

Abstract
Objective
To design an international standard to be used when reporting reliability of the implanted components of cochlear implant systems to appropriate governmental authorities, cochlear implant (CI) centers, and for journal editors in evaluating manuscripts involving cochlear implant reliability.
Study Design
The International Consensus Group for Cochlear Implant Reliability Reporting was assembled to unify ongoing efforts in the United States, Europe, Asia, and Australia to create a consistent and comprehensive classification system for the implanted components of CI systems across manufacturers.
Setting
All members of the consensus group are from tertiary referral cochlear implant centers.
Interventions
None.
Main Outcome Measure
A clinically relevant classification scheme adapted from principles of ISO standard 5841-2:2000 (1) originally designed for reporting reliability of cardiac pacemakers, pulse generators, or leads.
Results
Standard definitions for device failure, survival time, clinical benefit, reduced clinical benefit, and specification were generated. Time intervals for reporting back to implant centers for devices tested to be “out of specification,” categorization of explanted devices, the method of cumulative survival reporting, and content of reliability reports to be issued by manufacturers was agreed upon by all members. The methodology for calculating Cumulative survival was adapted from ISO standard 5841-2:2000 (1).
Conclusion
The International Consensus Group on Cochlear Implant Device Reliability Reporting recommends compliance to this new standard in reporting reliability of implanted CI components by all manufacturers of CIs and the adoption of this standard as a minimal reporting guideline for editors of journals publishing cochlear implant research results.
Keywords: Cochlear implant, Device failure, Reliability, Reporting standard
ISO standard 5841-2:2000 (1) is the current standard for reporting clinical performance of populations of pulse generators or leads for cardiac pacemakers and has been adapted by cochlear implant (CI) manufacturers to generate cumulative survival curves for reporting reliability of the implanted components of CI systems. Efforts to define a standard definition of internal CI device failure and to modify the ISO standard to more accurately reflect CI reliability were started in the United States and in Europe (1–4). Because of the global nature of the CI industry, The International Consensus Group for Cochlear Implant Reliability Reporting was formed on November 27, 2005 with the mission to create a clinically relevant reliability standard to be used by all CI manufacturers as they comply to reporting requirements to their respective governmental authority. An additional goal was to create a minimal reporting guideline for editors of journals who publish results of CI research related to reliability.
MATERIALS AND METHODS
Representatives from the United States, Europe, Japan, Australia, and Korea met to form the Global Consensus Group for Cochlear Implant Reliability Reporting in Hong Kong on November 27, 2005. An agenda was set to review independent efforts by researchers from each global region who were attempting to standardize internal device reliability reporting. A consensus group chairman was elected (R.D.B.). Subsequent meetings were held in Vienna, Austria on June 16, 2006; in Charlotte, North Carolina on April 12, 2007; in Sydney, Australia on November 1, 2007; in San Diego, California on April 11, 2008; and in Seattle, Washington on June 19, 2009. Each consensus group meeting was held in conjunction with a major international CI meeting with effort to solicit as much feedback from clinicians and scientists in the CI industry as possible. All members have approved the definitions and standard presented in this report.
RESULTS
The following basic definitions apply to the reporting of device reliability:
Manufacturer
The company who fabricated the internal CI device. According to current industry standards, this is also the company that will complete the analysis of explanted internal device components.
Device
Any internal CI component that is explanted or is left in vivo but is not within specification or providing expected clinical benefit according to ex vivo testing.
Competent authority or notified body
The respective national governing body accountable for device labeling and for consumer safety. Examples include the Communauté Européenne in Europe, the Food and Drug Administration in the United States, and the Therapeutic Goods Administration in Australia.
CI center
The clinical program that provided the CI evaluation, surgery, and subsequent longitudinal care to the patient in relation to their CI.
Survival time
The duration of internal CI device functioning within specifications as determined by the individual manufacturer and approved by the respective competent authority or notified body.
Clinical benefit
Patient performance is better than before the initial cochlear implantation, as demonstrated by speech reception test results, behavioral measures, pure-tone thresholds, and objective measures.
Reduced clinical benefit
Patient performance is materially below the previous stable performance with the CI as demonstrated by repeated performance measures over time.
Specification
Technical characteristics of a specific CI device that have been stated by the manufacturer to the notified body/competent authority when applying for market approval. Such technical characteristics can usually be measured objectively. Whether a device is “in” or “out” of’ specification will be determined by objective measures (e.g. Integrity Test, Bench Test) by the clinic and the manufacturer when appropriate.
The following principles should guide the reporting of device reliability:
When a device is explanted, it should be immediately returned by the CI Center to the appropriate manufacturer to be analyzed and assigned to one of the reporting categories (Fig. 1).
All explanted devices (category C) that test Bout[ of specification are considered internal device failures. These devices are to be included in the calculation of cumulative survival rate (CSR). Calculating and reporting of the CSR will be in accordance with the methodology outlined in ISO standard 5841-2:2000 (1).
Manufacturer’s reports of device reliability should indicate the sources of data, the sample size, and the time interval over which the data was collected. There must be no device exclusions.
Reports of CSR should give complete historic data of a given device, describing any technical modifications (that can be integrated into historic data by starting at Time 0).
The complete data set for internal device reliability of the “mother” product should always be supplied when presenting data on subsequent device modifications.
A “new internal device” can be attributed when there has been a change in the case and/or the electrodes that has been labeled by its own mark with a competent authority.
The overall CSR is reported and then further divided to separate adults and children (patients younger than 18 yr) and should be reported separately and with 95% confidence intervals as appropriate.
Survival time begins with closure of the wound at internal CI device surgical placement.
The manufacturer is required to notify, in writing, the CI center caring for the patient within 60 days of receipt of an explanted device as to whether the internal component was “in” or “out” of specification. Once root cause analysis is completed, which may take significant time to complete depending on the particular issue, the CI center should be notified within 60 days, in writing, to inform the patient of the outcome of device analysis.
At the 6-month interval from receipt of the explanted CI to the manufacturer, a device remaining under study for reliability should be reported as a Category C and included in CSR reporting until final analysis is completed. If the device is found to be “in specification” and no improved clinical performance is documented after internal device replacement (Category B2), than the device can be removed by the manufacturer from CSR reporting. The competent authority and the CI center should be notified in writing within 60 days of the change in category.
Devices determined to be “out of specification” by both clinical testing and reduced clinical benefit (resulting in device nonuse) and are not surgically removed at patient request are required to be reported as Category C and in CSR statistics.
Devices damaged due to trauma are categorized with all other explants. If the device is shown to be “out” of specification, they are categorized as C devices and will be listed in the CSR. If ipsilateral reimplatation produces improved clinical benefit, then the device would be listed as Category C and included in CSR reporting. If shown to be within specification, they are classified as Category D and not included in the CSR report.
FIG. 1.
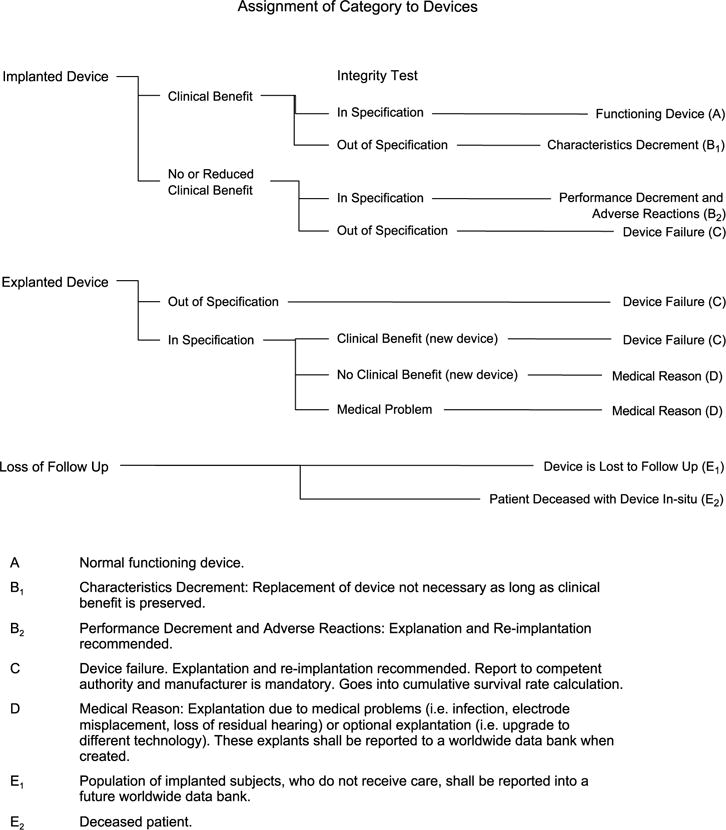
Categorization of explanted internal CI device components (adapted from Ref. 5). This figure was adapted from Figure A.1 (p.6) of ISO 5841-2:2000(E). This figure is not to be considered an official ISO figure nor was it authorized by ISO. Copies of ISO 5841-2:2000(E) can be purchased from ANSI at http://webstore.ansi.org.
The Internal Cochlear Implant Device Reliability Report, to be provided by the manufacturer, should include the following details and be sent to the competent authority/notified body and to CI clinic:
-
Header
Report header, manufacturer, type of device, serial number, date of manufacture, dates of implantation and explantation, institution (CI center), appropriate patient identifier, date of birth, sex, date of report to the competent authority/notified body, date of report to CI center.
-
Main Body
The report should contain the following sections:- Clinical summary;
- Test results: the result of each in vivo and ex vivo test performed on the implant should be described;
- Conclusion: the primary mode of failure according to the agreed classification (above) and should be described in one of the following categories:
- Impact failure
- Hermeticity failure
- Electronic failure
- Electrode array malfunction
- Other: specify
- No cause determined
- Corrective actions, schedule of implementation of corrective actions, safety notice, and information to customers;
- Clear documentation of inclusion of device in CSR reporting, pending inclusion or not included in CSR calculations (a simple check box format is suggested).
CONCLUSION
This new consensus statement provides a standard tool for reporting internal CI device reliability that is fair and consistent to all manufacturers, secures consistent notification to the relevant competent authorities, and provides the necessary information to CI centers for patient counseling. This standard also defines a minimal reporting guideline to ensure consistent reporting of device reliability in the medical literature.
References
- 1.ISO Standard: Implants for surgery—cardiac pacemakers. Part 2: Reporting of clinical performance of populations of pulse generators or leads: ISO 5841/2-2000. Available at: http://www.iso.org/iso/iso_catalogue/catalogue_ics/catalogue_detail_ics.htm?csnumber=31491&ics1=11&ics2=040&ics3=40.
- 2.European Consensus Statement on cochlear implant failures and explantations [Editorial] Otol Neurotol. 2005;26:1097–9. [PubMed] [Google Scholar]
- 3.Battmer RD, O’Donoghue GM, Lenarz T. A multicenter study of device failure in European cochlear implant centers. Ear Hear Suppl. 2007;0095S:95S–9. doi: 10.1097/AUD.0b013e3180315502. [DOI] [PubMed] [Google Scholar]
- 4.Backous DD, Watson SD. Standardization of cochlear implant device reliability reporting in the United States: an interim report. Ear Hear. 2007;28:91S–4. doi: 10.1097/AUD.0b013e31803154b0. [DOI] [PubMed] [Google Scholar]
- 5.Balkany TJ, Hodges AV, Buchman CA, et al. Cochlear implant soft failures consensus development conference statement. Otol Neurotol. 2005;26:815–8. doi: 10.1097/01.mao.0000178150.44505.52. [DOI] [PubMed] [Google Scholar]