

ToC Entry
A kinetic, single cell proteomic study of chemically-induced carcinogenesis is interpreted by treating the single cell data as fluctuations of an open system transitioning between different steady states. In analogy to a first-order transition, phase coexistence and the loss of degrees of freedom are observed. The transition is detected well before the appearance of the traditional biomarker of the carcinogenic transformation.
Keywords: single cell proteomics, microfluidics, carcinogenesis, phase transition, steady state
Cellular transitions between distinct steady states are fundamental in processes that range from cellular differentiation to the various steps of carcinogenesis. Such transitions are broadly studied, but general models for understanding them have been historically limited to qualitative descriptions.[1–4] This contrasts with phase transitions seen in physical systems, which are well characterized within the context of the physico-chemical laws, and can be partially understood within the context of, for example, simple Ising-type models of magnetization. A control parameter (such as temperature) is varied, and the fluctuations of the magnetic moments of lattice sites are analyzed as the system approaches and passes through a critical point. Those fluctuations can reveal statistical signatures of phase transition, which can be verified experimentally. These include signatures of stable states, signatures of the proximity to a critical point (increased fluctuations), or signatures of a critical point (loss of degrees of freedom or divergence of correlation length). The advantages of such models are that they are independent of many system-specific details; such details are captured in the interactions between the lattice sites.
Such physical models have been applied to in silico modelling of transitions in gene and protein regulatory networks within cells.[4, 5] Chemical kinetics[6] or a master equation formalism[7, 8] is used to model the regulatory networks as a set of elementary reactions, which can provide what are effectively the site interactions. Tuning specific kinetic or molecular parameters can push the model towards or through a critical point. These approaches can identify steady states, and provide insights into those parameters that can trigger transitions. For purely in silico models, or even for experimentally-calibrated models,[6] predictions near critical points (non-linear regimes) are challenging.
We describe a conceptually straightforward and potentially general approach for understanding cellular transitions. We begin with quantitative measurements of a panel of functional proteins from single cells. For each regulatory protein, we measure its single cell expression level for a statistically significant number of cells, thereby, determining the variations in expression levels. We interpret the experimental results using an information theoretic approach for resolving steady states, transitions between states, and a detailed analysis, at the molecular level, of how those transitions relate back to their control parameter(s).
The Single Cell Barcode Chip (SCBC) has been extensively described and validated previously.[9, 10] It is based on isolating single cells within nanoliter-volume microchambers for cell capture, lysis, and subsequent proteomic analysis (Figure S1 and Text ST1–7). Each microchamber contains a miniature antibody array for the capture and detection of a panel of proteins (Figure S1d). The cell determines the copy numbers of a given protein, while the microchamber volume determines the concentration. Sandwich ELISA-like assays with measurement error of <10%, permit full calibrations (Text ST7). The benchmarking of the SCBC assay with other single cell proteomics techniques such as FACS and mass cytometry has been reported.[11, 12]
Our theoretic approach starts with the statistical definition of a stable steady state, which is one in which the fluctuations (here, the measured protein copy numbers per cell, measured across many single cells) comprise a uniformly broadened distribution about an unchanging mean (a state of minimal free energy). The application of a chemical carcinogen to epithelial cells induces certain constraints within the cells that result in non-uniform fluctuations, which may be interpreted as deviations from the steady state. To analyze the fluctuations, we employ thermodynamics based Surprisal analysis.[13–15] This analysis was first applied to characterize the dynamics of non-equilibrium systems in chemical physics.[13] In biology, Surprisal analysis allows for the identification of the expected gene expression levels at the steady state,[16, 17] and deviations from the steady state due to constraints operating within the system.[15, 17] Here, we recognize the constraints by identifying groups of proteins associated with a given constraint, and so exhibit similar deviations from the steady state.[18] Thus, we relate a given constraint to an unbalanced process operating in the system. More than one unbalanced process may operate in the system. Since the experiments yield measurements of specific protein levels in copy numbers per cell, we can analyze the variations of free energy differences (albeit limited by the measured proteins) that exist between the cell populations at a particular time point of treatment, relative to the steady state (untreated) control cells.
Cells are finite systems. This means that cells from a clonal population will vary from one another in terms of the copy numbers of specific analytes.[19] It is this cell-to-cell variability that, comprises the fluctuations which, in turn, provide a critical input into the thermodynamics-inspired models used here. By contrast, bulk measurements just provide an average value. An additional set of parameters that is captured at the single cell level are the protein-protein correlations. In bulk assays, two proteins are correlated if their average levels increase or decrease together, when the system is perturbed. In this work, the measured correlations and anti-correlations depend upon the statistical relationship between any two proteins, as measured across many single cells. Two proteins may be correlated in the bulk, but have no statistically significant protein-protein correlation at the level of single cells (detailed example in Figure S2).
The basic system used for carcinogenic transformation is a non-transformed MCF-10F cell line, which is grown in a mitogenic factor-supplemented growth medium (Text ST8). Following literature protocols,[20–22] the CIC transition was triggered by periodically dosing a culture of MCF-10F human mammary epithelial cells with Benzo[a]pyrene (B[a]P), according to the timeline of Figure 1(a) and 1(b). Each 48 hour dose is followed by passaging in normal growth medium for ~2 weeks. After three dosages, the cells were maintained in the normal growth medium indefinitely. Untreated controls were cultured and passaged concurrently (Text ST8 and ST9). CiC kinetics was studied through quantification of a panel of 11 functional proteins from single cells, at 9 different time points (Figure 1(b)).
Figure 1. The experimental schema and results of colonogenic assay.

a. Protocol for the cycles of B[a]P treatment of the MCF 10F cells. b. Time line representing B[a]P dosing, and the 9 (including control) time points for the SCBC analysis. c. Colonogenic assays of cells treated with B[a]P, in complete growth medium under limited cell density. The numbers at the top right of the photographs presents the time point from which the treated cells were used to perform the assay. d. Colonogenic assay for characterizing the anchorage independence (AIG) of MCF-10F cells treated with BaP. e. quantification of colonogenic assay in complete growth medium under limiting cell density (Supporting information text ST 10). Plot shows number of colonies formed from 5000 cell (n=3). f. Quantification of colonogenic assay in reduced growth medium (Supporting information text ST 10; Figure S3). Plot shows number of colonies formed from 20000 cell (n=3). g. Quantification of colonogenic assay in Anchorage Independence assay (Supporting information text ST 10).
The selection of the protein panel to characterize the carcinogenic transformation was based on a literature analysis deigned to identify a readily managed number of functional phospho-proteins that best represent the signaling dynamics during carcinogenesis.[23–34] The panel of assayed proteins (Table S1) represent various cellular processes that are strongly influenced by the carcinogen treatment, including cell proliferation, cell-cycle regulation and apoptosis. A more comprehensive panel could permit a more statistically thorough investigation of the CiC and perhaps reveal more biology, but it would be unlikely to alter the basic conclusions drawn from this work.
The transformation phenotypes were analyzed using bulk colonogenic assays to assess changes in cell viability and cellular phenotypes. a.) limited cell density growth assay, b.) dependence on the mitotic factor for the cell survival. c.) ability of the cells to survive in 3D growth matrix (Anchorage Independence Assay). In all these assays, only the transformed cells demonstrated proliferative advantage. Four weeks following start of treatment, the cells exhibited minor morphology changes and, at some point after day 40, they began to exhibit a carcinogenic phenotype, which was clearly measured at day 96, by enhanced proliferation (Figure 1(d,e)), enhanced survival in low growth factor medium (Figure 1(f) and Figure S3), and enhanced ability to generate colonies in soft agar, (Figure 1(g,h)).
The fluctuations of most individual proteins are characterized by narrow distributions at day 0 (control; also see Figure S4 for control samples at different time points) and beyond day 40, where the cells in population are anticipated to be of pre-cancerous phenotype[20, 22]. Between day 0 and day 40, the fluctuations exhibit long, uneven tails towards high protein copy numbers (Figure 2(a) and Figure S5). The protein-protein correlation matrices are sparse at day 0 (untreated control; Figure S5a) and days 57–96, but exhibit nearly all-to-all correlations at intermediate times, with a temporary reduction in correlations at day 21 (Figure 2(b)). The all-to-all correlations indicate a divergence of correlation length, which is a statistical indicator of a critical point transition. Preliminary analysis of the single cell data using both the distribution variance and the protein-protein correlation indices indicate the existence of one or more critical points during the time point between days 8–28 (Figure 2).
Figure 2. Representative results of the SCBC analysis.
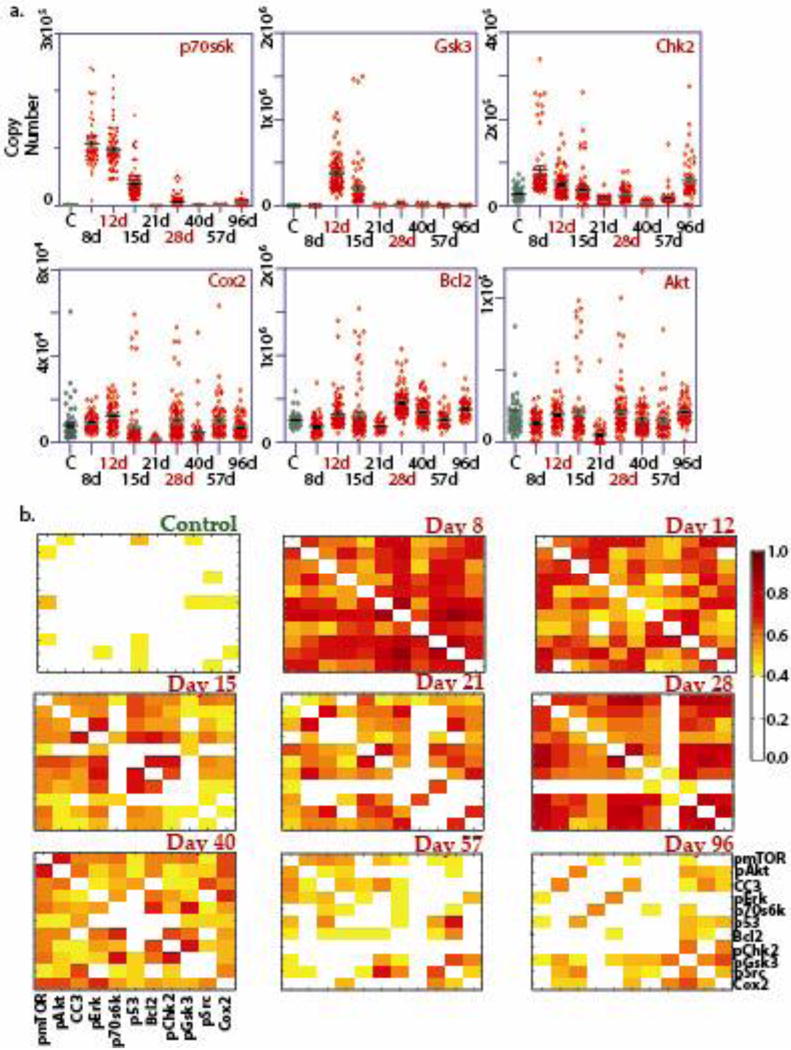
a. One-dimensional scatter plots of the single cell levels of few representative proteins across the kinetic timeline. b. Protein-protein covariance matrices, extracted from SCBC data of MCF-10F cells treated with BaP, at all the time points in the kinetic study.
To make a thermodynamic based characterization of the critical points during the carcinogenesis, we employed surprisal analysis.[13, 15] The analysis is based on the premise that all biological systems reach a state of minimal free energy under standard temperature and pressure given the existing environmental and genomic constraints. Importantly, this state is not necessarily the basal, most stable, steady state, which is reached when the system is free of constraints. Our analysis identifies the steady state protein expression levels, and the deviations in those levels due to the existing constraints. B[a]P treatment imposes a constraint but more than one constraint may be identified in the system. Each constraint is associated with an unbalanced biological process that deviates the system from the steady state and causes the coordinated deviations of a subset of proteins from their steady state expression level. For more details on surprisal analysis of non-equilibrium biological systems see [15–17].
Many complex biological processes, such as the initial transformation towards cancer, occur through the actions of individual cells.[35] Similarly, our analysis was implemented independently for each measured cell. The levels of different proteins for each cell at each time point t are represented as Equation 1 (details in Text ST11):
| (1) |
Here, the expected expression level of a protein i at the steady state in a measured cell at the time point t. This parameter corresponds to the steady state and is time-invariant. We do not constrain to a constant value but use any resulting variation as a check for the constancy of the parameter. The exponential term in Equation 1 represents the deviation from the steady value due to the constraints. For convenience, for each cell we express the steady state in a form analogous to the deviations, . and are weights of a protein i in the steady state and unbalanced processes respectively, while and are weights of the steady state and unbalanced processes in a cell i at time point t. (Details in Table S1 and Text ST12).
The untreated (control) cells, as well as cells at d40 – d96, were found to be close to the steady state, meaning that . This can be seen in Figure 3(a), in which we plot time dependent weights of the steady state and the first two constraints, for each cell. Note that and are near 0-valued for the control (C) and at days 40–96. Furthermore, note, in Figure 3(b), that the steady state variations ( ) for the control and for days 40–96 are well-fit by a single Gaussian. For these latter time points, the mean value of is slowly evolving, as the weights ( and ) of the constraints dissipate over time and the system settles to a steady state.
Figure 3. Analysis of single cell data through the CiC transition.
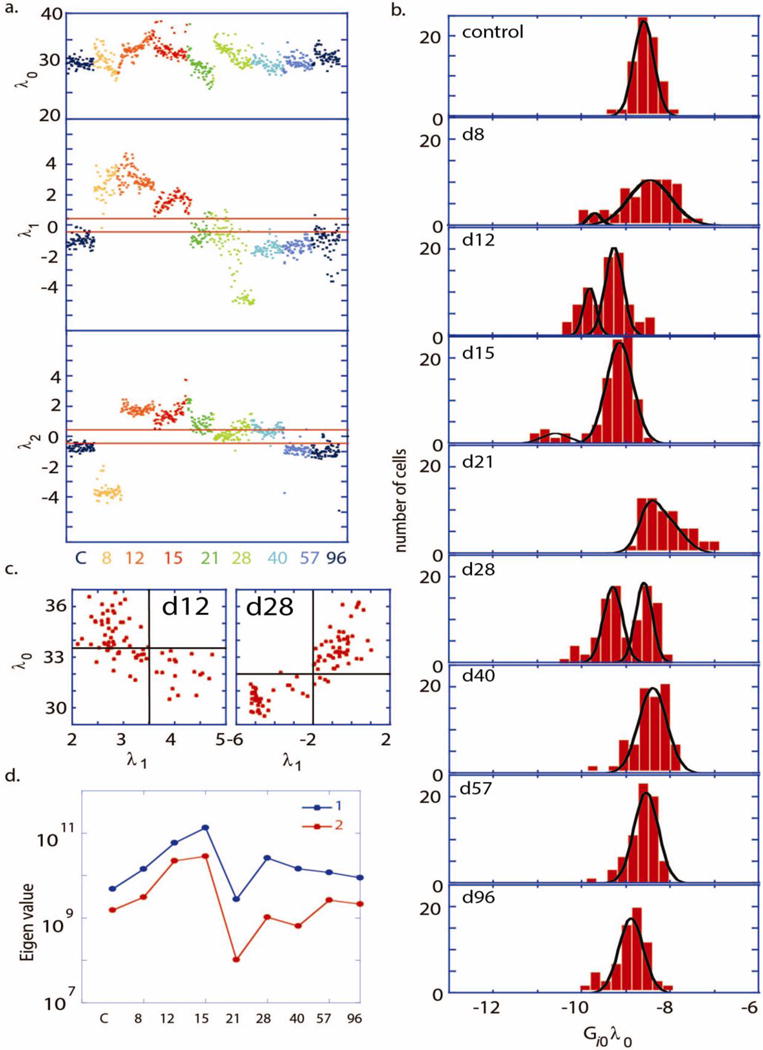
a. The weight of the steady state term ( ) and the first two constraints ( and ) plotted, for each cell and color-coded for each time point. b. Number of cells vs. , with different panels shown at different time points shown for the pS6K protein. Comparable distributions are obtained for other measured proteins since are similar (Supplementary figure S7). The distribution (b) at every time point was fitted to either unimodal or Bimodal Gaussian distributions. Bimodal Gaussian distributions appear as the best fitting distribution for the days 12, 15 and 28 (R2 >0.95), whereas unimodal Gaussian is the best fit for the control, d57 and d96 (R2 >0.95). c. Scatter plots of values vs at the day 12 and day 28. The unbalanced process has higher significance, , (as indicated by at the day 12 or at the day 28) in the cell subpopulations with lower protein abundance, as represented by lower values of . d. Principle eigenvalues (1 and 2) of the covariance matrix are plotted as a function of t.
At intermediate times, the picture is quite different. Shortly following carcinogen exposure (days 8–28), the two constraints take on significant amplitude. Figure 3(a)), implying strong deviations from the steady states. For a given time point, these constraints deviate in the same direction for all of the cells in the population, excepting at day 28 (Figure 3(a)). A third constraint ( ) was found to have significant weight only at day 21 (Figure S6). All other higher terms fluctuate about zero throughout the time course studied. Interestingly, the steady state variations (Figure 3(b)) yield clear bimodal distributions at day 12, either bimodal or strongly skewed distributions at days 15 and 21, and again a clear bimodal distribution at day 28. These bimodal distributions hint at a phase coexistence, which, in turn, hints at a phase transition that happens at day ~12 and then again at day 28.
We analyzed this potential phase coexistence by taking a cue from the ice/water transition system. We can distinguish between two phases (water and ice) based on different physical attributes such as different index of light reflection or differences in the shear modulus. In other words, we look for an observable parameter that differs in value between the two phases. Here we use the weight of the constraint (most significant constraint; ) as a distinguishing observable parameter for the CIC transition. Scatter plots of vs. for days 12 and 28 are shown in Figure 3(c). These plots reveal that the two coexisting cell populations differ in the significance of the first unbalanced process. (For more details see Text S12). A biological interpretation of the constraint is provided by analyzing the values of . This shows that the dominant constraint is largely characterized by an anti-correlation of the proteins p70S6K and pERK (Figure S7). If λ1 is positive then p70S6K is induced and pERK is repressed and vice versa. These two proteins are known to be involved in the process of CIC through the MAPK and p70S6K pathway.[36, 37] Protein p70S6k plays a special role in the analysis since it has the highest weight in the process (Figure S7). Therefore this process, which dominates the transition, is most clearly seen in the data for protein p70s6k (Figure 3b). Nevertheless the time trend, as shown by , is the same for all proteins (Text S12).
The constraint is important during days 8–15, and is largely described by an anti-correlation of pGSK with pCHK2 and, to a lesser extent, an anti-correlation between pGSK and Cox2. The constraint has measurable weight only at day 21, which is evidently the least stable point of the transition (Figure 3(b)). This constraint exhibits anti-correlations between the pair of proteins pAkt and pERK and the apoptotic proteins, pCHK2, BCL2 and p53 (Figure S7).
The above discussion implies one or more critical point transitions between days 8 and 28. If that transition is of first order, then changes in at least one or more extensive variables (i.e. energy) will not significantly influence the conjugated intensive variables (i.e. temperature).[38] In these experiments, we are particularly sensitive to the relationships between the extensive variables of protein copy numbers and the intensive variables of protein chemical potentials.[10, 39] This is because we directly measure protein copy numbers and protein-protein correlations. This allows us to estimate how changing the levels of one protein will influence other proteins through the relationship . Here, the components of the vectors and represent the changes in average protein copy numbers and protein chemical potentials, respectively, between any two time points, while and Σ is the measured protein-protein covariance matrix. For a stable state, or a weak perturbation, this matrix relation can be used as a quantitative statement of the principle of Le Chatelier.[10]
We examined the covariance matrix Σ at every time point. If Σ has a significant increase in its eigenvalues in comparison with other time points, this indicates that the inverse matrix, , has one or more near zero eigenvalues. The appearance of the near zero eigenvalues indicates that changes in protein copy numbers do not lead to changes in the chemical potential of those proteins, thus implying the loss of one or more degrees of freedom, as expected for a phase transition (the Gibbs Phase rule).[38] If one or more of the chemical potentials do not change, the implication is that no work is required to move those proteins from one phase to another.
We identified an onset of the increase in the eigenvalues of Σ at day 8, reaching a maximum at day 15, and then followed by a steep drop (Figure 3(d)), implying that a phase transition occurs at days 12–15. However, the overall transition between the two steady states at the endpoints of the study appears to sample multiple intermediate states, and in interesting ways. The steep drop in the eigenvalues at day 21 implies a transition at that point is the inverse of the ‘normal’ transition between days 12–15 (i.e. the eigenvalues of Σ approach 0 at 21). This still can be viewed as a singular point that is, in fact, similar to a hypoxia-induced transition previously reported within an mTOR signaling network for certain brain cancer tumor models.[39] A second sharp increase in the eigenvalues day 28 point to the occurrence of a second loss of degree of freedom. A key result is that the cellular transition, which is detected as early as days 12–15 following start of treatment, precedes the phenotypic change, (Figure 1 (c)–(g)), which is observed after day 40.
In this work, a microfluidic platform for single cell proteomic analysis, involving a panel of functional proteins relevant for carcinogenesis process, was used to investigate the existence of critical phase transitions in an epithelial system undergoing chemical induced carcinogenesis (CiC). Statistical fluctuations and protein-protein correlation matrices indicate the existence of tipping point bifurcation dynamics in the system undergoing CiC. A more detailed, quantitative Le Chatelier’s principle framework of Surprisal analysis, based on protein covariance matrices, was used to connect the measured fluctuations of proteins from single cells to a thermodynamics based approach for identifying the classical hallmarks of phase transition: phase coexistence, loss of degrees of freedom. This is conceptually similar to Ising-type model, where the CiC process may be viewed as a phase transition between two stable steady (Figure 3(b)). The transition, while broad, appears as almost a textbook example of a 1st order phase transition, with hallmarks that include, in the vicinity of the critical point, divergent correlations, phase coexistence, and loss of degrees of freedom.
A practical observation is that the actual CiC transition (around day 15), significantly precedes the actual emergence of the traditional precancerous phenotype, which appears between day 40 and day 96. In fact, statistical indicators anticipate the onset of a critical point transition as early as day 8 following start of treatment. There are a number of other cellular transitions associated with carcinogenesis, including the transition to an invasive and/or a metastatic phenotype, or the transition to a drug-resistant phenotype. Whether the approaches here can be generally applied towards these other transitions and, in particular, whether they can be used to anticipate those transitions, is an open question that we are actively pursuing.
Supplementary Material
Table 1.
Description of the Surpisal Analysis (Equation 1)
| Parameter | Description | |
|---|---|---|
| α | Labels the constraint and the associated unbalanced biological process | |
|
|
Weight/amplitude of the constraint in a particular cell at a time point t. | |
|
|
The extent of influence of the constraint on every measured protein i. | |
|
|
Deviations of the protein levels from the steady state levels due to the constraints Repeated for every protein i in every cell at the time point t | |
|
|
Expression levels of a protein i in every cell at the steady state at a time point t. These values are used to build histograms representing protein distributions at the steady state. |
Acknowledgments
We are grateful to Rudi Balling, Alexander Skupin and Yaron Antebi for their detailed and insightful discussion and comments on the contents of the manuscript. This work is funded by the National Cancer Institute through grant 5U54 CA119347, the Jean Perkins Foundation, the Ben and Catherine Ivy Foundation, and an Intermobility grant Sinc-Prot; 2013 from Fonds National de la Recherche Luxembourg, Grand Duchy of Luxembourg.
Footnotes
Contributions
S.K.P. and J.R.H. conceived and designed the project. S.K.P. planned and carried out the research. N.K.B. and R.D.L. developed the theory. N.K.B. performed the theoretical analysis with inputs from other authors. Y.S.S. contributed to reagent/analysis tools. S.K.P., N.K.B., R.D.L. and J.R.H., were involved in discussion of results and in preparing the manuscript.
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Suresh Kumar Poovathingal, Division of Chemistry and Chemical Engineering, California Institute of Technology, MC 127-72, 1200 E. California Blvd, Pasadena, CA 91125, USA; Luxembourg Centre for Systems Biomedicine, University of Luxembourg, Esch-sur-Alzette, L-4362, Luxembourg.
Nataly Kravchenko-Balasha, Division of Chemistry and Chemical Engineering, California Institute of Technology, MC 127-72, 1200 E. California Blvd, Pasadena, CA 91125, USA.
Young Shik Shin, Division of Chemistry and Chemical Engineering, California Institute of Technology, MC 127-72, 1200 E. California Blvd, Pasadena, CA 91125, USA.
R. D. Levine, Institute of Chemistry, The Hebrew University of Jerusalem, Jerusalem 91904, Israel Department of Molecular and Medical Pharmacology, David Geffen School of Medicine, University of California, Los Angeles, CA 90095.
James R. Heath, Division of Chemistry and Chemical Engineering, California Institute of Technology, MC 127-72, 1200 E. California Blvd, Pasadena, CA 91125, USA
References
- 1.Huang S. Bioessays. 2012;34(2):149–57. doi: 10.1002/bies.201100031. [DOI] [PubMed] [Google Scholar]
- 2.Slack JM. Nat Rev Genet. 2002;3(11):889–95. doi: 10.1038/nrg933. [DOI] [PubMed] [Google Scholar]
- 3.Waddington CH. The strategy of the genes ; a discussion of some aspects of theoretical biology. London. 1957:ix, 262. [Google Scholar]
- 4.Wang J, Zhang K, Xu L, Wang E. Proc Natl Acad Sci U S A. 2011;108(20):8257–62. doi: 10.1073/pnas.1017017108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang J, Xu L, Wang E. Proc Natl Acad Sci U S A. 2008;105(34):12271–6. doi: 10.1073/pnas.0800579105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krotov D, Dubuis JO, Gregor T, Bialek W. Proc Natl Acad Sci U S A. 2014;111(10):3683–8. doi: 10.1073/pnas.1324186111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gillespie DT. J Phys Chem. 1977;81(25):2340–2361. doi: 10.1021/j100540a008. [DOI] [Google Scholar]
- 8.Zhang B, Wolynes PG. Proc Natl Acad Sci U S A. 2014;111(28):10185–90. doi: 10.1073/pnas.1408561111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi Q, Qin L, Wei W, Geng F, Fan R, Shin YS, Guo D, Hood L, Mischel PS, Heath JR. Proc Natl Acad Sci U S A. 2012;109(2):419–24. doi: 10.1073/pnas.1110865109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shin YS, Remacle F, Fan R, Hwang K, Wei W, Ahmad H, Levine RD, Heath JR. Biophys J. 2011;100(10):2378–86. doi: 10.1016/j.bpj.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ma C, Fan R, Ahmad H, Shi Q, Comin-Anduix B, Chodon T, Koya RC, Liu CC, Kwong GA, Radu CG, Ribas A, Heath JR. Nat Med. 2011;17(6):738–43. doi: 10.1038/nm.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu J, Zhou J, Sutherland A, Wei W, Shin YS, Xue M, Heath JR. Annu Rev Anal Chem (Palo Alto Calif) 2014;7:275–95. doi: 10.1146/annurev-anchem-071213-020323. [DOI] [PubMed] [Google Scholar]
- 13.Levine RD, Bernstei Rb. Accounts Chem Res. 1974;7(12):393–400. doi: 10.1021/Ar50084a001. [DOI] [Google Scholar]
- 14.Levine RD, Tribus M. The maximum entropy formalism : a conference held at the Massachusetts Institute of Technology on May 2–4, 1978. MIT Press; Cambridge, Mass: 1979. p. xii.p. 498. [Google Scholar]
- 15.Remacle F, Kravchenko-Balasha N, Levitzki A, Levine RD. Proc Natl Acad Sci U S A. 2010;107(22):10324–9. doi: 10.1073/pnas.1005283107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kravchenko-Balasha N, Levitzki A, Goldstein A, Rotter V, Gross A, Remacle F, Levine RD. Proc Natl Acad Sci U S A. 2012;109(12):4702–7. doi: 10.1073/pnas.1200790109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kravchenko-Balasha N, Remacle F, Gross A, Rotter V, Levitzki A, Levine RD. BMC Syst Biol. 2011;5:42. doi: 10.1186/1752-0509-5-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kravchenko-Balasha N, Wang J, Remacle F, Levine RD, Heath JR. Proc Natl Acad Sci U S A. 2014;111(17):6521–6. doi: 10.1073/pnas.1404462111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walling MA, Shepard JR. Chem Soc Rev. 2011;40(7):4049–76. doi: 10.1039/c0cs00212g. [DOI] [PubMed] [Google Scholar]
- 20.Calaf G, Russo J. Carcinogenesis. 1993;14(3):483–92. doi: 10.1093/carcin/14.3.483. [DOI] [PubMed] [Google Scholar]
- 21.Luch A. Nat Rev Cancer. 2005;5(2):113–25. doi: 10.1038/nrc1546. [DOI] [PubMed] [Google Scholar]
- 22.Russo J, Calaf G, Russo IH. Crit Rev Oncog. 1993;4(4):403–17. [PubMed] [Google Scholar]
- 23.Bocca C, Ievolella M, Autelli R, Motta M, Mosso L, Torchio B, Bozzo F, Cannito S, Paternostro C, Colombatto S, Parola M, Miglietta A. Expert Opin Ther Targets. 2014;18(2):121–35. doi: 10.1517/14728222.2014.860447. [DOI] [PubMed] [Google Scholar]
- 24.Choo AY, Blenis J. Cancer Cell. 2006;9(2):77–9. doi: 10.1016/j.ccr.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 25.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Nature. 1995;378(6559):785–9. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 26.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Cell. 1997;91(2):231–41. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 27.Denissenko MF, Pao A, Tang M, Pfeifer GP. Science. 1996;274(5286):430–2. doi: 10.1126/science.274.5286.430. [DOI] [PubMed] [Google Scholar]
- 28.Gotoh J, Obata M, Yoshie M, Kasai S, Ogawa K. Carcinogenesis. 2003;24(3):435–42. doi: 10.1093/carcin/24.3.435. [DOI] [PubMed] [Google Scholar]
- 29.Hodgkinson CP, Sale EM, Sale GJ. Biochemistry. 2002;41(32):10351–9. doi: 10.1021/bi026065r. [DOI] [PubMed] [Google Scholar]
- 30.Shaw RJ, Cantley LC. Nature. 2006;441(7092):424–30. doi: 10.1038/nature04869. [DOI] [PubMed] [Google Scholar]
- 31.Sheppard K, Kinross KM, Solomon B, Pearson RB, Phillips WA. Crit Rev Oncog. 2012;17(1):69–95. doi: 10.1615/critrevoncog.v17.i1.60. [DOI] [PubMed] [Google Scholar]
- 32.Wyllie AH. Curr Opin Genet Dev. 1995;5(1):97–104. doi: 10.1016/s0959-437x(95)90060-8. [DOI] [PubMed] [Google Scholar]
- 33.Ye F, Xu XC. Mol Cancer. 2010;9:93. doi: 10.1186/1476-4598-9-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yook JI, Li XY, Ota I, Hu C, Kim HS, Kim NH, Cha SY, Ryu JK, Choi YJ, Kim J, Fearon ER, Weiss SJ. Nat Cell Biol. 2006;8(12):1398–406. doi: 10.1038/ncb1508. [DOI] [PubMed] [Google Scholar]
- 35.Navin NE. Genome Biol. 2014;15(8):452. doi: 10.1186/s13059-014-0452-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ding J, Ning B, Gong W, Wen W, Wu K, Liang J, He G, Huang S, Sun W, Han T, Huang L, Cao G, Wu M, Xie W, Wang H. J Biol Chem. 2009;284(48):33311–9. doi: 10.1074/jbc.M109.046417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shi Y, Hsu JH, Hu L, Gera J, Lichtenstein A. J Biol Chem. 2002;277(18):15712–20. doi: 10.1074/jbc.M200043200. [DOI] [PubMed] [Google Scholar]
- 38.Yeomans JM. Statistical mechanics of phase transitions. Clarendon Press; Oxford University Press; Oxford England New York: 1992. p. x.p. 153. [Google Scholar]
- 39.Wei W, Shi Q, Remacle F, Qin L, Shackelford DB, Shin YS, Mischel PS, Levine RD, Heath JR. Proc Natl Acad Sci U S A. 2013;110(15):E1352–60. doi: 10.1073/pnas.1303060110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.