Lin H, Gu J, Su H, et al. A deletion mutation in the betaA1/A3 crystallin gene (CRYBA1/A3) is associated with autosomal dominant congenital nuclear cataract in a Chinese family. Hum Genet. 2004;114(2):192–7. doi: 10.1007/s00439-003-1049-7. [DOI] [PubMed] [Google Scholar]
- 10.Graw J. Genetics of crystallins: cataract and beyond. Exp Eye Res. 2009;88(2):173–89. doi: 10.1016/j.exer.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 11.Ye X, Mehlen P, Rabizadeh S, VanArsdale T, Zhang H, Shin H, et al. TRAF family proteins interact with the common neurotrophin receptor and modulate apoptosis induction. J Biol Chem. 1999;274(42):30202–8. doi: 10.1074/jbc.274.42.30202. [DOI] [PubMed] [Google Scholar]
- 12.Regnier CH, Masson R, Kedinger V, Textoris J, Stoll I, Chenard MP, et al. Impaired neural tube closure, axial skeleton malformations, and tracheal ring disruption in TRAF4-deficient mice. Proc Natl Acad Sci U S A. 2002;99(8):5585–90. doi: 10.1073/pnas.052124799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim MH, Gunnersen JM, Tan SS. Localized expression of the seizure-related gene SEZ-6 in developing and adult forebrains. Mech Dev. 2002;118(1–2):171–4. doi: 10.1016/S0925-4773(02)00238-1. [DOI] [PubMed] [Google Scholar]
- 14.Shimizu-Nishikawa K, Kajiwara K, Sugaya E. Cloning and characterization of seizure-related gene, SEZ-6. Biochem Biophys Res Commun. 1995;216(1):382–9. doi: 10.1006/bbrc.1995.2635. [DOI] [PubMed] [Google Scholar]
- 15.Yu ZL, Jiang JM, Wu DH, Xie HJ, Jiang JJ, Zhou L, et al. Febrile seizures are associated with mutation of seizure-related (SEZ) 6, a brain-specific gene. J Neurosci Res. 2007;85(1):166–72. doi: 10.1002/jnr.21103. [DOI] [PubMed] [Google Scholar]
- 16.de Anda FC, Rosario AL, Durak O, Tran T, Graff J, Meletis K, et al. Autism spectrum disorder susceptibility gene TAOK2 affects basal dendrite formation in the neocortex. Nat Neurosci. 2012;15(7):1022–31. doi: 10.1038/nn.3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kikuno R, Nagase T, Waki M, Ohara O. HUGE: a database for human large proteins identified in the Kazusa cDNA sequencing project. Nucleic Acids Res. 2002;30(1):166–8. doi: 10.1093/nar/30.1.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu MF, Wang SG. Human TAO kinase 1 induces apoptosis in SH-SY5Y cells. Cell Biol Int. 2008;32(1):151–6. doi: 10.1016/j.cellbi.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 19.Draviam VM, Stegmeier F, Nalepa G, Sowa ME, Chen J, Liang A, et al. A functional genomic screen identifies a role for TAO1 kinase in spindle-checkpoint signalling. Nat Cell Biol. 2007;9(5):556–64. doi: 10.1038/ncb1569. [DOI] [PubMed] [Google Scholar]
- 20.Ramamoorthy S, Bauman AL, Moore KR, Han H, Yang-Feng T, Chang AS, et al. Antidepressant- and cocaine-sensitive human serotonin transporter: molecular cloning, expression, and chromosomal localization. Proc Natl Acad Sci U S A. 1993;90(6):2542–6. doi: 10.1073/pnas.90.6.2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adamsen D, Meili D, Blau N, Thony B, Ramaekers V. Autism associated with low 5-hydroxyindolacetic acid in CSF and the heterozygous SLC6A4 gene Gly56Ala plus 5-HTTLPR L/L promoter variants. Mol Genet Metab. 2011;102(3):368–73. doi: 10.1016/j.ymgme.2010.11.162. [DOI] [PubMed] [Google Scholar]
- 22.Bardoni B, Castets M, Huot ME, Schenck A, Adinolfi S, Corbin F, et al. 82-FIP, a novel FMRP (fragile X mental retardation protein) interacting protein, shows a cell cycle-dependent intracellular localization. Hum Mol Genet. 2003;12(14):1689–98. doi: 10.1093/hmg/ddg181. [DOI] [PubMed] [Google Scholar]
- 23.Pasmant E, de Saint-Trivier A, Laurendeau I, Dieux-Coeslier A, Parfait B, Vidaud M, et al. Characterization of a 7.6-Mb germline deletion encompassing the NF1 locus and about a hundred genes in an NF1 contiguous gene syndrome patient. Eur J Hum Genet. 2008;16(12):1459–66. doi: 10.1038/ejhg.2008.134. [DOI] [PubMed] [Google Scholar]
- 24.Zhang H, Webb DJ, Asmussen H, Niu S, Horwitz AF. A GIT1/PIX/Rac/PAK signaling module regulates spine morphogenesis and synapse formation through MLC. J Neurosci. 2005;25(13):3379–88. doi: 10.1523/JNEUROSCI.3553-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hong ST, Mah W. A critical role of GIT1 in vertebrate and invertebrate brain development. Exp Neurobiol. 2015;24(1):8–16. doi: 10.5607/en.2015.24.1.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets supporting the conclusions of this article are included within the article. More details are available on request.