

Abstract
Reinduction of fetal gene expression is characteristic of the failing adult heart. Han et al. (2014) demonstrate that a novel cluster of long noncoding RNAs, Myheart, protects the heart from pathological hypertrophy, involving a negative-feedback molecular circuit with the epigenetic Brg1 complex to inhibit fetal-gene reactivation.
Human hearts are capable of undergoing remarkable cardiac remodeling in response to various environmental stimuli. Cardiac hypertrophy is one of the primary responses of the heart to pathophysiological stress. While physiological hypertrophy enhances cardiac performance to match physiological demands, pathological hypertrophy results in a decrease in cardiac function, accompanied by reinduction of the fetal gene program and increased fibrosis, representing a maladaptive response to pathological stress/stimuli, which often progresses to heart failure if prolonged (Ahmad et al., 2005; Hill and Olson, 2008). Two myosin heavy-chain genes, Myh6 and Myh7, which encode the α- and β-MHC contractile proteins, respectively, are expressed in mammalian hearts in opposing patterns during cardiac development and remodeling. Cardiac injury shifts this ratio toward Myh7, with detrimental effects on cardiac functions and outcomes. Transcriptional and epigenetic regulation govern the expression of Myh6 and Myh7 genes (Hill and Olson, 2008), and genetic mutations and altered expression of these genes have been linked to cardiac hypertrophy and other cardiomyopathies (Ahmad et al., 2005). More recently, miR-208a and miR-208b, encoded by introns of Myh6 and Myh7, respectively, have also been involved in the regulation of α/β-MHC expression in pathological cardiac hypertrophy (Callis et al., 2009; van Rooij et al., 2007). Han and colleagues now report that a long noncoding RNA (lncRNA) transcript from the antisense strand of the Myh7 locus functions as a molecular switch in tipping the balance between α- or β-MHC expression by regulating the Brg1-HDAC-PARP chromatin remodeling complex (Han et al., 2014).
Although lncRNAs lack functional open reading frames, their critical roles in various biological systems have been increasingly recognized in recent years (Batista and Chang, 2013). Perhaps the most relevant examples of lncRNA function in cardiac lineage commitment would be the recently identified Braveheart and Fendrr (Grote et al., 2013; Klattenhoff et al., 2013). Mechanistically, Braveheart interacts with SUZ12, a component of the polycomb-repressive complex 2 (PRC2), to epigenetically activate the expression of mesoderm posterior 1 (Mesp1), a master regulator of cardiovascular progenitors. Interestingly, not only can Fendrr bind to the PRC2 complex and regulate its activity, it can also associate with the TrxG/MLL complex to control cardiac lineage commitment.
Previous work from Dr. Chang's group identified a Brg1-HDAC-PARP complex that cooperatively controls the change in the α/β-MHC ratio in failing hearts (Hang et al., 2010). Han and colleagues now describe a novel lncRNA, Myheart (Mhrt), enriched in adult cardiomyocytes and able to protect adult hearts from pathological hypertrophy. The authors initiated this elegant study by identifying a cluster of alternatively spliced antisense transcripts of the Myh7 gene. Intriguingly, these lncRNAs, which are spliced isoforms of Mhrt, appeared to have a tight correlation with the Myh6/Myh7 ratio during cardiac development and in hypertrophic hearts. Indeed, upon transverse aortic constriction (TAC) in a mouse model of pathological hypertrophy, cardiomyocytes showed a progressive loss of Mhrt expression along with the development of pathological hypertrophy. Remarkably, restoring Mhrt expression to the prestress level attenuated pathological hypertrophic responses and restored cardiac function, demonstrating the cardioprotective role of Mhrt in vivo.
ATP-dependent chromatin remodeling complexes are important controllers of chromatin dynamic structure, and their biological specificity arises, at least partly, from the combinatory assembly of the complex (Ho and Crabtree, 2010). In order to uncover the molecular mechanism by which Mhrt regulates Myh expression and cardiac hypertrophy, Han et al. tested whether the Brg1 chromatin remodeling complex is responsible for the downregulation of Mhrt expression in pathological hypertrophy (Han et al., 2014). The authors defined specific DNA elements responsible for Brg1-mediated regulation of Mhrt and Myh6. Perhaps the most intriguing result of the paper is that Mhrt can directly interact with the helicase domain of Brg1 to regulate its occupancy and activity, suggesting that Mhrt may function as a decoy for the chromatin remodeler to prevent its occupancy on target gene promoters. Thus, Han et al. uncover a negative feedback loop between a lncRNA and the Brg1 chromatin remodeling protein complex, providing a molecular switch for fine-tuning the dynamic regulation of chromatin remodeling complex function in the heart (Figure 1).
Figure 1. Regulation of Cardiac Gene Expression by Epigenetic Regulators and lncRNAs during Heart Development and Hypertrophy.
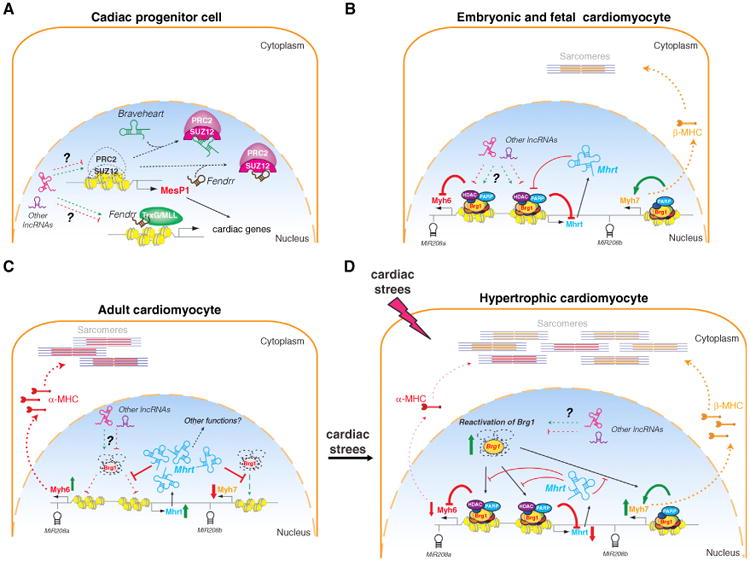
(A) In cardiac progenitor cells, lncRNA Braveheart functions as a decoy for PRC2 complex by binding to Suz12 to exclude the PRC2 complex from the promoter of MesP1, a master cardiac transcription factor, thus leading to the activation of MesP1 and downstream cardiac specification transcription network. Similarly, Fendrr, another cardiac lncRNA, associates with TrxG/MLL complex to regulate the activation of a set of cardiac specification genes in cardiac progenitor cells.
(B) In embryonic cardiomyocytes, where Brg1 expression remains relatively high, the involvement of lncRNAs is unclear. Mhrt may participate in the regulation of Myh6 and Myh7 expression.
(C) In adult cardiomyocytes, Brg1 protein levels are low; thus the repression of Myh6 and Mhrt expression is released and Myh7 transcription is inhibited.
(D) Under cardiac stress, Brg1 is reactivated, which triggers the tipping of a mutual inhibitory balance between Mhrt and Brg1 complexes, resulting in the reactivation of fetal gene program with high Myh7 expression and low Myh6 and Mhrt level in hypertrophic cardiomyocytes.
While this study unveils an important novel lncRNA in cardiomyopathy, many questions remain to be answered. For example, it is still unclear how the binding of Mhrt to the helicase domain of Brg1 can modulate the assembly of the SWI/SNF complex and how individual components of this complex are specifically recruited. Nor is it completely clear at this time how a lncRNA can define the specificity of such a regulatory complex to achieve appropriate activation/repression of target genes. More specifically, while the Brg1 complex mediates Myh6 and Mhrt inhibition, it also mediates the activation of Myh7 in the same cell. It remains elusive if the primary function of Mhrt is to regulate Brg1-mediated Myh6 inhibition in cis or to regulate a plethora of pathological hypertrophic genes in trans. It should be pointed out that while most of the functional studies conducted here came from a gain-of-function approach, a genetic loss-of-function approach for Mhrt would be essential to establish its biological function. It will also be interesting to determine whether mutations in Mhrt are associated with human cardiomyopathy, especially hypertrophic cardiomyopathy. Another fascinating fact about Mhrt is that it is encoded partially by the antisense strand of the Myh7 gene. Thus, to date, the DNA loci encoding Myh6 and Myh7 have been found to encompass not only major cardiac contractile protein genes, but also micro-RNAs (miR-208a and miR-208b) and now lncRNA (Mhrt). It is amazing to perceive how such rich biological information is incorporated into such a condensed genomic region and raises an interesting question not addressed in the paper: are there physical and functional interactions among mRNAs (Myh6, Myh7), miRNAs (miR-208a, miR-208b), and lncRNA (Mhrt) encoded by the Myh6 and Myh7 gene loci? Answers to these questions will certainly reveal new mechanisms of how different coding and non-coding RNAs arising from same locus harmonize their functions, in cis or in trans, during cardiac development and in heart disease.
In summary, Han et al. identify Mhrt as the first cardioprotective lncRNA in the adult heart and also as the first lncRNA regulating ATP-dependent chromatin remodeling complexes in vivo. Evidently, we are still at the beginning of lncRNA discovery; understanding their functional roles and molecular mechanisms in a variety of biological systems warrants future investigation. Nevertheless, the finding that an lncRNA directly regulates chromatin remodeling and targets gene expression undoubtedly offers great opportunities (and challenges) in developing therapeutic interventions to treat cardiovascular diseases.
Acknowledgments
The authors are supported by the March of Dimes Foundation (FY-11-426), Muscular Dystrophy Association (186548, 294854), and the NIH (HL085635, HL116919).
References
- Ahmad F, Seidman JG, Seidman CE. Annu Rev Genomics Hum Genet. 2005;6:185–216. doi: 10.1146/annurev.genom.6.080604.162132. [DOI] [PubMed] [Google Scholar]
- Batista PJ, Chang HY. Cell. 2013;152:1298–1307. doi: 10.1016/j.cell.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callis TE, Pandya K, Seok HY, Tang RH, Tatsuguchi M, Huang ZP, Chen JF, Deng Z, Gunn B, Shumate J, et al. J Clin Invest. 2009;119:2772–2786. doi: 10.1172/JCI36154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grote P, Wittler L, Hendrix D, Koch F, Währisch S, Beisaw A, Macura K, Bläss G, Kellis M, Werber M, Herrmann BG. Dev Cell. 2013;24:206–214. doi: 10.1016/j.devcel.2012.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han P, Li W, Lin CH, Yang J, Shang C, Nuernberg ST, Jin KK, Xu W, Lin CY, Lin CJ, et al. Nature. 2014 doi: 10.1038/nature13596. Published online August 10, 2014. http://dx.doi.org/10.1038/nature13596. [DOI] [PMC free article] [PubMed]
- Hang CT, Yang J, Han P, Cheng HL, Shang C, Ashley E, Zhou B, Chang CP. Nature. 2010;466:62–67. doi: 10.1038/nature09130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JA, Olson EN. N Engl J Med. 2008;358:1370–1380. doi: 10.1056/NEJMra072139. [DOI] [PubMed] [Google Scholar]
- Ho L, Crabtree GR. Nature. 2010;463:474–484. doi: 10.1038/nature08911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klattenhoff CA, Scheuermann JC, Surface LE, Bradley RK, Fields PA, Steinhauser ML, Ding H, Butty VL, Torrey L, Haas S, et al. Cell. 2013;152:570–583. doi: 10.1016/j.cell.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Science. 2007;316:575–579. doi: 10.1126/science.1139089. [DOI] [PubMed] [Google Scholar]