

Abstract
Objectives
To evaluate anthropometric measures of growth and development (height, weight, body mass index (BMI)) in a group of children, adolescents, and young adults diagnosed with Juvenile Onset Huntington’s Disease (JHD).
Methods
Growth measures for 18 JHD patients, documented prior to or shortly after diagnosis, were obtained through medical records. JHD growth measures were compared to a large sample (n=274) of healthy children, as well as the Center for Disease Control (CDC) growth norms.
Results
After controlling for sex and age, the JHD subjects had no significant differences in height. However, they were an average of 10% lower than controls in weight and BMI. Using CDC norms, the JHD subjects had the same pattern of normal height but decrement in weight. Length of cytosine-adenine-guanine (CAG) repeat in the huntingtin gene was significantly correlated to measures of weight with longer CAG repeats being associated with more severe weight reduction. A subset of 4 subjects had measures that pre-dated onset of any symptom and were therefore prodromal JHD (preJHD). These subjects also had a significant decrement in BMI compared to CDC norms.
Conclusions
Children with JHD have normal height, but significantly reduced weight and BMI, indicative of a specific deficit in body weight. As the preJHD subjects were also low in BMI, this suggests that these changes are directly due to the effect of the mutated gene on development, rather than symptom manifestation of the disease itself. Potential mechanisms of the weight decrement include energy deficiency due to mitochondrial dysfunction during development.
Keywords: Huntington’s disease, Juvenile, Body Weight, Growth
Introduction
Huntington’s disease (HD) is a neurodegenerative disease, inherited in autosomal dominant fashion, and typically manifests with cognitive decline, neuropsychiatric disturbances and involuntary movements [1]. While the typical age range of disease onset is between 30 and 50 years old, approximately 5° of all HD patients [2-4] occur under the age of 21 years. These earlier-onset cases are classified as juvenile huntington’s disease (JHD).
HD is caused by an expansion of cytosine-adenine-guanine (CAG) repeats in the huntingtin (HTT) gene [5]. Although the mechanism by which mutant HTT (mHTT) leads to neurodegeneration has not been fully understood, evidence suggest that defects in energy metabolism may play a key role in the pathophysiology [6-8]. In HD, skeletal muscle has been shown to have an energy deficit [9-11], which is in line with the most common ‘peripheral’ manifestation of HD: lower than normal body mass index (BMI) and weight loss. The lower than normal BMI has been documented in prodromal HD (preHD) subjects as well [12-14].
Energy deficiency may preferentially affect tissues of highest metabolic demand namely the brain and skeletal muscle [15, 16]. During childhood growth and development - an epoch in time of high metabolic demand [17-19], these tissues might be particularly vulnerable to potential energy deficits related to the lifelong HTT expression [20].
Previously, we reported that preHD children (6-18 years of age, tested for the gene expansion for research purposes only) to have normal height but decreased weight, resulting in lower than normal BMI [21]. The current study was designed to extend our earlier findings by evaluating growth and development measures (height, weight, BMI) in a group of children diagnosed with JHD.
Materials and Methods
This study was approved by our local Institutional Review Board (IRB). Subjects with JHD were identified by retrospective chart review. By accessing hospital database, we identified 535 patients diagnosed with Huntington’s disease at the University of Iowa Hospitals and Clinics between the years of 1966 and 2013. Since no unique billing code exists for JHD, we reviewed all of the charts with the HD diagnosis.
A total of 18 subjects (n=9 females, n=9 males), diagnosed between 1970 and 2010 fulfilled the inclusion criteria of (1) symptom onset before 21 years old, (2) HD diagnosis confirmed by documented genetic testing in the patient or clinical diagnosis of HD based on the accepted criteria prior to the availability of genetic testing [22-24], and (3) height, weight, and BMI data available at time of diagnosis or prior to HD diagnosis. Age at symptom onset was defined as the age when the first significant change was noticed, either in cognitive, psychiatric or motor symptoms [24, 25]. Clinical characteristics of 12 of the 18 had been reported previously [23], however growth measures were not documented in that case series.
The average age of symptom onset was 11.9 years (range 4.0-19.4 years). The most common presenting symptom was behavioral change (n=8), with the beginning symptom being motor in 7 patients, cognitive change as presenting symptom in 2 patients, and onset of seizures as presenting symptom for one patient. Age at diagnosis was determined by the age at which the clinical diagnosis of HD was made. All diagnoses were made before the age of 21 with the exception of one patient in which formal diagnosis was made soon after she turned 22 years old. The average age at diagnosis was 15.0 years (range 6.3-22.4 years). Time between symptom onset and formal diagnosis in JHD can be quite long and in this sample, the average time was 3.2 years with a range of 0.9 - 10.9 years. Molecular confirmation with CAG repeat length was available for 9 of the cases with an average of 75.1 repeats (range 53-130).
Healthy children (n=274) were recruited from the community via local newspaper advertisements for separate studies on normal brain development. All children in the healthy control group had no major medical, neurologic, or psychiatric illness, nor any history of learning disability as disclosed by parents during the screening process. The Institutional Review Board (IRB) of the University of Iowa approved the study.
Growth Measures: For the healthy control sample, height (cm) and weight (kg) were obtained by a trained research nurse at the University of Iowa Institute for Clinical and Translational Sciences, and BMI was calculated as weight (kg) / height (m2). For the JHD subjects these measures were obtained from the medical records. Additionally, standardized Z-scores for height, weight and BMI were calculated for the JHD subjects using CDC growth charts database [26], controlling for age and sex. See Table 1 for demographic and growth measurement data.
Table 1.
Demographics, unadjusted growth and genetic data.
| JHD | Healthy Controls | |||
|---|---|---|---|---|
| 9 males, 9 females | 135 males, 139 females | |||
| Mean (SD) | Min - Max | Mean (SD) | Min - Max | |
| Growth Data Age (years) | 13.3 (5.4) | 3.1-20.7 | 12.6 (3.6) | 6.0-21.9 |
| Height (cm) | 149.2 (29.2) | 92.7-185.8 | 153.2 (17.2) | 113.8-190.4 |
| Weight (kg) | 44.3 (19.3) | 12.7-67.1 | 50.3 (19.0) | 20.4-130.8 |
| BMI (kg/m2) | 18.7 (3.7) | 14.6-29.2 | 20.7 (4.4) | 13.3-39.1 |
| Symptom Onset Age (years) | 11.9 (4.7) | 4.0–19.4 | -- | -- |
| JHD Diagnosis Age (years) | 15.0 (5.3) | 6.29-22.4 | -- | -- |
| CAG Repeat : Mutated Allele (n=9) | 75.1 (25.0) | 53-130 | -- | -- |
In order to minimize the direct effects of the disease process on growth measures, growth measures were recorded as far from either symptom onset or diagnosis as possible. Most but not all JHD subjects had multiple visits with recorded growth data. Growth data were analyzed using the earliest time point available after birth, where it was possible to compute Z-scores for height, weight and BMI at the same age according to CDC guidelines. All 18 JHD subjects had growth measures that were obtained either prior to their diagnosis (n=16) or within 9 months of their diagnosis (n=2). Among the 16 with pre-diagnosis growth data, the average elapsed time between measurement of growth and diagnosis was 2.15 years (SD=2.36, range 0.54 to 7.49). Therefore, these measures reflect the subject in the earliest phase of disease (before or shortly after diagnosis).
Four of the 18 JHD subjects had growth measures that preceded symptom onset. In this sub-group, the elapsed time between the growth measures and onset of symptoms was an average of 2.90 years (SD=2.30, range 0.81 to 5.77). These measures would be considered to reflect the child in a preJHD phase.
Statistical Analysis
Statistical analysis was performed using software R, version 3.01[27] and SAS. Means, standard deviations, minimums, and maximums were calculated for numeric variables. The SAS GLM Procedure was used to compare all growth measures (height, weight, and BMI) between JHD and healthy control groups, adjusting for sex, age, age2, and appropriate interactions. For example, interactions between sex and the age terms were included for modeling height and growth, since female growth tends to plateau around age 16 years, whereas males continue to grow at this age, resulting in non-parallel growth curves for males and females [21, 26]. However, exploratory analysis showed no evidence of age-by-sex interactions for BMI, so group differences for BMI were only adjusted for sex, age, and age2. Residual plots of the models suggested that the data may not have been normal, so we also transformed the growth measures to improve normality, and compared the groups on the transformed scales to confirm our findings. In addition to the above modeling, we also performed one-sample t-tests of the Z-scores of the growth measures, to compare the 18 JHD subjects to national CDC norms. Z-scores are standardized accounting for age and sex and are on a scale such that 0 represents a score equal to the normative values where positive scores indicate a weight that is higher than the normative value and negative scores indicate a weight that is lower than the normative score. Finally, we used Spearman correlations to assess whether CAG repeats were associated with age at symptom onset and diagnosis, as well as with the Z-score growth measures among the nine subjects for whom we had CAG repeat data.
Results
JHD vs. Controls
Descriptive statistics for age and unadjusted measures of growth are presented in Table 1. Tables 2 and 3 contain a summary of our adjusted comparisons. Table 2 shows the results of our model-based approach based on our entire dataset, while Table 3 compares the 18 JHD subjects to CDC normative data via Z-scores. The consistent message is that there is no significant difference in height of JHD subjects vs. controls, while weight and BMI tend to be approximately 10° lower in JHD subjects, after adjusting for age and sex. In further analyses, we also repeated the t-tests shown in Table 3, based on only the 4 subjects for whom we had data prior to the onset of symptoms (preJHD). In this analysis, the mean Z scores were 0.11 for height (p=0.791), −0.49 for weight (p=0.171), and −0.71 for BMI (p=0.025).
Table 2.
Results of growth measure comparisons between the JHD group (n=18) and the healthy control group (n=274).
| Growth Measure | JHD |
Healthy
Controls |
JHD vs.
Healthy Controls |
||
|---|---|---|---|---|---|
| Mean (SE) | Mean (SE) | Mean Difference (95% CI) |
P-value
(original scale) |
P-value
(transformed scale) |
|
| Height (cm)a | 151.4 (1.8) | 153.1 (0.4) | −1.7 (−5.3, 2.0) | 0.373 | 0.114 |
| Weight (kg)a | 44.9 (2.9) | 50.3 (0.7) | −5.4 (−11,3, 0.6) | 0.071 | 0.018 |
| BMI (kg/m2)b | 18.6 (0.9) | 20.7 (0.2) | −2.0 (−3.9, −0.1) | 0.036 | 0.027 |
Adjusted for age, age2, sex, age × sex and age2 × sex.
Adjusted for age, age2, and sex.
Abbreviations: SE=Standard Error, CI=Confidence Interval.
Table 3.
Results of growth measure comparisons between the JHD group and CDC standards.
| N | Μean | SD | T Statistics | P-value | |
|---|---|---|---|---|---|
| Height (Z-score) | 18 | −0.18 | 1.13 | −0.67 | 0.512 |
| Weight (Z-score) | 18 | −0.36 | 0.67 | −2.29 | 0.035 |
| BMI (Z-score) | 18 | −0.4 | 0.92 | −1.83 | 0.084 |
Abbreviations: SD= Standard Deviation
Relationship between CAG Repeats and Disease Progression
For this analysis, all children with genetic information (n = 9) were included. The mutated allele repeats have a highly significant negative relationship with both age at JHD symptom onset (rs = −0.95, p < 0.0001) and at disease diagnosis (rs = −0.97, p < 0.0001). Figure 1 shows the data relating CAG repeats to age at diagnosis.
Figure 1.
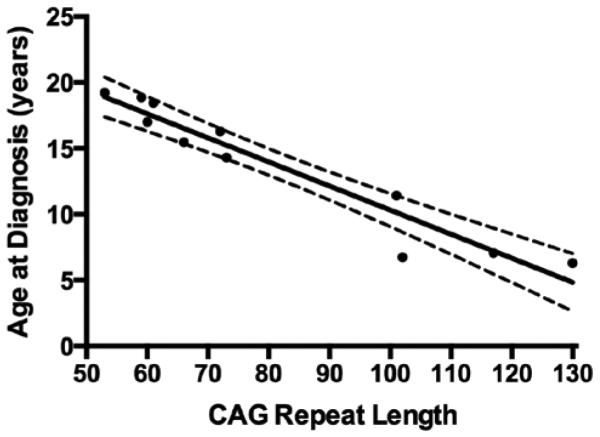
Relationship between CAG repeat length of the mutated allele and age at JHD diagnosis. The dashed lines indicate the 95° confidence limits for the fitted line.
Relationship between CAG Repeats and Growth Measures
Spearman correlations were used to assess the relationship between standardized Z-scores of weight, BMI, and CAG repeat length. Weight Z-scores and CAG repeats had a significant negative relationship (rs = −0.77, p = 0.016), indicating that the largest CAG repeat sizes were associated with the greatest reductions in weight. The BMI Z-scores had a negative, yet not significant relationship with CAG repeat length (rs = −0.52, p = 0.154).
Discussion
In this first ever study of anthropometric measures of subjects with JHD, measures of height taken either prior or near the time of diagnosis were not different from typically developing controls. However, weight and the proportional measure of BMI were significantly lower in the JHD group. These findings are consistent with previous studies that demonstrate diminished weight and BMI in early and late HD patients [28, 29], pre-HD adults [14], and pre-HD children [21].
Although the cause of weight change in HD is yet to be elucidated, it has been suggested that changes in motor activity, caloric intake or metabolic rate [30] may underlie the unintended weight loss. Given that JHD patients are, on average, much slower in their movements due to bradykinesia and dystonia [23, 31] the suggested higher energy expenditure due to excessive involuntary movements is unlikely to explain the current findings. However, especially for those whose heralding complaint was oropharyngeal dysfunction, the patients could have experienced difficulty chewing and slowing which would affect adequate food consumption to achieve normal range of weight.
Could the decreased weight and BMI be due to environmental factors? Significant disruption in the families of JHD patients likely exist; especially if one of the parents is in the symptomatic stages of HD. This type of environment could lead to factors that could impinge on weight such as lower socioeconomic status and its accompanying risk for poor health care and poor nutrition. In addition, having an ill parent can lead to significant emotional and behavioral issues for these children that could affect them long before they manifest their own symptoms. However, in our previous study on growth measures in at-risk children [21], all of the subjects had a parent with HD and therefore shared, at some level, the same environmental hardships. Yet the weight and BMI measures of the gene-expanded (pre-HD; referred to as gene-positive) children and those who were gene non-expanded (referred to as gene-negative) was dramatically different. Compared to healthy controls, the children that were raised in a home where a parent had HD, but were gene-negative had significantly larger weight and BMI measures compared to both the gene-positive group and the healthy controls. Conversely, the group of children who also come from a home where one parent had HD but were gene-positive had the exact opposite pattern with significantly lower weight and BMI compared to the gene-negative or healthy control groups. This is strong support for the notion that the growth measures in that study were more likely related to the effects of the gene rather than the environment. For the current study, support for the notion that the lower weight in the JHD sample is due to gene effects rather than environment effects is the strong correlation between CAG repeat length, or degree of gene mutation, and the z-score of weight.
Although the current study did not seek to identify the cause of the anthropometric measure change, one proposed mechanism by which mutant HTT leads to lower than normal BMI is an energy deficit due to mitochondrial dysfunction [32, 33]. Evidences of low ATP/ADP ratio in cell lines from HD patients [34], where higher CAG repeat length was associated with greater energy disruption and energetic disturbances found in HD adult skeletal muscle [9-11] together, point to a systemic peripheral energy deficit in HD. Conformingly, while weight and BMI were significantly different, the JHD patients did not differ in height from healthy controls. This highlights the absence of a generalized growth defect where both height and weight would be proportionately low, without the pronounced decrement of weight and BMI. Whilst the exact change in body composition is unknown, inadequate muscle bulk could account for the specific weight decrement as such tissues with very high metabolic demand are particularly susceptible to the presence of systemic energy deficit [15, 16].
The brain, like skeletal muscle, has a high energy demand and this is particularly true during childhood growth and development [18, 19]. Previous work has shown that pre-HD adults [35] and pre-HD children [21] have smaller than normal brain size. Whether or not this brain growth deficiency is due to an energy deficit is not yet clear. However, the brain is entirely dependent on energy substrates provided by the peripheral organs [36, 37]. Thus, a strain on brain energy demands could prompt compensatory mechanisms in the periphery and the need to shunt energy to the brain may subsequently exacerbate any potential primary systemic energy deficit. Future studies clarifying the change in energy allocation during development are necessary to understand the lifelong systemic impact of mHTT.
Early, pre-disease systemic metabolic defect is well documented by the studies that show both lower than normal BMI [12-14] and abnormal energy metabolism [38] in PreHD subjects. This is supported by our previous study of preHD children who show lower than normal head circumference, weight, and BMI who are, on average, 30 years from expected motor onset [21]. The lower BMI in 4 JHD children who had measures obtained prior to symptom onset may extend upon these earlier findings. These measures were obtained, on average, nearly 2 years prior to the time in which any symptom was identified, reflecting measures within the preJHD phase. Although the reported age at symptom onset may not be precise owing to the retrospective nature, collectively, the results support the notion that a systemic metabolic defect is present prior to disease onset.
In the context of adults with HD, deficits in weight are due to weight loss, which can be severe in the later stages of the disease. However reduced weight and BMI in children may not be a loss of weight, but instead a lack of muscle, adipose tissue growth or even change in bone density. It is possible that for some of our children, weight and BMI were comparable to the healthy controls, but then dropped when measured in the early phase of disease. Unfortunately limited by recorded information available on charts, we do not have enough longitudinal data that crosses the span of pre-symptomatic through later phase of the disease, therefore it is unclear whether the expected weight was achieved at some point then decreased over time. The lower than normal BMI in the preJHD children are more likely to reflect poor body mass growth rather than a loss of normally developed muscle.
The findings of low BMI in JHD subjects is similar to the findings of adult onset HD, providing another clinical feature that is shared by both. While the hallmark symptom indicates that an overlapping pathophysiological deficit linked to CAG expansion is in place, extreme CAG expansion may have a different pathogenic mechanism. However, limited by the small sample size and unrecorded dietetic data of this cohort it is difficult to determine the discrepancy. Many studies have displayed inverse correlations between CAG length and age of onset, and have subsequently created moderately predictive equations [39]. It has been estimated that repeat length only accounts for 67° of the variation of age at onset [40], and other genetic modifiers and environmental condition play major roles [41, 42]. In addition, it has been reported that the relationship between CAG repeat and age of onset is stronger for adult onset HD compared to JHD [43]. The findings in our study, on the other hand, suggest a much stronger predictive correlation in our JHD sample, with CAG length explaining about 92° variation in JHD age at diagnosis [89° for symptom]. While environmental and genetic factors have a strong effect in adult onset, these results suggest the biological drive of the mutant allele in extreme CAG lengths may become more dominant against other factors. Further accumulation of evidence showing the lack of or the presence of amplified genetic influence of extreme CAG repeats on well-recognized pathological features of HD may help establish a pathoetiological model potentially unique to JHD.
In the current sample of subjects, growth measures were obtained on average 2.29 years prior to the formal diagnosis of JHD with a focused effort to characterize the early course of illness. During this early stage, the majority of the BMI and weight measures of the JHD children remained within the normal percentile range (defined as 5th percentile to 85th percentile) when compared to the CDC norms, however on average were lower than the healthy controls. Therefore, it is important to note that, although the drop in weight and BMI in the JHD patients are statistically significant, they are subtle in a clinical sense. Nevertheless, the current findings not only extend the documentation of phenotypic features of JHD but could also further our understanding of the effect of CAG expansion on development that could potentially influence the pathogenic process of HD.
Our report provides an additional characterization of a relatively uniform and objective feature of this cohort. This may aid recognition of this rare form of HD, which presenting clinical symptom and age at onset vary between individuals. Certainly the presence of lower body weight and BMI should not hasten diagnostic genetic testing of children at risk for HD [3]. Importantly, the impact of maintaining adequate caloric intake and weight on JHD disease progression is worth further investigation.
Acknowledgments
This study was sponsored by National Institutes of Health (NIH), National Heart, Lung, and Blood Institute (NHLBI) T35 HL007485 which supported the time for Michael McHugh.
Footnotes
Financial Disclosure
No other disclosures.
Conflict of interest
The authors have no conflict of interest.
Author Roles
1 Alexander Tereshchenko Contributed equally as first authors. Involved in concept, medical chart review, statistical analysis and manuscript writing.
2 Michael McHugh Contributed equally as first authors. Involved in concept, medical chart review, statistical analysis and manuscript writing.
3 Jessica K. Lee Involved in concept, design, interpretation, and manuscript writing and review
4 Pedro Gonzalez-Alegre Involved in medical chart review, interpretation, and manuscript review
5 Kaitlin Crane Involved in design and execution of statistical analysis, interpretation and manuscript review.
6 Jeffrey Dawson Involved in design and execution of statistical analysis, interpretation and manuscript review.
7 Peg Nopoulos Senior author. Involved in all aspects of the paper’s concept through manuscript review
References
- [1].Martin JB, Gusella JF. Huntington's disease. Pathogenesis and management. N Engl J Med. 1986;315(20):1267–76. doi: 10.1056/NEJM198611133152006. [DOI] [PubMed] [Google Scholar]
- [2].Falstein EI. The uncontrollable child: some general sociopsychological observations. South Med J. 1966;59(2):228–9. doi: 10.1097/00007611-196602000-00024. [DOI] [PubMed] [Google Scholar]
- [3].Nance MA. Genetic testing of children at risk for Huntington's disease. US Huntington Disease Genetic Testing Group. Neurology. 1997;49(4):1048–53. doi: 10.1212/wnl.49.4.1048. [DOI] [PubMed] [Google Scholar]
- [4].Quarrell O, O'Donovan KL, Bandmann O, Strong M. The Prevalence of Juvenile Huntington's Disease: A Review of the Literature and Meta-Analysis. PLoS Curr. 2012;4:e4f8606b742ef3. doi: 10.1371/4f8606b742ef3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Duyao M, Ambrose C, Myers R, Novelletto A, Persichetti F, Frontali M, Folstein S, Ross C, Franz M, Abbott M, Gray J, Conneally P, Young A, Penney J, Hollingsworth Z, Shoulson I, Lazzarini A, Falek A, Koroshetz W, Sax D, Bird E, Vonsattel J, Bonilla E, Alvir J, Bickham Conde J, Cha J-H, Dure L, Gomez F, Ramos M, Sanchez-Ramos J, Snodgrass S, de Young M, Wexler N, Moscowitz C, Penchaszadeh G, MacFarlane H, Anderson M, Jenkins B, Srinidhi J, Barnes G, Gusella J, MacDonald M. Trinucleotide repeat length instability and age of onset in Huntington's disease. Nat Genet. 1993;4(4):387–92. doi: 10.1038/ng0893-387. [DOI] [PubMed] [Google Scholar]
- [6].Aziz NA, Pijl H, Frolich M, Snel M, Streefland TC, Roelfsema F, Roos RA. Systemic energy homeostasis in Huntington's disease patients. J Neurol Neurosurg Psychiatry. 2010;81(11):1233–7. doi: 10.1136/jnnp.2009.191833. [DOI] [PubMed] [Google Scholar]
- [7].Labbadia J, Morimoto RI. Huntington's disease: underlying molecular mechanisms and emerging concepts. Trends Biochem Sci. 2013;38(8):378–85. doi: 10.1016/j.tibs.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Mehrotra A, Sandhir R. Mitochondrial cofactors in experimental Huntington's disease: behavioral, biochemical and histological evaluation. Behav Brain Res. 2014;261:345–55. doi: 10.1016/j.bbr.2013.12.035. [DOI] [PubMed] [Google Scholar]
- [9].Ciammola A, Sassone J, Sciacco M, Mencacci NE, Ripolone M, Bizzi C, Colciago C, Moggio M, Parati G, Silani V, Malfatto G. Low anaerobic threshold and increased skeletal muscle lactate production in subjects with Huntington's disease. Mov Disord. 2011;26(1):130–7. doi: 10.1002/mds.23258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Koroshetz WJ, Jenkins BG, Rosen BR, Beal MF. Energy metabolism defects in Huntington's disease and effects of coenzyme Q10. Ann Neurol. 1997;41(2):160–5. doi: 10.1002/ana.410410206. [DOI] [PubMed] [Google Scholar]
- [11].Lodi R, Schapira AH, Manners D, Styles P, Wood NW, Taylor DJ, Warner TT. Abnormal in vivo skeletal muscle energy metabolism in Huntington's disease and dentatorubropallidoluysian atrophy. Ann Neurol. 2000;48(1):72–6. [PubMed] [Google Scholar]
- [12].Aziz NA, van der Burg JM, Landwehrmeyer GB, Brundin P, Stijnen T, Group ES. Roos RA. Weight loss in Huntington disease increases with higher CAG repeat number. Neurology. 2008;71(19):1506–13. doi: 10.1212/01.wnl.0000334276.09729.0e. [DOI] [PubMed] [Google Scholar]
- [13].Farrer LA, Meaney FJ. An anthropometric assessment of Huntington's disease patients and families. Am J Phys Anthropol. 1985;67(3):185–94. doi: 10.1002/ajpa.1330670304. [DOI] [PubMed] [Google Scholar]
- [14].Marder K, Zhao H, Eberly S, Tanner CM, Oakes D, Shoulson I, Huntington Study G Dietary intake in adults at risk for Huntington disease: analysis of PHAROS research participants. Neurology. 2009;73(5):385–92. doi: 10.1212/WNL.0b013e3181b04aa2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ritter AM, Robertson CS. Cerebral metabolism. Neurosurg Clin N Am. 1994;5(4):633–45. [PubMed] [Google Scholar]
- [16].Wilson DF. Energy metabolism in muscle approaching maximal rates of oxygen utilization. Med Sci Sports Exerc. 1995;27(1):54–9. [PubMed] [Google Scholar]
- [17].Tonson A, Ratel S, Le Fur Y, Vilmen C, Cozzone PJ, Bendahan D. Muscle energetics changes throughout maturation: a quantitative 31P-MRS analysis. J Appl Physiol (1985) 2010;109(6):1769–78. doi: 10.1152/japplphysiol.01423.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Brooks GA. Cell-cell and intracellular lactate shuttles. J Physiol. 2009;587:5591–600. doi: 10.1113/jphysiol.2009.178350. Pt 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Erecinska M, Cherian S, Silver IA. Energy metabolism in mammalian brain during development. Prog Neurobiol. 2004;73(6):397–445. doi: 10.1016/j.pneurobio.2004.06.003. [DOI] [PubMed] [Google Scholar]
- [20].Bhide PG, Day M, Sapp E, Schwarz C, Sheth A, Kim J, Young AB, Penney J, Golden J, Aronin N, DiFiglia M. Expression of normal and mutant huntingtin in the developing brain. J Neurosci. 1996;16(17):5523–35. doi: 10.1523/JNEUROSCI.16-17-05523.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lee JK, Mathews K, Schlaggar B, Perlmutter J, Paulsen JS, Epping E, Burmeister L, Nopoulos P. Measures of growth in children at risk for Huntington disease. Neurology. 2012;79(7):668–74. doi: 10.1212/WNL.0b013e3182648b65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Folstein SE, Leigh RJ, Parhad IM, Folstein MF. The diagnosis of Huntington's disease. Neurology. 1986;36(10):1279–83. doi: 10.1212/wnl.36.10.1279. [DOI] [PubMed] [Google Scholar]
- [23].Gonzalez-Alegre P, Afifi AK. Clinical characteristics of childhood-onset (juvenile) Huntington disease: report of 12 patients and review of the literature. J Child Neurol. 2006;21(3):223–9. doi: 10.2310/7010.2006.00055. [DOI] [PubMed] [Google Scholar]
- [24].Barker RA, Squitieri F. The clinical phenotype of juvenile Huntington's disease. In: Quarrell O, Brewer HM, Squitieri F, Barker RA, Nance MA, Landwehrmeyer GB, editors. Juvenile Huntington's disease: (and other trinucleotide repeat disorders) Oxford University Press; New York: 2009. pp. 39–50. [Google Scholar]
- [25].Ribai P, Nguyen K, Hahn-Barma V, Gourfinkel-An I, Vidailhet M, Legout A, Dode C, Brice A, Durr A. Psychiatric and cognitive difficulties as indicators of juvenile huntington disease onset in 29 patients. Arch Neurol. 2007;64(6):813–9. doi: 10.1001/archneur.64.6.813. [DOI] [PubMed] [Google Scholar]
- [26].Centers for Disease Control and Prevention NCfHS. Vol. 30. CDC growth charts; United States: May, 2000. [Google Scholar]
- [27].R Core Team . R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2013. [Google Scholar]
- [28].Djousse L, Knowlton B, Cupples LA, Marder K, Shoulson I, Myers RH. Weight loss in early stage of Huntington's disease. Neurology. 2002;59(9):1325–30. doi: 10.1212/01.wnl.0000031791.10922.cf. [DOI] [PubMed] [Google Scholar]
- [29].Robbins AO, Ho AK, Barker RA. Weight changes in Huntington's disease. Eur J Neurol. 2006;13(8):e7. doi: 10.1111/j.1468-1331.2006.01319.x. [DOI] [PubMed] [Google Scholar]
- [30].Aziz NA, Swaab DF, Pijl H, Roos RA. Hypothalamic dysfunction and neuroendocrine and metabolic alterations in Huntington's disease: clinical consequences and therapeutic implications. Rev Neurosci. 2007;18(3-4):223–51. doi: 10.1515/revneuro.2007.18.3-4.223. [DOI] [PubMed] [Google Scholar]
- [31].Ruocco HH, Lopes-Cendes I, Laurito TL, Li LM, Cendes F. Clinical presentation of juvenile Huntington disease. Arq Neuropsiquiatr. 2006;64(1):5–9. doi: 10.1590/s0004-282x2006000100002. [DOI] [PubMed] [Google Scholar]
- [32].Browne SE, Beal MF. The energetics of Huntington's disease. Neurochem Res. 2004;29(3):531–46. doi: 10.1023/b:nere.0000014824.04728.dd. [DOI] [PubMed] [Google Scholar]
- [33].Mochel F, Haller RG. Energy deficit in Huntington disease: why it matters. J Clin Invest. 2011;121(2):493–9. doi: 10.1172/JCI45691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Seong IS, Ivanova E, Lee JM, Choo YS, Fossale E, Anderson M, Gusella JF, Laramie JM, Myers RH, Lesort M, MacDonald ME. HD CAG repeat implicates a dominant property of huntingtin in mitochondrial energy metabolism. Hum Mol Genet. 2005;14(19):2871–80. doi: 10.1093/hmg/ddi319. [DOI] [PubMed] [Google Scholar]
- [35].Nopoulos PC, Aylward EH, Ross CA, Mills JA, Langbehn DR, Johnson HJ, Magnotta VA, Pierson RK, Beglinger LJ, Nance MA, Barker RA, Paulsen JS, Investigators P-H, Coordinators of the Huntington Study G Smaller intracranial volume in prodromal Huntington's disease: evidence for abnormal neurodevelopment. Brain. 2011;134:137–42. doi: 10.1093/brain/awq280. Pt 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ames A., 3rd CNS energy metabolism as related to function. Brain Res Brain Res Rev. 2000 Nov 22;:42–68. doi: 10.1016/s0165-0173(00)00038-2. 2000. [DOI] [PubMed] [Google Scholar]
- [37].Bauernfeind AL, Barks SK, Duka T, Grossman LI, Hof PR, Sherwood CC. Aerobic glycolysis in the primate brain: reconsidering the implications for growth and maintenance. Brain Struct Funct. 2013 doi: 10.1007/s00429-013-0662-z. [DOI] [PubMed] [Google Scholar]
- [38].Saft C, Zange J, Andrich J, Muller K, Lindenberg K, Landwehrmeyer B, Vorgerd M, Kraus PH, Przuntek H, Schols L. Mitochondrial impairment in patients and asymptomatic mutation carriers of Huntington's disease. Mov Disord. 2005;20(6):674–9. doi: 10.1002/mds.20373. [DOI] [PubMed] [Google Scholar]
- [39].Langbehn DR, Hayden MR, Paulsen JS, Group P-HIotHS CAG-repeat length and the age of onset in Huntington disease (HD): a review and validation study of statistical approaches. Am J Med Genet B Neuropsychiatr Genet. 2010;153B(2):397–408. doi: 10.1002/ajmg.b.30992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Gusella JF, MacDonald ME. Huntington's disease: the case for genetic modifiers. Genome Med. 2009;1(8):80. doi: 10.1186/gm80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Li JL, Hayden MR, Warby SC, Durr A, Morrison PJ, Nance M, Ross CA, Margolis RL, Rosenblatt A, Squitieri F, Frati L, Gomez-Tortosa E, Garcia CA, Suchowersky O, Klimek ML, Trent RJ, McCusker E, Novelletto A, Frontali M, Paulsen JS, Jones R, Ashizawa T, Lazzarini A, Wheeler VC, Prakash R, Xu G, Djousse L, Mysore JS, Gillis T, Hakky M, Cupples LA, Saint-Hilaire MH, Cha JH, Hersch SM, Penney JB, Harrison MB, Perlman SL, Zanko A, Abramson RK, Lechich AJ, Duckett A, Marder K, Conneally PM, Gusella JF, MacDonald ME, Myers RH. Genome-wide significance for a modifier of age at neurological onset in Huntington's disease at 6q23-24: the HD MAPS study. BMC Med Genet. 2006;7:71. doi: 10.1186/1471-2350-7-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wexler NS, Lorimer J, Porter J, Gomez F, Moskowitz C, Shackell E, Marder K, Penchaszadeh G, Roberts SA, Gayan J, Brocklebank D, Cherny SS, Cardon LR, Gray J, Dlouhy SR, Wiktorski S, Hodes ME, Conneally PM, Penney JB, Gusella J, Cha JH, Irizarry M, Rosas D, Hersch S, Hollingsworth Z, MacDonald M, Young AB, Andresen JM, Housman DE, De Young MM, Bonilla E, Stillings T, Negrette A, Snodgrass SR, Martinez-Jaurrieta MD, Ramos-Arroyo MA, Bickham J, Ramos JS, Marshall F, Shoulson I, Rey GJ, Feigin A, Arnheim N, Acevedo-Cruz A, Acosta L, Alvir J, Fischbeck K, Thompson LM, Young A, Dure L, O'Brien CJ, Paulsen J, Brickman A, Krch D, Peery S, Hogarth P, Higgins DS, Jr., Landwehrmeyer B, Project US-VCR Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington's disease age of onset. Proc Natl Acad Sci U S A. 2004;101(10):3498–503. doi: 10.1073/pnas.0308679101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Andresen JM, Gayan J, Djousse L, Roberts S, Brocklebank D, Cherny SS, Group US-VCR. Group HMCR. Cardon LR, Gusella JF, MacDonald ME, Myers RH, Housman DE, Wexler NS. The relationship between CAG repeat length and age of onset differs for Huntington's disease patients with juvenile onset or adult onset. Ann Hum Genet. 2007;71:295–301. doi: 10.1111/j.1469-1809.2006.00335.x. Pt 3. [DOI] [PubMed] [Google Scholar]