

Abstract
Binding of divalent metal ions with intrinsically disordered fibrillogenic proteins, such as amyloid-β (Aβ), influences the aggregation process and the severity of neurodegenerative diseases. The Aβ monomers and oligomers are the building blocks of the aggregates. In this work, we report the structures and free energy landscapes of the monomeric zinc(II)-bound Aβ40 (Zn:Aβ40) and zinc(II)-bound Aβ42 (Zn:Aβ42) intrinsically disordered fibrillogenic metallopeptides in an aqueous solution by utilizing an approach that employs first principles calculations and parallel tempering molecular dynamics simulations. The structural and thermodynamic properties, including the secondary and tertiary structures and conformational Gibbs free energies of these intrinsically disordered metallopeptide alloforms, are presented. The results show distinct differing characteristics for these metallopeptides. For example, prominent β-sheet formation in the N-terminal region (Asp1, Arg5, and Tyr10) of Zn:Aβ40 is significantly decreased or lacking in Zn:Aβ42. Our findings indicate that blocking multiple reactive residues forming abundant β-sheet structure located in the central hydrophobic core and C-terminal regions of Zn:Aβ42 via antibodies or small organic molecules might help to reduce the aggregation of Zn(II)-bound Aβ42. Furthermore, we find that helix formation increases but β-sheet formation decreases in the C-terminal region upon Zn(II) binding to Aβ. This depressed β-sheet formation in the C-terminal region (Gly33–Gly38) in monomeric Zn:Aβ42 might be linked to the formation of amorphous instead of fibrillar aggregates of Zn:Aβ42.
Keywords: Intrinsically disordered metalloprotein, Molecular dynamics simulations, Amyloid-β, Zinc
Introduction
Certain transition metal ion bound intrinsically disordered proteins have been proposed to play crucial roles in the mechanisms of severe diseases whereby the metal ion binds to fibrillogenic intrinsically disordered proteins and impacts their aggregation process [1–3]. The mechanism of Alzheimer’s disease has been linked to various factors, including the formation of toxic amyloid- β (Aβ) aggregates, tau dysfunction, inflammation, oxidative stress, neuron death, and synaptic impairment [4, 5]. Recently, it has been proposed that the monomeric and oligomeric forms of Aβ are toxic to neurons [5–7] and that binding of specific transition metal ions, such as zinc, copper, or iron, to Aβ influences the toxicity [8–14].
In general, proteolytic cleavages of the amyloid precursor protein release Aβ into the extracellular medium [15]. The γ-cleavage results in various fragments of Aβ that have 38–43 residues [15]. Among these various fragments, Aβ40 and Aβ42 are the most dominant ones and possess different features from each other: faster kinetics for fibrillar aggregation with Aβ42 than with Aβ40, different aggregation mechanisms, and increased toxicity of Aβ42 in comparison with Aβ40 have been reported [16–19]. These different features depend on the differing aggregation propensities, which in turn are a result of structural differences at the monomeric and oligomeric levels. Under physiological conditions, monomeric Aβ binds to Zn(II), and amorphous instead of fibrillar aggregation is promoted when the Zn(II) and Aβ concentration ratio reaches 1 [20]. Surprisingly, the structural differences between monomeric zinc(II)-bound Aβ40 (Zn:Aβ40) and zinc(II)-bound Aβ42 (Zn:Aβ42) in an aqueous solution at the molecular level with dynamics are currently poorly understood. Such an understanding can aid in the design of efficient treatments, i.e., antibodies, metal ion chelators, or small organic drugs. For instance, the formation of β-sheet in monomeric and oligomeric Aβ has been directly linked to the toxicity and aggregation process [21]. Therefore, characterization of residues forming certain secondary structures, including β-sheet in Zn(II)-bound Aβ (Zn:Aβ), aids in understanding which residues should be blocked or inactivated to change or prevent the aggregation process.
Extensive spectrometric and spectroscopic measurements have been performed on Zn:Aβ, and the coordination chemistry as well as the structures of Zn:Aβ are debated in the current literature because of challenges provided by solvent effects, fast conformational changes, and rapid aggregation [20, 22–35]. For example, the N-terminus, Asp, Glu, Tyr, Arg, and H2O have been proposed to be possible coordinating ligands in conjunction with the three His residues of Aβ (Table 1). Interestingly, recent isothermal calorimetry and theoretical studies have reported that Zn(II) binding to residues His6, Glu11, His13, and His14 is preferred in comparison with Zn(II) binding to the other reported probable binding sites [31, 36]. A consensus needs to be reached regarding the full-length Zn:Aβ40 and Zn:Aβ42 alloform structures. Theoretical studies complement experiments and provide molecular-level information with dynamics. Even though these studies provide interesting insights that are difficult to obtain otherwise, theoretical studies and in vitro experiments currently face challenges in fully capturing the impact of the in vivo environment on the predicted structures. However, theoretical studies can help to gain insights into fibrillogenic monomeric protein and metalloprotein structures via capturing solvent effects at the molecular level without the addition of nonbiological solvents or compounds for controlling the aggregation process. A limitation in metalloprotein theoretical studies is related to the lack of metal–ligand moiety potential functions that are required in molecular simulations. As a result, previous molecular simulations performed on full-length Zn:Aβ used either geometry constraints for the metal–ligand moiety or parameters that were actually for the reactive sites of the human carbonic anhydrase, in which the Zn(II) ion is coordinated to three His residues and a water molecule or an OH group and not to the Glu residue [23, 37, 38]. Furthermore, the large size and fast conformational changes that override the large energy barriers associated with the intrinsically disordered nature require the use of special sampling techniques that help to overcome the multiple-minima problem. It is difficult to study microsecond timescales in both the quantum mechanical and the coupled molecular mechanical regions in quantum mechanical/molecular mechanical simulations using special sampling techniques; therefore, the application of classical molecular simulations is crucial.
Table 1.
Experimentally proposed amyloid-β (Aβ) residues for binding to Zn(II)
| Proposed coordination chemistry | References |
|---|---|
| His6–His13–His14–H2O | [34] |
| Nter–His6–His13–His14 | [20, 23–26] |
| Asp1–His6–His13–His14 | [20, 32] |
| His13–His14–His13–His14 | [27, 30] |
| His6–Glu11–His13–His14 | [22, 23, 31, 32] |
| Nter–Glu11–His13–His14 | [23] |
| Arg5–His6–His13–His14 | [33] |
| His6–Tyr10–His13–His14 | [35] |
Nter N-terminus of the protein
Our previous studies have shown that the electrostatic interactions and charge transfer effects between transition metal ions and biomolecules govern the resulting structure of biometallic species [39–45]. Therefore, we expect that the electrostatic interactions capturing charge transfer effects of the metal–ligand moiety are crucial in determining metalloprotein structures. Potential function development has been an active area of research [46–55]. In general, previously developed potential functions use either nonbonded or bonded models. The first utilizes the full ionic charge of the metal ion, which often results in less accurate geometries around the metal [47, 48]. The bonded model has been used to generate the metal–ligand moiety parameters that are compatible with widely established protein force field parameters [49, 50]. Useful information has been gained from such metalloprotein simulations [49–52]. Here, we use the bonded model for a detailed study of the structural and thermodynamic properties of the monomeric Zn:Aβ40 and Zn:Aβ42 metallopeptide alloforms in aqueous solution. Our results show that these metalloprotein alloforms are disordered, but we also find interesting structural characteristics that differ from each other for Zn:Aβ40 and Zn:Aβ42. In addition, we compare our results for Zn:Aβ40 and Zn:Aβ42 with those reported for free Aβ40 and Aβ42 using the same techniques under the same conditions to gain insights into the impact of Zn(II) binding on the structures of the free alloforms.
Materials and methods
To simulate Zn:Aβ using a bonded model for the Zn:His3Glu moiety that captures the electrostatic interactions, we performed first principles calculations and different sets of parallel tempering molecular dynamics simulations along with thermodynamic calculations. The force constant values for the model Zn:His3Glu moiety were recently derived from the eigenvalues and eigenvectors initially calculated based on Seminario’s method [49]. These parameters were found to be compatible with the Amber force field parameters [49]. However, the electrostatic interaction parameters are missing for the Zn:His3Glu model. To derive the electrostatic interaction parameters via capturing the charge transfer effect and to provide parameters that are compatible with those mentioned above, we performed structure optimization and partial charge calculations utilizing the B3LYP/6-31G*//B3LYP/6-31G* method using the Gaussian 03 software package [56] (see the electronic supplementary material). To ensure compatibility with Amber parameters, we rescaled the partial charges utilizing the restrained electrostatic potential (RESP) method; first, the heavy-atom equivalence was taken into account with the RESP method and then the hydrogen-atom equivalence was obtained via the two-stage RESP partial charge optimization procedure [57]. The presence of the metal ion requires use of the B3LYP instead of the Hartree–Fock method to avoid underestimation of the charge transfer effect [58]. The calculated energies, minimum energy structural parameters, and electrostatic partial charges are given in the electronic supplementary material. The optimized minimum energy structures obtained from these first principles calculations are in agreement with the NMR measurements of Zn:Aβ16 (see the electronic supplementary material) [22]. Following previous studies, the van der Waals parameters σ and ɛ for Zn(II) were set to 1.10 Å and 0.0125 kcal mol−1, respectively [49].
The initial Zn:Aβ40 and Zn:Aβ42 structures were generated by connecting the NMR-derived Zn:Aβ16 structure (Protein Data Bank ID 1ZE9) with the Leu17–Val40 and Leu17–Ala42 fragments, respectively [22]. Sets of replica-exchange molecular dynamics simulations [59] were performed utilizing the electrostatic interaction parameters that we calculated along with the force field parameters reported for the Zn:His3Glu metal–ligand moiety [49] and the Amber ff99SB parameters [60], which are currently one of the best parameters for the simulations of secondary structures due to the modified backbone parameters. The Onufriev–Bashford–Case generalized Born implicit solvent model [61] was used and the long-range interactions were treated with the particle mesh Ewald method using a cutoff value of 25 Å [62, 63]. An implicit model for water was chosen to avoid the impact of the confined aqueous volume in simulations performed using an explicit model for water. The temperature was controlled utilizing Langevin dynamics with a collision factor frequency of 2 ps−1 [62]. The initial structures were first equilibrated for 500 ps for each replica. The integration time step for each replica was 2 fs and trajectories were saved every 500 steps. The time interval for exchange attempts between replicas was set to 5 ps and 16 replicas were used in each simulation with exponentially distributed temperatures between 280 and 408 K [64]. The production total simulation time was 6.4 μs. The convergence was tested in various ways via calculating the time-dependent secondary structure component abundances and thermodynamic properties (see the electronic supplementary material). The results showed that the systems require the first 120 ns of the simulation time to converge. The reported results belong to the physiological temperature replica. Simulations utilizing an implicit water model—overcoming the confined aqueous volume effects—do not capture the impact of intermolecular hydrogen-bonding interactions between the solute and solvent molecules. Furthermore, parallel-tempering replica-exchange molecular dynamics simulations using an explicit model water performed by Prakash et al. [65] showed that the specific heat value of pure liquid water does not remain constant. Structures from the replica-exchange molecular dynamics simulations of Zn:Aβ40 and Zn:Aβ42 that presented an extended N-terminal region were selected to investigate the impact of intermolecular hydrogen-bonding interactions using an explicit modified TIP5P model for water on the simulated structures [66, 67]. The modified TIP5P model captures the temperature and pressure dependence of the thermodynamic properties of liquid water more accurately than the TIP3P and TIP4P potential functions. These structures were solvated with approximately 9,100 water molecules, yielding a volume of 286,188 Å3, and were then simulated for an additional 50 ns using the isothermal–isobaric ensemble at a temperature of 310 K and a pressure of 1.0 bar (see the electronic supplementary material). Langevin dynamics was used in these simulations [62].
Following our recent studies [68], the thermodynamic properties were studied using the molecular mechanics/Poisson–Boltzmann surface area method [69, 70] as well as the potential of mean force (PMF) surfaces [68]. Specifically, all converged structures belonging to the physiological temperature were used in the conformational Gibbs free energy (G) calculations utilizing the molecular mechanics/Poisson–Boltzmann surface area method, which is widely utilized for predicting the thermodynamic properties of macromolecular biological systems. For the calculations of the PMF surfaces, we used the coordinates of the end-to-end distance (RE–E), which is defined as the distance between the Asp1 peptide backbone nitrogen atom and the Val40 or Ala42 peptide backbone carbonyl carbon atom, and the radius of gyration (Rg). The convergence was verified by calculating the PMF surfaces at different simulation timescales. These methods are described in detail in the electronic supplementary material. The thermodynamic properties were directly linked to the analyses of the properties of the secondary and tertiary structures. The secondary structure components were analyzed using the program DSSP [71], which uses the hydrogen-bond criteria. Following our recent studies [68], intramolecular metallopeptide interactions were considered to exist when the centers of mass of two residues were within a distance of 9.0 Å. A hydrogen bond is defined to exist when the distance between donor hydrogen and acceptor atoms is less than or equal to 2.5 Å and the hydrogen-bond angle is larger than 113°. A salt bridge occurs between hydrogen-bonded atoms with opposite electrostatic charges.
Results and discussion
The calculated conformational Gibbs free energy surfaces based on enthalpic and entropic contributions for Zn:Aβ40 and Zn:Aβ42 are shown in Fig. 1. Both metallopeptide structures override small and large free energy barriers and there is no clear relationship between the enthalpic or entropic contribution and the conformational Gibbs free energy. These findings further support the disordered nature of these metallopeptides. The calculated average thermodynamic properties, i.e., conformational enthalpy, entropy, and Gibbs free energy, for both disordered metallopeptides are listed in Table 2. These results indicate that the structures of Zn:Aβ40 are favorable owing to the enthalpic rather than the entropic contributions in comparison with the favorability of the Zn:Aβ42 structures in aqueous solution. On the other hand, the structures of Zn:Aβ42 are more favorable than those of Zn:Aβ40 owing to a larger entropic contribution. Overall, the structures of Zn:Aβ42 are about 21 kJ mol−1 more stable than those of Zn:Aβ40 in aqueous solution. Despite these thermodynamic differences, it is interesting to note that both Zn:Aβ40 and Zn:Aβ42 have an average Rg of approximately 11 ± 1.5 Å, which is about 13 % smaller than that reported for the wild-type Aβ42 peptide [68]. This finding indicates that Zn(II) binding results in a more compact peptide structure. In parallel, a comparison of our thermodynamic values for Zn:Aβ42 with those that we recently reported for Aβ42 shows that the Zn(II) binding stabilizes the structures of free Aβ42 by 310 kJ mol−1 in an aqueous solution [68].
Fig. 1.
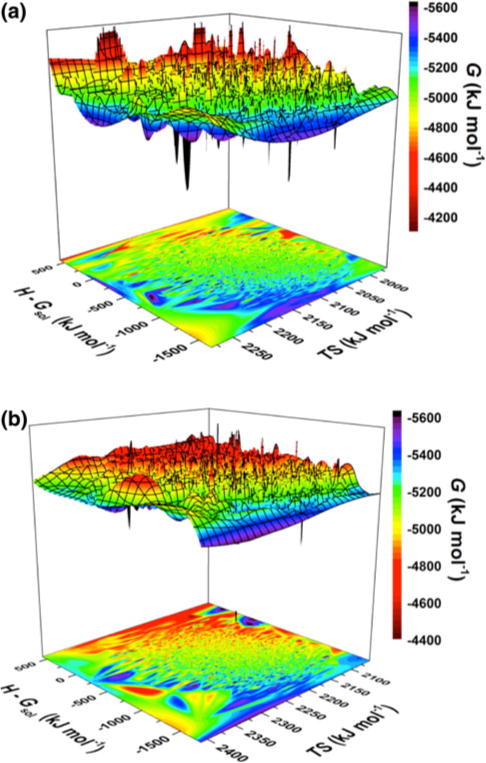
Calculated conformational Gibbs free energy (G) surfaces of the a Zn(II)-bound amyloid-β(1–40) (Zn:Aβ40) and b Zn(II)-bound amyloid-β(1–42) (Zn:Aβ42) metallopeptides based on the enthalpic (H − Gsol) and entropic (TS) contributions in an aqueous solution
Table 2.
The calculated enthalpy (H), entropy (−TS), and Gibbs free energy (G) of the simulated Zn(II)-bound Aβ40 (Zn:Aβ40) and Zn(II)-bound Aβ42 (Zn:Aβ42) structures in an aqueous solution
| 〈H〉 (kJ mol−1) | 〈Gsol〉 (kJ mol−1) | 〈−TS〉 (kJ mol−1) | 〈G〉 (kJ mol−1) | |
|---|---|---|---|---|
| Zn:Aβ40 | −2,979.8 (±16.3) | −2,235.9 (±24.9) | −2,096.2 (±28.5) | −5,076.0 (±20.6) |
| Zn:Aβ42 | −2,908.3 (±12.1) | −2,183.2 (±28.8) | −2,189.1 (±29.3) | −5,097.4 (±21.0) |
The PMF surfaces in the coordinates of RE–E and Rg are shown in Fig. 2. There is only one most favorable basin (basin I, within the 1kBT range) for the Zn:Aβ40 structures, and it is located at Rg values ranging between 9.5 and 11.1 Å and RE–E values ranging between 11.7 and 22.6 Å (Fig. 2a). The most preferred structures of Zn:Aβ42 are also located in one basin (basin I) with Rg values ranging between 9.6 and 10.7 Å and RE–E values ranging between 10.0 and 24.3 Å (Fig. 2b). Our recent studies showed that the favorable structures of aqueous Aβ42 are located in two basins (basins IA and IB) with different RE–E and Rg values but the same PMF values [68]. These results showed that transitions between the Aβ42 structures located in the most stable basins require the overriding of large energy barriers (more than 1kBT). As shown in Fig. 2, this characteristic changes upon Zn(II) binding with Aβ42. Altogether, these findings show that Zn(II) binding impacts the thermodynamic properties of the free aqueous Aβ peptide.
Fig. 2.
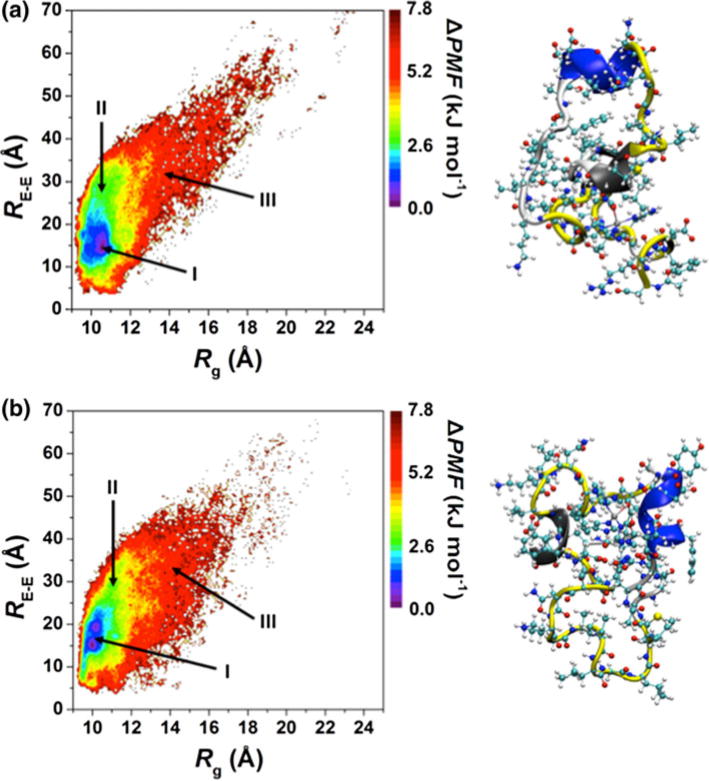
Potential of mean force surfaces (ΔPMF) of the a Zn:Aβ40 and b Zn:Aβ42 metallopeptide structures along the coordinates of the radius of gyration (Rg) and end-to-end distance (RE–E)
Secondary structure properties of the intrinsically disordered Zn:Aβ40 and Zn:Aβ42 metallopeptides
The secondary structure components per residue along with their abundances exhibit significant differences between Zn:Aβ40 and Zn:Aβ42 (Fig. 3). The N-terminal (Asp1–Ser8) and the central hydrophobic core (CHC) regions of Zn:Aβ42 adopt a more prominent helical (α-helix and/or 310-helix) structure (by up to 20 %) in comparison with Zn:Aβ40, except for 310-helix formation at Asp7–Gly9 and Val18. A more abundant helical structure (up to 20 %) occurs at Glu11–Gln15, Asp23–Lys28, and Ile32–Val36 of Zn:Aβ40 in comparison to Zn:Aβ42, except for the 310-helix at His14, Gln15, and Lys28. Prominent 310-helix formation is detected (up to 12 %) at Lys16, Gly29, Ala30, and Gly38–Val40 of Zn:Aβ42 in comparison with the structures of Zn:Aβ40. In contrast, β-sheet formation at Asp1, Arg5, and Tyr10 is more abundant (up to 12 %) in Zn:Aβ40 than in Zn:Aβ42. Furthermore, residues located in parts of the CHC (Leu17–Ala21) and in the adjacent Glu22–Ala30 regions form more prominent β-sheet in Zn:Aβ40 in comparison with Zn:Aβ42, except for Leu17–Phe19 and Asp23. In addition, Ile31, Val36, and Val39 in the structures of Zn:Aβ40 adopt a slightly more abundant β-sheet conformation than in Zn:Aβ42, whereas the opposite trend is observed for Ile32 and Val40. Interestingly, the reported larger β-sheet abundance in the C-terminal region of free Aβ42 in comparison with free Aβ40 [72] is not detected for Zn:Aβ40 and Zn:Aβ42, except for Ile32 and Val40 (Fig. 3). This finding suggests that increased structuring in the C-terminal region of Aβ42 in comparison with Aβ40 due to β-sheet formation is depressed upon Zn(II) binding.
Fig. 3.
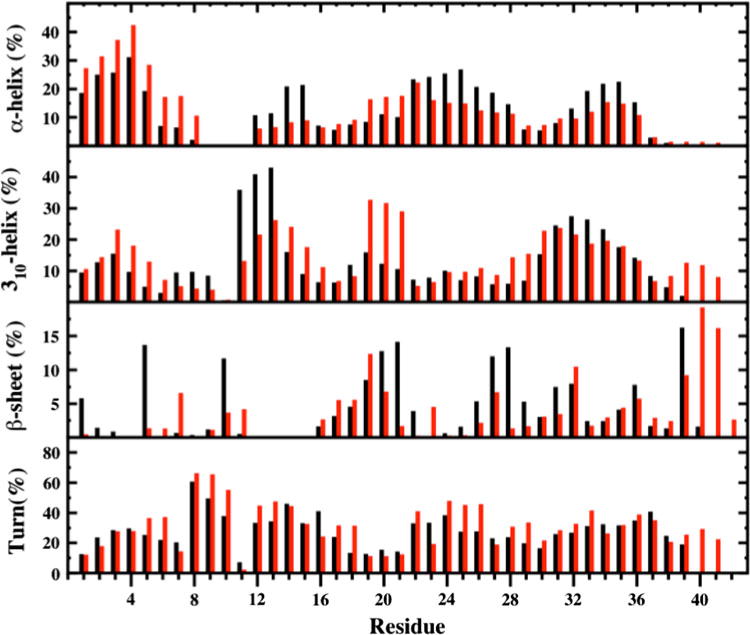
Secondary structures per residue along with their abundances in the Zn:Aβ40 (black) and Zn:Aβ42 (red) metallopeptide structures in an aqueous solution. The π-helix and coil structures are not displayed
Residues Ser8–Tyr10 adopt a more abundant turn structure in Zn:Aβ42 than in Zn:Aβ40 (Fig. 3). This trend was reported to be the opposite between the structures of free Aβ40 and Aβ42 [72], indicating that Zn(II) binding affects the turn structure formation trend in this part of the N-terminal region. Lys16 and Asp23 adopt an about 10 % more abundant turn structure in Zn:Aβ40 in comparison with Zn:Aβ42. Overall, the most prominent turn structure formation (more than 30 %) occurs in parts of the N-terminal, C-terminal, and mid-domain regions of Zn:Aβ42 (Fig. 3). Except for the Asn27 residue, Yang and Teplow [72] reported a slightly greater turn structure formation at Ala21–Ala30 for free Aβ42 than for Aβ40. We found that the turn structure formation at Asp23 and Asn27 is slightly less prominent in Zn:Aβ42 in comparison with Zn:Aβ40. Except for these variations, the residues located in the Ala21–Ala30 decapeptide region of Zn:Aβ42 adopt a more prominent (up to 15 %) turn structure than does the same region in Zn:Aβ40.
Given that the increased β-sheet propensity has been linked to the toxicity and aggregation process [21], it is crucial to gain insights into the residual tendencies for β-sheet formation with dynamics. Asp1, Arg5, Tyr10, Phe19–Ala21, Ser26–Gly29, Ile31, Ile32, Val36, and Val39 adopt a β-sheet conformation (more than 5 %) in the structures of Zn:Aβ40 (Fig. 3). For Zn:Aβ42, Asp7, Leu17–Phe20, Asn27, Ile32, Val36, and Val39–Ile41 adopt the β-sheet conformation with abundances greater than 5 %. Interestingly, β-strand formation with abundances greater than 5 % was reported at Arg5 and Glu11–His14 in free Aβ40 [72]. The N-terminal region residues (Asp1–Lys16) of the free Aβ42 peptide were reported not to adopt a β-strand structure with an abundance that is greater than 5 % [72]. In other words, the N-terminal region was shown to possess a larger propensity for β-strand formation in the free Aβ40 peptide structures than in the free Aβ42 peptide structures. Residues Glu11–His13 do not form an abundant β-sheet structure; however, Asp1, Arg5, and Tyr10 adopt a more prominent (up to 10 %) β-sheet structure upon Zn(II) binding to Aβ40 (Fig. 3). For the CHC region, the abundance of β-sheet increases by up to 12 % at Phe20 and Ala21 in the structures of Zn:Aβ40 in comparison with those of Zn:Aβ42. Instead, for the free Aβ40 and Aβ42 peptides, no such trend was obtained [72]. Another interesting impact of Zn(II) binding on the structures of Aβ occurs in the Ala21–Ala30 decapeptide region. Specifically, β-sheet formation in this region is more prominent for Zn:Aβ40 than for Zn:Aβ42 (Fig. 3). However, an opposite trend for the β-sheet structure formation in this region was detected for free Aβ40 and Aβ42 [72]. The C-terminal region of the free Aβ42 peptide (Ile31–Val36, Val39, Val40) and residues Ile31–Val36 of the free Aβ40 peptide adopt a β-strand structure with an abundance greater than 5 % (Fig. 3). Overall, the C-terminal region was found to be more structured based on the β-strand conformation in free Aβ42 in comparison with Aβ40 [72]. No such trend was found for the Zn:Aβ40 and Zn:Aβ42 alloforms, except at the C-terminus of Zn:Aβ42.
A comparison of our data with those of reported experiments yields interesting insights. Danielsson et al. [20] reported increased structuring in the N-terminal region of Aβ40 upon Zn(II) binding via NMR measurements. Our results indicate that this increased structuring might be further related to the increased helical structure formation in the N-terminal region (Asp1–Arg5) of Aβ40 upon Zn(II) binding. We found the same trend for Zn:Aβ42 in comparison with the reported values for Asp1–Arg5 in the structures of the free Aβ42 peptide [72]. Even though NMR measurements of Zn:Aβ16 reported 310-helix formation at Asp7–Gly9, Glu11–His14 in Zn:Aβ40 and Val12–His14 in Zn:Aβ42 exhibit a 10–40 % increase in the abundance of adopted 310-helical structure in comparison with the structures of the free Aβ40 and Aβ42 peptides [22, 68, 72]. This discrepancy might be attributed to Aβ structure variations due to fragment size differences as reported previously [68]. Furthermore, NMR measurements reported deviations in the chemical shifts of Asp23–Lys28 for Zn:Aβ40 in comparison with free Aβ40 that were attributed to either an increase in the turn structure or the presence of an additional Zn(II) binding site [20, 26]. Based on our results, we expect that these experimental chemical shift deviations result from an additional binding site, or the helical and β-sheet structure rather than the turn structure might be affected. Specifically, we detected a decrease in abundance of turn structure at Asp23–Lys28 of Zn:Aβ40 and at Asp23 and Val24 of Zn:Aβ42. However, the α-helix abundance for these residues and the β-sheet abundance at Ser26–Lys28 of Aβ40 and Asp23 of Aβ42 increase upon Zn(II) binding in comparison with the results reported for the free Aβ40 and Aβ42 (Fig. 2) [68, 72].
Previous simulations using the parameters of Hoops et al. [53] for the Zn:His3Glu region of Zn:Aβ40 resulted in increased β-sheet formation at Leu17–Gly22 and Val36–Val40 and the opposite trend for the N-terminal region [37]. In contrast, we found that the abundance of β-sheet structure increases only at Phe20, Ala21, and Val39 upon Zn(II) binding in the structures of Aβ40 (Fig. 3) [72]. We also found prominent β-sheet formation at Val24–Met35, which is in accord with NMR measurements that reported transient β-sheet formation at Lys28–Gly37 in Zn:Aβ40 [26]. This trend was not observed in the previous simulations of Zn:Aβ40 [37]. These differences are due to variations in the force field parameters for Zn:His3Glu [37]. As described above, our results show that Asp1, Arg5, and Tyr10 in the N-terminal region of Zn:Aβ40 adopt β-sheet structure (more than 5 %). These are missing in the structures of Zn:Aβ42, except for Tyr10, but the abundance is less than 5 %. Interestingly, quenched H/D-exchange NMR measurements suggested involvement of the N-terminal region of Zn:Aβ40 rather than Zn:Aβ42 in the aggregation process due to decreased solvent exposure of the Asp1–His14 region [28]. Since β-sheet formation has been associated with the aggregation likelihood of Aβ, we expect that Asp1, Arg5, and Tyr10 of the monomeric Zn:Aβ40 are reactive towards aggregation. Parallel to the N-terminal region, the reported β-sheet formation differences in the mid-domain and C-terminal regions between Zn:Aβ40 and Zn:Aβ42 from our simulations (see above) might be related to the decreased solvent exposure of Phe20–Asp23 and Ile31–Val36 in Zn:Aβ40 and of Leu17–Glu22 and Ile31–Ile41 in the Zn:Aβ42 aggregates as reported by experiments [28]. It is interesting to note that the C-terminal region of Zn:Aβ42 adopts prominent β-sheet structure at Val40–Ala42, which might be associated with the reduced solvent exposure of the C-terminus reported by NMR measurements for the aggregates [28]. To gain insights into the impact of intermolecular hydrogen-bonding interactions with the water molecules on the exposure of the N-terminal region, we also performed additional molecular dynamics simulations utilizing the modified TIP5P model for water (see “Materials and methods” and the electronic supplementary material). These simulations show that the N-terminal region of Zn:Aβ42 further extends into the solution in comparison with the same region in Zn:Aβ40, which in turn illustrates that this reported trend is not affected by the use of an explicit model for water.
Tertiary structure properties of the intrinsically disordered Zn:Aβ40 and Zn:Aβ42 metallopeptides
Prominent interactions occur between the mid-domain and N-terminal or C-terminal regions of Zn:Aβ40 and Zn:Aβ42 (Fig. 4). Specifically, abundant interactions between Arg5–His6 and Phe20–Asp23 as well as between Phe19–Phe20 and Ile32–Gly37 are detected in the structures of Zn:Aβ40 (Fig. 4a). Interactions between these residues may also correlate with the decreased solvent exposure of Phe20–Asp23 and Ile31–Val36 observed in the Zn:Aβ40 aggregates via NMR experiments [28]. In comparison with the tertiary structures of free Aβ40, the interactions between the N-terminal (His6–Glu11) and mid-domain (Asp23-Lys28) regions almost disappear upon Zn(II) binding (Fig. 4a) [72]. This finding supports the NMR measurements that revealed less abundant N-terminal and mid-domain interactions in the structures of Aβ40 upon Zn(II) binding [26]. Zn:Aβ42 displays abundant interactions between Arg5–His6 and Asp23–Ser26, Gly33–Leu34 and Val39–Val40, and Phe20 and Gly38–Ile41 (Fig. 4b). Interestingly, the interactions between Arg5–His6 and Asp23–Ser26 in Zn:Aβ42 are more abundant than in the structures of free Aβ42 [68], whereas the opposite trend holds true for Aβ40 and Zn:Aβ40. Furthermore, interactions between His13 or His14 and Val18–Phe20 are more abundant in Zn:Aβ42 in comparison with Zn:Aβ40. These findings might also relate to the decreased solvent exposure of Leu17–Glu22 and Ile31–Ile41 in the aggregates of Zn:Aβ42 [28]. The calculated ΔG values indicate that the N-terminal and mid-domain interactions are more stable by 32 and 18 kJ mol−1 than those between the C-terminal and mid-domain regions of Zn:Aβ40 and Zn:Aβ42, respectively. Even though the C-terminal interacts with the mid-domain region, these interactions are less favorable than the N-terminal and C-terminal region interactions in the structures of Zn:Aβ42 (ΔG = 52 kJ mol−1). The opposite trend was found for Zn:Aβ40.
Fig. 4.
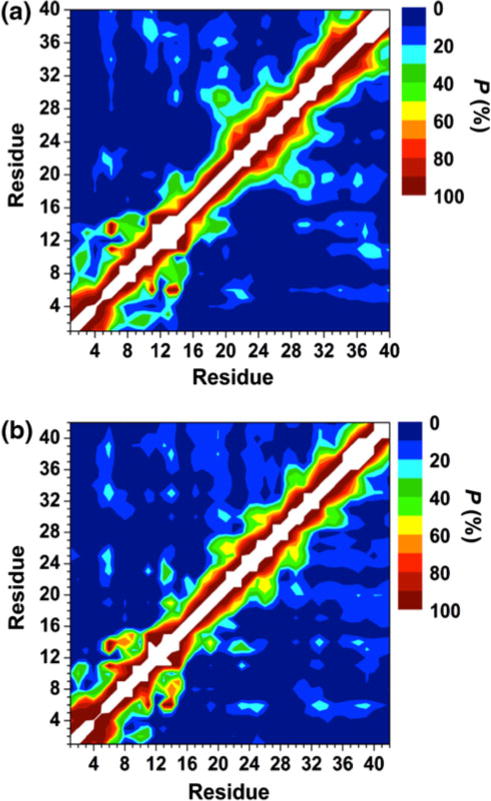
Calculated intramolecular metallopeptide interactions for the structures of the a Zn:Aβ40 and b Zn:Aβ42 metallopeptides in an aqueous solution. The color scale corresponds to the computed probability (P) for these interactions
The PMF analysis shows that the Zn:Aβ40 and Zn:Aβ42 structures become more stable with more abundant intramolecular interactions between the N-terminal and C-terminal, as well as the mid-domain and N-terminal or C-terminal regions (Fig. 5; see also the electronic supplementary material). Moreover, the interactions within the CHC region are more abundant with more favorable PMF values in the structures of both metallopeptides. Previous simulations reported that the abundance of the interactions between Arg5–Glu22 and Lys28–Val40 increases upon Zn(II) binding in the structures of Aβ40 [37]. When we compare our results for Zn:Aβ40 with those reported for monomeric Aβ40 [72], we find the same trend (Fig. 4). The N-terminal and Val24–Ala30 regions were reported to be more compact in Aβ40 in comparison with Aβ42 due to increased contacts in these regions [72]. We observe the same trend between the alloforms upon Zn(II) binding (Fig. 4). A more extended N-terminal region for Aβ42 in comparison with Aβ40 has also been reported in Aβ alloforms via molecular dynamics simulations performed by Urbanc et al. [73]. Overall, our results suggest that the N-terminal of Zn:Aβ42 is more extended than the N-terminal region of Zn:Aβ40 in aqueous solution, which agrees with the differences in the solvent exposure of the N-terminal found by experiments (see above) [28]. Thus, our results indicate that this trend is not affected by Zn(II) binding to the Aβ alloforms. Additionally, we obtained the same trend with the use of an explicit water model as explained above.
Fig. 5.
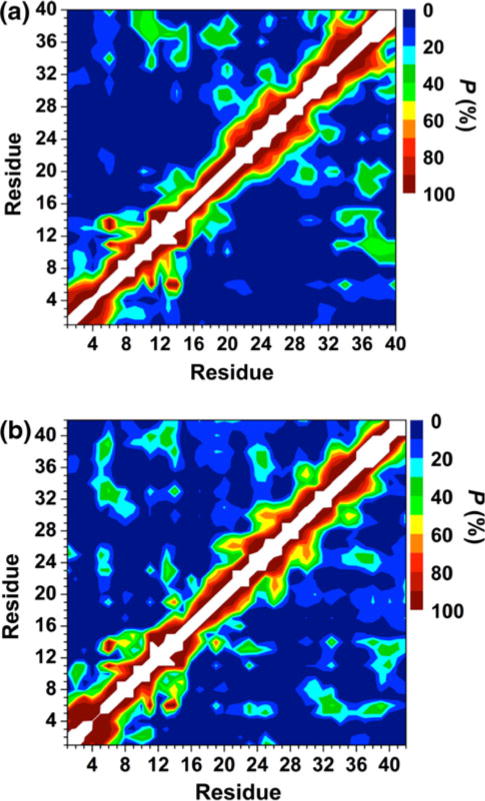
Calculated intramolecular metallopeptide interactions in the a Zn:Aβ40 and b Zn:Aβ42 metallopeptide structures located in each basin I on the potential of mean force surfaces (see Fig. 2). The color scale corresponds to the probability (P) of these calculated intramolecular interactions
Interestingly, Arg5 forms salt bridges (more than 10 %) with Asp1, Glu22, and Asp23 in both metalloalloforms (Table 3). The probabilities of Arg5 forming salt bridges with Asp1 and Glu22 increase upon Zn(II) binding to Aβ42 (Table 3) [68]. However, the abundances of Arg5 and Glu11 or Asp23 decrease in free Aβ42 upon Zn(II) binding. The most stable Glu22 and Lys28 salt bridge in the structures of Zn:Aβ40 occurs at a distance of 4.0 Å between Cγ and Nζ, whereas the stability of the salt bridge between Asp23 and Lys28 is negligible at the same distance (Fig. 6a). The opposite trend was reported for Aβ40 upon Zn(II) binding by Li et al. [37] using the force field parameters that were generated for a model in which Zn(II) is coordinated to three His residues and one water molecule. In the structures of Zn:Aβ42, the difference in stability between the Lys28 and Glu22 or Asp23 salt bridges is negligibly small at a distance of 4.0 Å between the Cγ and Nζ atoms (Fig. 6b). A comparison of these findings with our recent results indicates that the stability of the Lys28 and Glu22 salt bridge in Aβ42 decreases upon Zn(II) binding (Fig. 6) [68]. Further comparison also shows that the abundance of the turn structure formed in the Ala21–Val24 region of Aβ42 decreases by up to 20 % upon Zn(II) binding (Fig. 3) [68]. The probabilities of formation of the Arg5 and Asp1 or Glu22 salt bridges increase with decreasing PMF values (see the electronic supplementary material), indicating that these salt bridges play a role in the stabilization of the monomeric Zn:Aβ40 and Zn:Aβ42 structures in aqueous solution.
Table 3.
The salt bridges formed in the structures of Zn:Aβ40 and Zn:Aβ42 along with their probabilities
| Donor | Acceptor | Probability (%)
|
|||||
|---|---|---|---|---|---|---|---|
| Zn:Aβ40
|
Zn:Aβ42
|
||||||
|
R(C–N) ≤4 Å |
R(C–N) ≤5 Å |
R(C–N) ≤6 Å |
R(C–N) ≤4 Å |
R(C–N) ≤5 Å |
R(C–N) ≤6 Å |
||
| Arg5 | Asp1 | 55.1 | 63.5 | 65.1 | 82.8 | 93.3 | 94.3 |
| Arg5 | Glu22 | 13.1 | 13.4 | 13.5 | 14.9 | 15.2 | 15.2 |
| Arg5 | Asp7 | 23.2 | 29.6 | 34.1 | 26.1 | 29.7 | 32.1 |
| Arg5 | Glu3 | 20.2 | 21.4 | 21.6 | 5.1 | 5.7 | 5.9 |
| Arg5 | Asp23 | 14.0 | 15.3 | 16.3 | 12.9 | 12.9 | 13.0 |
| Lys16 | Glu22 | 1.0 | 1.3 | 1.4 | 19.7 | 26.5 | 27.7 |
| Arg5 | Glu11 | 0.1 | 0.7 | 1.3 | 2.0 | 7.1 | 22.5 |
| Lys28 | Glu3 | 1.3 | 1.8 | 1.9 | 0.2 | 0.4 | 0.5 |
R(C–N) is the distance between the carboxylate carbon atom and the side-chain or N-terminal nitrogen atom
Fig. 6.
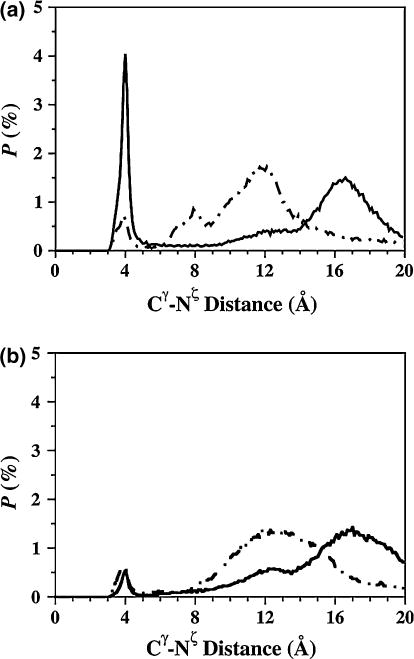
The calculated probability distribution of the distance between the Cγ atom of the Glu22 (solid line) and Asp23 (dashed line) residues and the Nζ atom of the Lys28 residue in the simulated a Zn:Aβ40 and b Zn:Aβ42 metallopeptide structures in an aqueous solution
Overall, this study shows the specific structural differences and thermodynamic properties of Zn:Aβ40 and Zn:Aβ42 in aqueous solution that appear to underlie the pathogenesis of Alzheimer’s disease. Although the results presented here offer the first comparison of the monomeric Zn:Aβ40 and Zn:Aβ42 metallopeptide structures in aqueous solution with dynamics, these simulations depend on the chosen force field parameters. The inclusion of dynamic effects that capture polarizability derived from various quantum mechanical methods (Hartree–Fock, density functional theory, and multireference methods) can influence the outcome of molecular simulations. Currently, we are developing these force field parameters for a large class of metalloproteins. Nevertheless, the microsecond timescale simulation results obtained for the structural properties are in overall agreement with the findings of reported experimental studies for the full-length metallopeptides. This agreement further indicates the biological relevance of our theoretical studies.
In summary, the thermodynamics shows that the structures of Zn:Aβ42 are more favorable than those of Zn:Aβ40 and that Zn(II) binding stabilizes the free Aβ42 structures in aqueous solution. More prominent α-helix is formed at Asp1–Ser8 located in the N-terminal region of Zn:Aβ42 in comparison with Zn:Aβ40. Despite, residues Asp1, Arg5, and Tyr10 adopt abundant β-sheet structure in the N-terminal region of Zn:Aβ40, which disappears or is significantly depressed in Zn:Aβ42. Previous studies reported a larger structuring in the C-terminal region of free Aβ42 on the basis of β-sheet formation in comparison with the same region of free Aβ40. Surprisingly, such a clear trend is not detected for Zn:Aβ40 and Zn:Aβ42, indicating that Zn(II) binding impacts this C-terminal structuring trend. It is also interesting to note that Zn(II) binding decreases the abundance of the adopted turn structure in the Ala21–Ala30 decapeptide region of free Aβ40 and Aβ42. We should recall that the salt bridge formations between Lys28 and Glu22 or Asp23 have been linked to the stable turn structure formation at Ala21–Ala30 in the conformations of free Aβ in an aqueous medium. The decreased abundance of the salt bridge between Glu22 and Lys28 upon Zn(II) binding to Aβ42 might be associated with the depressed turn structure formation at Ala21–Ala30 in the structures of Zn:Aβ42 in comparison with those reported for free Aβ42. The Gibbs free energy calculations show that the C-terminal region of Zn:Aβ42 interacts more favorably with the mid-domain rather than with the N-terminal region. The opposite thermodynamic trend is detected for Zn:Aβ40. A comparison of the tertiary structures with those reported for the free Aβ40 and Aβ42 peptides reveals that the intramolecular interactions between His6–Glu11 and Asp23–Lys28 are less abundant in Aβ40 upon Zn(II) binding. However, Arg5 and His6 interact more abundantly with Asp23–Ser26 in Aβ42 upon Zn(II) binding. Separate simulations utilizing either the implicit or explicit water models showed that the N-terminal region of Zn:Aβ42 is more extended into the solution than the same region of Zn:Aβ40. Our thermodynamic calculations further support this finding and show that the N-terminal region of Zn:Aβ40 interacts more favorably with the C-terminal region in comparison with Zn:Aβ42. Interestingly, Arg5 forms stable salt bridges with various residues in both metallopeptides.
In comparison with free Aβ40 and Aβ42 peptides [72], our findings show that the helical structure formation in the C-terminal region of free Aβ increases upon Zn(II) binding. Despite, β-sheet formations at Val12–Gln15, Ala21–Ala30 and Gly33–Gly38 in the structures of free Aβ are depressed upon Zn(II) binding. Since β-sheet formation at the monomeric and early oligomeric levels has been proposed to be linked to the aggregation process, these reduced or lacking β-sheet formations might be associated with the formation of less toxic Zn:Aβ amorphous aggregates instead of fibrillar aggregates. Furthermore, the N-terminal region extends more into the solution in the structures of monomeric Zn:Aβ42 in comparison with Zn:Aβ40. Given that experiments reported a more extended N-terminal region for the aggregates of Zn:Aβ42 in comparison with Zn:Aβ40, we propose that this trend in aggregation characteristic is related to the structural differences between monomeric Zn:Aβ42 and Zn:Aβ40. Antibodies blocking parts of the mid-domain and C-terminal regions instead of the N-terminal region of Zn:Aβ42 might help to further depress the aggregation process and to decrease the toxicity.
Supplementary Material
Acknowledgments
This research was supported by an allocation and computing resources provided with the help of the National Institute for Computational Sciences (grant TG-CHE110044). The calculations and simulations were performed on Kraken at the National Institute for Computational Sciences and Ranger at the Texas Advanced Computing Center. G.P. is thankful for the financial support provided by the National Institutes of Health (G12-RR013646), the RCMI Center for Interdisciplinary Health Research, and the Alzheimer’s Association. O.C. is thankful for support from the University of Texas at San Antonio and the Neuroscience Institute of the San Antonio Life Sciences Institute (Charles Wilson) at the University of Texas at San Antonio. We also thank Carlos Gonzalez (NIST) for helpful discussions.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00775-012-0909-9) contains supplementary material, which is available to authorized users.
Contributor Information
Olivia Wise-Scira, Department of Chemistry, The University of Texas at San Antonio, One UTSA Circle, San Antonio, TX 78249, USA.
Liang Xu, Department of Chemistry, The University of Texas at San Antonio, One UTSA Circle, San Antonio, TX 78249, USA.
George Perry, Neurosciences Institute, The University of Texas at San Antonio, One UTSA Circle, San Antonio, TX 78249, USA.
Orkid Coskuner, Email: orkid.coskuner@utsa.edu, Department of Chemistry, The University of Texas at San Antonio, One UTSA Circle, San Antonio, TX 78249, USA; Neurosciences Institute, The University of Texas at San Antonio, One UTSA Circle, San Antonio, TX 78249, USA.
References
- 1.Breydo L, Uversky VN. Metallomics. 2011;3:1163–1180. doi: 10.1039/c1mt00106j. [DOI] [PubMed] [Google Scholar]
- 2.Tõugu V, Palumaa P. Coord Chem Rev. 2012 doi: 10.1016/jcc.r.2011.12.008. [DOI] [Google Scholar]
- 3.Brown DR. Dalton Trans. 2009;(21):4069–4076. doi: 10.1039/b822135a. [DOI] [PubMed] [Google Scholar]
- 4.Walsh DM, Selkoe DJ. J Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- 5.Hardy J, Selkoe DJ. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 6.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 7.Walsh DM, Lomakin A, Benedek GB, Condron MM, Teplow DB. J Biol Chem. 1997;272:22364–22372. doi: 10.1074/jbc.272.35.22364. [DOI] [PubMed] [Google Scholar]
- 8.Chen TT, Wang XY, He YF, Zhang CL, Wu ZY, Liao K, Wang JJ, Guo ZJ. Inorg Chem. 2009;48:5801–5809. doi: 10.1021/ic900025x. [DOI] [PubMed] [Google Scholar]
- 9.Garai K, Sahoo B, Kaushalya SK, Desai R, Maiti S. Biochemistry. 2007;46:10655–10663. doi: 10.1021/bi700798b. [DOI] [PubMed] [Google Scholar]
- 10.Lovell MA, Xie CS, Marksbery WR. Brain Res. 1999;823:88–95. doi: 10.1016/s0006-8993(99)01114-2. [DOI] [PubMed] [Google Scholar]
- 11.Cardoso SM, Rego AC, Pereira C, Oliveira CR. Neurotox Res. 2005;7:273–281. doi: 10.1007/BF03033885. [DOI] [PubMed] [Google Scholar]
- 12.Huang XD, Cuajungco MP, Atwood CS, Hartshorn MA, Tyndall JDA, Hanson GR, Stokes KC, Leopold M, Multhaup G, Goldstein LE, Scarpa RC, Saunders AJ, Lim J, Moir RD, Glabe C, Bowden EF, Masters CL, Fairlie DP, Tanzi RE, Bush AI. J Biol Chem. 1999;274:37111–37116. doi: 10.1074/jbc.274.52.37111. [DOI] [PubMed] [Google Scholar]
- 13.Yoshiike Y, Tanemura K, Murayama O, Akagi T, Murayama M, Sato S, Sun XY, Tanaka N, Takashima A. J Biol Chem. 2001;276:32293–32299. doi: 10.1074/jbc.M010706200. [DOI] [PubMed] [Google Scholar]
- 14.Liu B, Moloney A, Meehan S, Morris K, Thomas SE, Serpell LC, Hider R, Marciniak SJ, Lomas DA, Crowther DC. J Biol Chem. 2011;286:4248–4256. doi: 10.1074/jbc.M110.158980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reinhard C, Hébert SS, De Strooper B. EMBO J. 2005;24:3996–4006. doi: 10.1038/sj.emboj.7600860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jarrett JT, Berger EP, Lansbury PT. Biochemistry. 1993;32:4693–4697. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- 17.El-Agnaf OM, Mahil DS, Patel BP, Austen BM. Biochem Biophys Res Commun. 2000;273:1003–1007. doi: 10.1006/bbrc.2000.3051. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y, McLaughlin R, Goodyer C, LeBlac A. J Cell Biol. 2002;156:519–529. doi: 10.1083/jcb.200110119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mucke LE, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Danielsson J, Pierattelli R, Banci L, Graslund A. FEBS J. 2007;274:46–59. doi: 10.1111/j.1742-4658.2006.05563.x. [DOI] [PubMed] [Google Scholar]
- 21.Sato T, Kienlen-Campard P, Mahiuddin A, Liu W, Li H, Elliott JI, Aimoto S, Constantinescu SN, Octave JN, Smith SO. Biochemistry. 2006;45:5503–5516. doi: 10.1021/bi052485f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zirah S, Kozin SA, Mazur AK, Blond A, Cheminant M, Segalas-Milazzo I, Debey P, Rebuffat S. J Biol Chem. 2006;281:2151–2161. doi: 10.1074/jbc.M504454200. [DOI] [PubMed] [Google Scholar]
- 23.Gaggelli E, Janicka-Klos A, Jankowska E, Koxlowski H, Migliorini C, Molteni E, Valensin D, Valensin G, Wieczerzak E. J Phys Chem B. 2008;112:100–109. doi: 10.1021/jp075168m. [DOI] [PubMed] [Google Scholar]
- 24.Mekmouche Y, Coppel Y, Hochgrafe K, Guilloreau L, Tallmard C, Mazarguil H, Faller P. ChemBioChem. 2005;6:1663–1671. doi: 10.1002/cbic.200500057. [DOI] [PubMed] [Google Scholar]
- 25.Syme CD, Viles JH. Proteins Proteomics. 2006;1764:246–256. doi: 10.1016/j.bbapap.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 26.Rezaei-Ghaleh N, Giller K, Becker S, Zweckstetter M. Biophys J. 2011;101:1202–1211. doi: 10.1016/j.bpj.2011.06.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Minicozzi V, Stellato F, Comai M, Dalla Serra M, Potrich C, Meyer-Klaucke W, Morante S. J Biol Chem. 2008;283:10784–10792. doi: 10.1074/jbc.M707109200. [DOI] [PubMed] [Google Scholar]
- 28.Olofsson A, Lindhagen-Persson M, Vestling M, Sauer-Eriksson AE, Ohman A. FEBS J. 2009;276:4051–4060. doi: 10.1111/j.1742-4658.2009.07113.x. [DOI] [PubMed] [Google Scholar]
- 29.Liu ST, Howlett G, Barrow CJ. Biochemistry. 1999;38:9373–9378. doi: 10.1021/bi990205o. [DOI] [PubMed] [Google Scholar]
- 30.Miura T, Suzuki K, Kohata N, Takeuchi H. Biochemistry. 2000;39:7024–7031. doi: 10.1021/bi0002479. [DOI] [PubMed] [Google Scholar]
- 31.Tsvetkov PO, Kulikova AA, Golovin AV, Tkachev YV, Archakov AI, Kozin SA, Makarov AA. Biophys J. 2010;99:L84–L86. doi: 10.1016/j.bpj.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang DS, McLaurin J, Qin KF, Westaway D, Fraser PE. Eur J Biochem. 2000;267:6692–6698. doi: 10.1046/j.1432-1327.2000.01767.x. [DOI] [PubMed] [Google Scholar]
- 33.Zirah S, Rebuffat S, Kozin SA, Debey P, Fournier F, Lesage D, Tabet JC. J Mass Spectrom. 2003;228:999–1016. [Google Scholar]
- 34.Curtain CC, Ali F, Volitakis I, Cherny RA, Norton RS, Beyreuther K, Barrow CJ, Masters CL, Bush AI, Barnham KJ. J Biol Chem. 2001;5:20466–20473. doi: 10.1074/jbc.M100175200. [DOI] [PubMed] [Google Scholar]
- 35.Miura T, Suzuki K, Takeuchi H. J Mol Struct. 2001;598:79–84. [Google Scholar]
- 36.Furlan S, La Penna G. Phys Chem Chem Phys. 2009;11:6468–6481. doi: 10.1039/b822771c. [DOI] [PubMed] [Google Scholar]
- 37.Li WF, Zhang J, Su Y, Wang J, Qin M, Wang W. J Phys Chem B. 2007;111:13814–13821. doi: 10.1021/jp076213t. [DOI] [PubMed] [Google Scholar]
- 38.Miller Y, Ma BY, Nussinov R. Proc Natl Acad Sci USA. 2010;107:9490–9495. doi: 10.1073/pnas.0913114107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bergeron DE, Coskuner O, Hudgens JW, Gonzalez CA. J Phys Chem C. 2008;112:12808–12814. doi: 10.1021/jp711759q. [DOI] [PubMed] [Google Scholar]
- 40.Coskuner O, Bergeron DE, Rincon L, Hudgens JW, Gonzalez CA. J Phys Chem A. 2008;112:2940–2947. doi: 10.1021/jp711759q. [DOI] [PubMed] [Google Scholar]
- 41.Coskuner O, Bergeron DE, Rincon L, Hudgens JW, Gonzalez CA. J Phys Chem A. 2009;113:2491–2499. doi: 10.1021/jp805747f. [DOI] [PubMed] [Google Scholar]
- 42.Coskuner O, Jarvis EAA. J Phys Chem A. 2008;112:2628–2633. doi: 10.1021/jp7098575. [DOI] [PubMed] [Google Scholar]
- 43.Coskuner O, Allison TC. In: Metallic systems: a quantum chemist’s perspective. Allison TC, Coskuner O, Gonzalez CA, editors. CRC; Boca Raton: 2011. pp. 107–134. [Google Scholar]
- 44.Coskuner O, Gonzalez CA. In: Metallic systems: a quantum chemist’s perspective. Allison TC, Coskuner O, Gonzalez CA, editors. CRC; Boca Raton: 2011. pp. 83–106. [Google Scholar]
- 45.Wise O, Xu L, Coskuner O. In: Metallic systems: a quantum chemist’s perspective. Allison TC, Coskuner O, Gonzalez CA, editors. CRC; Boca Raton: 2011. pp. 29–82. [Google Scholar]
- 46.Shi SH, Yan L, Yang Y, Fisher-Shaulsky J, Tacher T. J Comput Chem. 2003;24:1059–1076. doi: 10.1002/jcc.10171. [DOI] [PubMed] [Google Scholar]
- 47.Koca J, Zhan CG, Rittenhouse RC, Ornstein RL. J Am Chem Soc. 2001;123:817–826. doi: 10.1021/ja000439r. [DOI] [PubMed] [Google Scholar]
- 48.Suarez D, Merz KM. J Am Chem Soc. 2001;123(3759):3770. doi: 10.1021/ja003796a. [DOI] [PubMed] [Google Scholar]
- 49.Lin F, Wang RX. J Chem Theory Comput. 2010;6:1852–1870. doi: 10.1021/ct900454q. [DOI] [PubMed] [Google Scholar]
- 50.Peters MB, Yang Y, Wang B, Fusti-Molnar L, Weaver MN, Merz KM. Chem Theory Comput. 2010;6:2935–2947. doi: 10.1021/ct1002626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Banci L. Curr Opin Chem Biol. 2003;7:143–149. doi: 10.1016/s1367-5931(02)00014-5. [DOI] [PubMed] [Google Scholar]
- 52.Comba P, Remenyi R. Coord Chem Rev. 2003;238:9–20. [Google Scholar]
- 53.Hoops SC, Anderson KW, Merz KM. J Am Chem Soc. 1991;113:8262–8270. [Google Scholar]
- 54.Vedani A, Huhta DW. J Am Chem Soc. 1990;112:4759–4767. [Google Scholar]
- 55.Wu R, Lu Z, Cao Z, Zhang Y. J Chem Theory Comput. 2011;7:433–443. doi: 10.1021/ct100525r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery J, Vreven JAT, Kuden KN, Burant JC, Milliam JM, Iyengar SS, Tomasi J, Barone V, Munnucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham A, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez CA, Pople JA. Gaussian 03. Gaussian; Wallingford: 2004. [Google Scholar]
- 57.Bayly CI, Cieplak P, Cornell WD, Kollman PA. J Phys Chem. 1993;97:10269–10280. [Google Scholar]
- 58.Sousa SF, Fernandes PA, Ramos MJ. In: Kinetics and dynamics: from nano- to bioscale. Paneth P, Dybala-Defratyka A, editors. Springer; New York: 2010. pp. 299–330. [Google Scholar]
- 59.Sugita Y, Okamoto Y. Chem Phys Lett. 1999;314:141–151. [Google Scholar]
- 60.Simmerling C, Hornak V, Abel R, Okur A, Strockbine B, Roitberg A. Proteins. 2006;65:712. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Case DA, Onufriev A, Bashford D. Proteins. 2004;55:383–394. doi: 10.1002/prot.20033. [DOI] [PubMed] [Google Scholar]
- 62.Allen MP, Tildesley DJ. Computer simulations of liquids. Oxford University Press; Oxford: 1999. [Google Scholar]
- 63.Frenkel D, Smit B. Understanding molecular simulation: from algorithms to applications. Academic; San Diego: 2002. [Google Scholar]
- 64.van der Spoel D, Patriksson A. Phys Chem Chem Phys. 2008;10:2073–2077. doi: 10.1039/b716554d. [DOI] [PubMed] [Google Scholar]
- 65.Prakash MK, Barducci A, Parrinello M. J Chem Theory Comput. 2011;7:2025. doi: 10.1021/ct200208h. [DOI] [PubMed] [Google Scholar]
- 66.Coskuner O, Deiters UK. Z Phys Chem. 2006;220:349–369. [Google Scholar]
- 67.Coskuner O, Deiters UK. Z Phys Chem. 2007;221:785–799. [Google Scholar]
- 68.Wise-Scira O, Xu L, Kitahara T, Perry G, Coskuner O. J Chem Phys. 2011;135:205101. doi: 10.1063/1.3662490. [DOI] [PubMed] [Google Scholar]
- 69.Kollman PA, Massova I, Reyes C, Kuhn B, Huo SH, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, Case DA, Cheatham TE. Acc Chem Res. 2000;33:889–897. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 70.Lee MR, Duan Y, Kollman PA. Proteins Struct Funct Genet. 2000;39:309–316. [PubMed] [Google Scholar]
- 71.Kabsch W, Sander C. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 72.Yang MF, Teplow DB. J Mol Biol. 2008;384:450–464. doi: 10.1016/j.jmb.2008.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Urbanc B, Cruz L, Yun S, Buldyrev SV, Bitan G, Teplow DB, Stanley HE. Proc Natl Acad Sci USA. 2004;101:17345–17350. doi: 10.1073/pnas.0408153101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.