

ABSTRACT
The genome of an invasive skin-tropic strain (AP53) of serotype M53 group A Streptococcus pyogenes (GAS) is composed of a circular chromosome of 1,860,554 bp and carries genetic markers for infection at skin locales, viz., emm gene family pattern D and FCT type 3. Through genome-scale comparisons of AP53 with other GAS genomes, we identified 596 candidate single-nucleotide polymorphisms (SNPs) that reveal a potential genetic basis for skin tropism. The genome of AP53 differed by ∼30 point mutations from a noninvasive pattern D serotype M53 strain (Alab49), 4 of which are located in virulence genes. One pseudogene, yielding an inactive sensor kinase (CovS−) of the two-component transcriptional regulator CovRS, a major determinant for invasiveness, severely attenuated the expression of the secreted cysteine protease SpeB and enhanced the expression of the hyaluronic acid capsule compared to the isogenic noninvasive AP53/CovS+ strain. The collagen-binding protein transcript sclB differed in the number of 5′-pentanucleotide repeats in the signal peptides of AP53 and Alab49 (9 versus 15), translating into different lengths of their signal peptides, which nonetheless maintained a full-length translatable coding frame. Furthermore, GAS strain AP53 acquired two phages that are absent in Alab49. One such phage (ΦAP53.2) contains the known virulence factor superantigen exotoxin gene tandem speK-slaA. Overall, we conclude that this bacterium has evolved in multiple ways, including mutational variations of regulatory genes, short-tandem-repeat polymorphisms, large-scale genomic alterations, and acquisition of phages, all of which may be involved in shaping the adaptation of GAS in specific infectious environments and contribute to its enhanced virulence.
IMPORTANCE Infectious strains of S. pyogenes (GAS) are classified by their serotypes, relating to the surface M protein, the emm-like subfamily pattern, and their tropicity toward the nasopharynx and/or skin. It is generally agreed that M proteins from pattern D strains, which also directly bind human host plasminogen, are skin tropic. We have sequenced and characterized the genome of an invasive pattern D GAS strain (AP53) in comparison to a very similar strain (Alab49) that is noninvasive and developed a genomic rationale as to possible reasons for the skin tropicity of these two strains and the greater invasiveness of AP53.
INTRODUCTION
Group A Streptococcus pyogenes (GAS) is a beta-hemolytic, Gram-positive, human-pathogenic bacterium that infects multiple epithelial surfaces and, in some cases, is able to invade deeper soft tissue and cause severe disease (1). An estimated 700 million cases of GAS infection occur worldwide per year (2), ranging from benign treatable clinical conditions, e.g., pharyngitis and impetigo, to more invasive and potentially lethal infections, including necrotizing fasciitis (NF), streptococcal toxic shock syndrome (STSS), acute rheumatic fever (ARF), and acute glomerulonephritis (AGN) (3, 4). The wide range of infectious niches and broad variance in the severity of GAS are largely associated with the diverse genetic repertoire of this organism, including numerous virulence factors, complex regulatory systems, and extensive recombination mechanisms, which assist the bacteria in circumventing the host innate immune response in a highly strain- or disease-specific manner (5).
The characterization of the genetic properties of different GAS strains has been employed to gain insights into their pathophysiological relationships. M protein, a surface protein encoded by the emm gene that is present in all GAS strains, is a known essential virulence determinant. This gene has also been used for serotyping of GAS strains and has distinguished >250 subtypes of GAS. In addition to the emm gene product, M protein, other M-like cell surface gene products, e.g., IgG Fc receptor FcR, IgA Fc receptor Enn, and fibronectin-binding protein Fba, are responsible for specific cell surface binding of host proteins, e.g., IgG, IgA, C4-binding protein, factor H, and fibronectin. The genes encoding these M-like proteins are present in the same regulon as the emm gene, under the transcriptional control of a multigene activator protein (Mga) (6). Based on the presence or absence of these emm-like cell surface genes, their chromosomal arrangements, and their 3′ sequence similarities, GAS strains have been further categorized into different emm patterns, viz., patterns A to E (7). In addition, previous population surveys showed that emm patterns are strongly associated with the tissue tropicity of GAS subtypes. Pattern A to C strains are more closely related to pharyngitis, pattern D strains are mainly agents for skin infections, and pattern E strains are equally adapted to both throat and skin environments (8, 9).
Another genomic locus encoding multiple surface proteins, i.e., the FCT region, contains fibronectin (F)- and collagen (C)-binding proteins as well as T antigens (T) involved in pilus assembly. This FCT region plays an important role in GAS pathogenesis by mediating the adherence to, and colonization of, human tissues (10, 11). The composition of the FCT region is highly variable and has been classified into at least nine subtypes, viz., FCT1 to -9, based on gene arrangements and sequence divergence of genes within this region (11, 12). The variability of the FCT region has been linked to the diversity of tissue tropism in GAS infection, although direct associations remain to be elucidated (13).
During the past decade, the full genomes of >40 GAS strains that span a wide range of serotypes have been derived by using high-throughput sequencing technologies. This complete information has allowed comparisons of GAS strain-specific genomic properties at the single-gene level and even the single-nucleotide level, along with a broader understanding from a genomic viewpoint of the mechanisms of GAS infection as well as its evolutionary adaptation capacity in its epidemiological history.
In this work, we present a genomic analysis of a skin-tropic serotype M53 GAS isolate, AP53, and analyzed the data in comparison with those for another serotype M53 strain, Alab49, which was sequenced previously. The serotype M53 GAS strains are considered to be the most common types that cause superficial skin diseases (14). However, in contrast to Alab49, the AP53 strain is highly pathogenic. The comparative genome study of AP53 and Alab49 presented here revealed specific genomic distinctions that are likely key in mediating host environment tropism. The results obtained shed light on evolutionary adaptations that pattern D strains employ to cause mild or lethal infections.
MATERIALS AND METHODS
Bacterial strain and isolation of genomic DNA.
GAS strain AP53 was provided by G. Lindahl (Lund, Sweden), and its genomic DNA was isolated as previously described (15).
Genome sequencing and gene annotation.
The genome of AP53 was sequenced by using Illumina MiSeq (Illumina, San Diego, CA), with paired-end reads of 250 bp. A draft genome assembly was performed by using Velvet (16), followed by gap closure using PCR primer walking. The protein-coding sequences were predicted by using Glimmer3.02b (17), the rRNA sequences were predicted by using RNAmmer (18), and the tRNA sequences were detected by tRNAScan-SE (18). Genome annotation was performed by using the automated RAST annotation server (19) and subsequently curated by manual inspection.
Comparative analysis of GAS genomes.
Genome sequences of other fully sequenced GAS strains were downloaded from the NCBI database (20) for comparative analysis. The genome sequence map of S. pyogenes AP53, in comparison to the available whole-genome sequences, was generated by using BRIG (21). The local sequence comparisons of these genomes were viewed and examined by using the Artemis Comparison Tool (ACT) (22).
Polymorphism analyses.
The fragmented genomes were aligned against a modified genome of AP53 by excluding repetitive regions, phage regions, and mobile elements. Thus, a “core” genome of 1,662,906 bp was created for further single-nucleotide polymorphism (SNP) discoveries. The polymorphisms were detected by using the Variant Ascertainment Algorithm (23) and annotated with customized scripts. Regions of high-density SNPs likely reflected horizontal genetic transfer and therefore were excluded. The SNPs falling within the genes carried by only some of the GAS strains were not considered. A matrix for the alleles of all GAS strains at each polymorphic locus was generated, and the alleles at all loci for each GAS genome were concatenated for further distance estimation and phylogeny inference by using MEGA (24) and SplitsTree (25). The pairwise distance of strains was estimated based on the model of maximum composite likelihood, and the clustering relationships were inferred by using the maximum likelihood method with a bootstrap value of 1,000.
Quantitative real-time PCR (qRT-PCR).
Bacteria were cultured overnight at 37°C in the Todd-Hewitt broth (BD Biosciences, San Jose, CA) supplemented with 1% yeast extract (THY). The cells were collected from a single colony grown to mid-log phase (A600 = 0.5 to 0.6) and stationary phase (A600 = 0.9 to 1.0) and digested with mutanolysin in 300 μl of spheroplasting buffer (20 mM Tris-HCl, 10 nM MgCl2, 56% raffinose [pH 6.8]). Total RNA was extracted by using the RNeasy minikit (Qiagen, Valencia, CA). RNA was treated twice with DNase from the DNA-free kit. An amount of 250 ng extracted RNAs was reverse transcribed to cDNA by using an iScript cDNA synthesis kit (Bio-Rad Laboratories, Inc.) in a 20-μl reaction mixture. Each reaction was repeated three times from independent RNA extractions. Quantitative PCRs (qPCRs) were performed with 12.5 μl of 2× SYBR green PCR master mix (Applied Biosystems) and 100 nM forward and reverse primers (see Table S1 in the supplemental material). The plr (gapdh) gene was used as a control.
Mouse survival assays.
The humanized plasminogen transgenic (hPgTg) helicobacter-free mouse model fully backcrossed into the C57BL/6 strain was employed for survival studies (26). The mice were anesthetized with isoflurane and subcutaneously injected with 1.7 × 108 to 2.9 × 108 CFU of GAS/mouse. Mice were monitored for 10 days for survival status, and the survival differences were evaluated by using the log rank test.
All animal experiments were performed under the approval of the University of Notre Dame (Notre Dame, IN) Institutional Animal Care and Use Committee.
Nucleotide sequence accession number.
The complete genomic sequence and annotation information have been deposited in the NCBI GenBank database with accession number CP013672.
RESULTS
Genomic properties of GAS strain AP53.
The genome of S. pyogenes AP53 consists of a circular chromosome of 1,860,554 bp, with an average G+C content of 38.6%. The genome harbors 1,841 predicted open reading frames (ORFs), six rRNA operons, 73 tRNAs, and 15 pseudogenes. While no clustered regulatory interspaced short palindromic repeat (CRISPR) sequences were detected, five prophage regions were found in the genome of AP53, with lengths of ∼14.9 kb to ∼41.6 kb. The prophages account for 8.8% of the total AP53 genome. Compared with other known GAS genomes with diverse M serotypes, all of the genes from AP53 essentially have orthologs in at least one other GAS genome with >90% amino acid homology (Fig. 1). As illustrated in Fig. 1, the sequences are conserved in GAS genomes and are mosaic in the prophage regions. Short mobile element stretches (or transposons) are present, reinforcing the viewpoint that phages and transposons are the major sources of genetic diversifying factors among GAS genomes.
FIG 1.

Circular representation of the AP53 genome in comparison with other known GAS genomes. The genomic sequences are generally conserved across the GAS genomes, with mosaicism being concentrated in regions encoding prophages, transposons, and several hypothetical proteins (black bars, red arrowheads, and blue arrowheads, respectively, in the outer circle). The white areas represent high-level divergence of the genomes. The emm-like gene locus and FCT locus with mosaicism are also indicated. The circle representing the comparison between AP53 and Alab49 is highlighted with a black arrow.
Genotypic markers of AP53 for infection preference for skin tissue sites.
A previous population-based survey revealed that GAS pattern D strains are more likely to cause infections in the skin than in the throat, and the pattern D strains exhibited a linkage with the genes in the FCT locus (27). Therefore, the emm and FCT loci are considered to be important genotypic markers for tissue-specific infections. This observation prompted us to examine the presence of these genetic markers in strain AP53 isolated from skin. It is readily seen that its emm-like gene locus conforms to the pattern D subclass, with a gene arrangement of mga-fcR-pam-enn-scpA-fbaA-lmb-htpA (15), in comparison with patterns A to C and pattern E for other fully sequenced GAS genomes (9, 12) (Fig. 2A). It has been shown that the pattern D isolates are generally found in skin and also possess the FCT type 3 locus, with the gene organization nra-cpa-fctA-srtB-fctB-msmR-prtF2 (11) (Fig. 2B). However, the full correspondence between emm pattern A-B-C-E and FCT types is not fully clear, thus signifying possible genetic recombinations during GAS evolution (28). The FCT locus encodes five distinct cell wall-anchored surface proteins, viz., collagen-binding protein (cpa), fibronectin-binding proteins (prtF1 and prtF2), and pilus structural proteins (fctA and fctB). However, gene compositions and sequence similarities between different types are highly variable, and the individual types are assigned to multiple emm subfamily patterns. Overall, the complexity of the FCT regions suggests that this region has evolved to become highly specific and adapted to the process of host adherence to diverse infectious factors, such as tissue specificity, immune modulation, and human infection niches. The divergence in gene contents and organizations of the emm-like and FCT gene loci was also observed in the whole-genome comparison (Fig. 1).
FIG 2.

Gene organizations for different emm patterns and FCT types in known GAS genomes. (A) The gene members for emm patterns A to E are depicted, and the M serotypes are grouped for each pattern. All pattern D strains are serum opacity factor negative (SOF−) and do not carry sof and its associated alternative fibronectin-binding protein-encoding gene sfbX. The strains of serotype M12 contain the sequences of sof and sfbX but encode a defective serum opacity factor. The corresponding FCT types for each M serotype are labeled ❶ to ❻. (B) Gene members for six FCT types for all known GAS genomes along with their M serotypes. The corresponding emm patterns for each M serotype are indicated in blue for patterns A to C, red for pattern D, and purple for pattern E. Both AP53 and Alab49 have pattern D and FCT type 3. Note that serotype M59 strains are classified as having emm pattern E in our study, different from the emm pattern D classification reported previously by McGregor et al. (8). We obtained the same classification as that reported previously by Fittipaldi et al. (51).
SNPs as genetic markers of skin tropism.
SNPs have been frequently used in many species as high-resolution markers for genetic studies related to adaptive evolution or disease genotyping. In this work, we conducted single-nucleotide variation detection for all known GAS genomes in order to identify candidate SNP markers for tissue-associated infection. Collectively, 71,558 distinct SNP loci were identified for 44 GAS genomes, with altered alleles in at least one of the genomes. The majority of the loci (82%) fall into coding regions. A total of 21,579 loci (30%) involve nonsynonymous substitutions causing amino acid changes.
The allelic distance between strains of the same emm type was limited to several hundred unique substitutions, corresponding to a genetic distance of 0.04, much lower than that observed for strains of different emm types, where up to 17,000 substitutions, corresponding to a genetic distance of 0.34, were found to occur. This reinforces the usefulness of emm genes as a basic genotypic marker for GAS disease phenotypes. The distance between strains of different emm types varies in a broad range, from 9,400 to 17,000, corresponding to a genetic distance of 0.15 to 0.34 (Fig. 3A). Surprisingly, the skin infection-associated strains, including AP53, M53 Alab49, M101 NGAS638, serotype M83 strains, serotype M3 strains, ATCC 19615, and M14 HSC5, are closely related to each other by genetic distance and exclusively clustered together (Fig. 3A; see also Fig. S1A in the supplemental material). The distance pattern based on the SNP loci with nonsynonymous changes does not differ from that based on all SNP loci (data not shown). These strains are presumed to be skin tropic due to the presence of emm pattern D in the genomes, except for the serotype M14 and M3 strains, which were also found to be capable of causing skin diseases in animal models. The clustering of the strains capable of causing skin infection is in contrast to the clustering pattern inferred from the allelic changes in the seven housekeeping genes, where the skin infection-associated strains grouped in Fig. 3A are subdivided into distinct clusters (8) (Fig. 3B; see also Fig. S1B in the supplemental material). The flattened pattern of the genetic distance of the housekeeping genes (Fig. 3B) is consistent with natural selection acting on the housekeeping genes (29, 30). Therefore, we hypothesize that the clustering of the skin infection-associated strains is attributed to SNP loci shared by these strains, which may serve as a determinant for the skin specificity of GAS infections.
FIG 3.

GAS strain clustering based on pairwise distances of all GAS genomes measured by base substitutions per site. (A) The substitution rate was determined for the core genome of GAS, containing 71,558 SNP loci. The skin-tropic strains, i.e., AP53, M53 Alab49, M101 NGAS638, M83 strains, M3 strains, ATCC 19615, and M14 HSC5, are clustered together and highlighted in blue. (B) The substitution rates for seven housekeeping genes, viz., gki, gtr, murI, mutS, recP, xpt, and yqiL, were determined, generating 434 SNP loci. The strains clustered in panel A are subdivided into distinct clusters in panel B. The pairwise distance was estimated based on the model of maximum composite likelihood. The clustering relationship was inferred by using the maximum likelihood method with a bootstrap value of 1,000.
We next identified the SNP loci shared within the skin infection-associated strains and the corresponding functional genes. To accomplish this, we used hierarchical clustering to partition the nonsynonymous SNP loci to different groups based on the presence or absence of the alleles in the GAS genomes. We isolated four clusters of interest, totaling 595 SNP loci (see Fig. S2 and Table S1 in the supplemental material). The 595 SNPs in the four clusters were present in at least two distinct emm types of strains capable of causing skin infection (see Fig. S2 in the supplemental material) and were highly represented in genes for four primary functional categories, (i) membrane transport; (ii) cell wall and capsule; (iii) amino acid synthesis; and (iv) cofactors, vitamins, and prosthetic groups, in comparison to the functional representation among the complete core GAS genome locations (P value of <10−5, as determined by a hypergeometric test) (Table 1). For example, the genes with the most frequent mutations include proA for proline synthesis, murF for cell wall peptidoglycan biosynthesis, and zntA for metal cation export. Surprisingly, the virulence-associated functions were not enriched in the SNPs in strains with a preference for skin. The overrepresented functional categories manifest the potential niche-specific requirement for nutrient metabolism and protective activities during interaction with hosts. Therefore, we propose that the potential SNPs in strains with a preference for skin provide alternative genetic markers for the GAS preference for infection of skin. In contrast, the total nonsynonymous SNPs detected among all compared GAS genomes are enriched in distinct functional categories (Table 1). Specifically, virulence-associated functions were overrepresented, with a high level of significance for the total number of genes in all strains, highlighting the role of virulence genes as a reservoir for accumulating mutations during GAS evolution.
TABLE 1.
Functional classification of the 595 candidate skin-specific SNPs and enrichment of each functional category in the SNPs
| Functional category | Nonsynonymous SNPs in skin strainsa |
Total nonsynonymous SNPsb |
||
|---|---|---|---|---|
| No. of SNPs | P valuec | No. of SNPs | P valuec | |
| Membrane transport | 74 | 6.2E−10 | 1,295 | 0.0008 |
| Cell wall and capsule | 58 | 3.5E−07 | 955 | 0.8610 |
| Amino acids and derivatives | 43 | 2.4E−06 | 721 | 0.0823 |
| Cofactors, vitamins, and prosthetic groups | 39 | 8.4E−07 | 710 | 1.6E−09 |
| Carbohydrates | 53 | 0.8337 | 2,710 | 0.0000 |
| Protein metabolism | 53 | 0.6798 | 1,992 | 0.1159 |
| DNA metabolism | 38 | 0.9354 | 1,628 | 0.6507 |
| RNA metabolism | 28 | 0.6135 | 987 | 0.7967 |
| Regulation and signaling | 23 | 0.4128 | 828 | 0.0025 |
| Nucleosides and nucleotides | 23 | 0.5974 | 970 | 0.0000 |
| Virulence, disease, and defense | 30 | 0.5666 | 1,497 | 0.0000 |
| Fatty acids and lipids | 14 | 0.0665 | 585 | 0.0000 |
| Stress response | 12 | 0.1667 | 350 | 0.0032 |
| Cell division and cell cycle | 8 | 0.7924 | 336 | 0.8012 |
| Iron acquisition and metabolism | 7 | 0.6153 | 260 | 0.4677 |
| Respiration | 5 | 0.8627 | 219 | 0.9937 |
| Miscellaneous | 3 | 0.9285 | 215 | 0.1622 |
| Sulfur metabolism | 3 | 0.3166 | 88 | 0.0129 |
| Phosphorus metabolism | 1 | 0.9258 | 52 | 1.0000 |
| Potassium metabolism | 1 | 0.8496 | 83 | 0.0210 |
| Hypothetical protein | 36 | 0.0900 | 1,800 | 0.0000 |
| Other | 16 | 0.1526 | 760 | 0.0000 |
| Unclassified | 27 | 1.0000 | 1,649 | 1.0000 |
| Total | 595 | 20,690 | ||
Nonsynonymous SNPs detected in the clustered strains associated with skin infection.
Total detected nonsynonymous SNPs among all the compared GAS genomes.
P values were estimated by using the hypergeometric test by comparison with the functional distribution among the core GAS genome locations.
In summary, the SNP data show that (i) virulence genes have few SNPs among the skin-tropic strains, compared to some of the other functional categories, and (ii) genes with many SNPs are present in specific functional categories, such as nutrient acquisition or cell wall synthesis, and thus are revealed to be important determinants or genetic markers for skin tropism.
Genomic comparisons between GAS strains AP53 and Alab49.
With respect to major GAS genetic determinants, AP53 is highly similar to another serotype M53 isolate, i.e., Alab49. Both genomes share emm pattern D and FCT type 3, and the candidate skin-specific SNPs are present. However, in vivo studies using both GAS strains revealed that infection with GAS strain AP53 caused a more severe invasive outcome than did infection with the less virulent isolate Alab49 (Fig. 4). A comparison of two strains with different disease phenotypes is ideal for revealing the genetic changes underlying the emergence of virulent clones. A reciprocal BLAST comparison of the two genomes showed that the gene contents of the two strains are highly syntenic, and 1,716 (>92% of the total) of the ORFs are shared by the two strains, based on a minimum homology of 90% and a minimum gene coverage of 90%. The genes that differ between each genome are mainly those that encode phage proteins, transposons, and several hypothetical proteins. The high similarity between these two strains at the genomic level suggests that they are closely related phylogenetically. However, there are also key differences in the nonphage regions between the two genomes.
FIG 4.
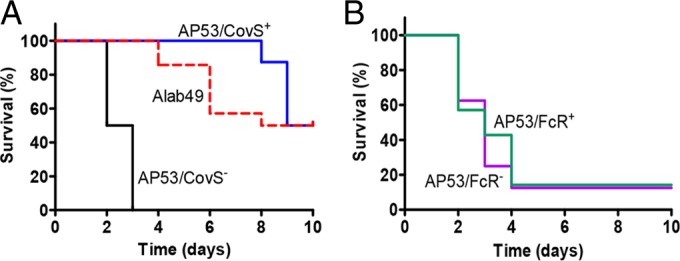
Mouse survival curves for GAS strains. (A) Survival curves for two isogenic strains of AP53 (AP53/CovS+ and AP53/CovS−) and another emm53 strain, Alab49. (B) Survival curves for two isogenic strains, AP53/FcR+ and AP53/FcR−. The mice used were fully backcrossed into the C57BL/6 strain. Mice were subcutaneously injected with 1.7 × 108 to 2.9 × 108 CFU/mouse of the relevant GAS strain and were monitored for 10 days. The differences between curves were evaluated by the log rank test. The differences in survival rates are significant in panel A, with a P value of <0.0008, and insignificant in panel B, with a P value of 0.786. The mice used contained the human plasminogen transgene (hPgTg) (n = 10 to 12 for each group).
First, two genes were interrupted, resulting in truncated proteins, by frameshift deletion or an early stop codon. The covS gene, which encodes the sensor kinase of a two-component regulatory system, covRS, was inactivated in AP53 by a deletion of T at nucleotide 1403, causing a frameshift and resulting in a truncated 467-amino-acid peptide (15). A survival study of a plasminogen-humanized mouse model showed that the invasiveness and virulence of AP53 were attenuated in an isogenic strain complemented with a fully translatable covS gene (AP53/CovS+) (Fig. 4A). The fcR gene, which encodes an IgG-binding protein located in the mga regulon, was interrupted in AP53 by an early stop codon that was a result of a single-nucleotide change, C253T, in the gene (15). However, the inactivated fcR gene in AP53 does not induce significant changes in virulence in comparison to an isogenic strain with a complemented fcR gene (AP53/FcR+) (Fig. 4B).
Furthermore, we deduced from nucleotide sequence analysis that a single-amino-acid substitution (I30T) occurred in the hyaluronic acid capsule synthesis protein HasA as well as in the transcriptional regulator protein RopB (K38Q), which regulates the transcription of speB and other virulence factors. These mutations were present only in AP53. Other naturally occurring mutations were also found to exist in 31 nonvirulence genes, involved mainly in core functions such as basic molecule biosynthesis, metabolism, and the stress response (see Table S2 in the supplemental material). By examining the alleles at the 31 loci in other GAS strains, we found that they are essentially divided into two groups, one that is unique to AP53 and another that is unique to Alab49 (see Table S2 in the supplemental material). They do not overlap the SNP clusters of potential skin preference genes (see Fig. S2 in the supplemental material) and are probably strain specific.
AP53 encodes a collagen-binding cell surface protein, SclB, with the only difference from that of Alab49 being the number of pentanucleotide repeat sequences (AACAA) that are immediately downstream of the start codon (GTG) in the amino terminus. The smaller number of repeats in AP53 produces a shortened polar region, viz., NKTKQ, in the signal peptides (Fig. 5A and B). Further examination of the nucleotide sequences of sclB across all GAS genomes showed high variability in the number of these repeats between strains of different serotypes and even between strains of the same serotypes. The variation in the number of repeats was proposed to serve as a switch to determine the full translation of the protein via the mechanism of slipped-strand mispairing (31–34) and is believed to contribute to pathogenic adherence to host cells. Interestingly, we observed that the GTG start sites of sclB genes from the strains with emm pattern D, including AP53 and Alab49, are all in frame with the ORFs and should be fully translated, perhaps with different efficiencies. Meanwhile, the translation of sclB from the strains with emm patterns A to C or pattern E is mosaic. This indicates that sclB could be related to skin adherence, and repeat-sequence-controlled translation may represent an alternative mechanism evolved by GAS to adapt to various infectious conditions.
FIG 5.

Domain architecture of the collagen-binding protein SclB from AP53 in comparison with SclB proteins from all known GAS genomes. (A) The surface protein SclB has an N-terminal signal peptide, a variable A domain, a GXY triple-helix-repeat region, and a conserved cell wall membrane region at the C terminus. (B) The translation of sclB mRNA is influenced by the number of pentanucleotide (AACAA) repeats immediately downstream of the GTG start codon in the N terminus. Improper numbers of repeats will induce premature translation termination, indicated by “X” framed with red in panel A. The full translation of the protein contains a signal peptide that is extended by stretches of polar residues, NKTKQ. (C) The surface protein SclB has a conserved C terminus, with the cell wall-anchoring motif LPATG (yellow) separating a Pro-rich region (gray) from a hydrophobic transmembrane region (blue), a positively charged tail (red), and an intracellular tripeptide. The sclB genes from strains AP53 and Alab49 are distinguished only by the number of AACAA repeats in the N terminus.
The protein domain structure of SclB also suggests that the bacterial strain expressing this protein has a skin affinity. SclB contains a hypervariable A domain, followed by triple contiguous Gly-X-Y repeats, characteristic of human collagen. Thus, SclB may allow the binding of GAS strains that express this protein to human collagen receptors (33). There is a total of 93 such repeats in SclB from both AP53 and Alab49. The numbers of repeats of this domain appear to be strain specific, with AP53 and Alab49 being among the strains with the highest numbers of repeats found in SclB proteins (Fig. 5A). Finally, SclB contains an LPATG cell wall-anchoring motif near the carboxy terminus, suggesting that it is covalently bound to the cell wall through a sortase-dependent mechanism (Fig. 5C). Overall, SclB manifests multifaceted adaptation capabilities in the recognition of and interaction with host proteins and has important implications in antigenic potential.
Genomic variations in phage elements and virulence implications for AP53.
In addition to the nucleotide-level differences between AP53 and Alab49, AP53 is strongly differentiated from Alab49 and other GAS strains by the number and nature of its phage elements. We identified the phages in AP53 by locating the integration sites and compared them with those from Alab49 by BLAST searches. Five prophages were detected in AP53, designated ΦAP53.1 to ΦAP53.5, with lengths ranging from 14.8 to 41.6 kb (Fig. 6A). Two novel phages (ΦAP53.1 and ΦAP53.2) were present in AP53, and one phage that was present in Alab49 was absent (ΦAlab49.1). With the acquisition of phage ΦAP53.2, AP53 also obtained the pyrogenic exotoxin gene pair speK-slaA, which are adjacent genes in ΦAP53.2 but transcribed independently (35), and lost the superantigens speL-speM due to the absence of ΦAlab49.1 in AP53. A qRT-PCR assay showed that speK and slaA were significantly upregulated, by 2.3-fold and 2.4-fold, respectively, in the naturally mutated strain (AP53/CovS−) at the stationary growth phase, in comparison with its nonvirulent isogenic counterpart (AP53/CovS+) (Fig. 6B). In contrast, the virulence genes speC-spd1, carried by ΦAP53.3, were downregulated in the AP53/CovS− strain, whereas the expression of spd3, carried by ΦAP53.4, was not significantly altered (Fig. 6B). The exclusive upregulation of speK-slaA in AP53 suggests that speK-slaA may contribute to the lethality of the AP53/CovS− strain and that the acquisition of ΦAP53.2 may have provided a mechanism to increase the overall survival fitness and pathogenicity of GAS strain AP53.
FIG 6.

Unique genomic content of AP53 in comparison with that of Alab49. (A) Whole-genome comparison of AP53 and Alab49 reveals the acquisition of two novel phages in AP53, viz., ΦAP53.1 and ΦAP53.2, and the loss of one phage, ΦAlab49.1. Otherwise, the gene contents of the two genomes are highly syntenic. The blue and red lines represent forward and reverse alignments, and the purple boxes depict the locations of phage elements, with the virulence genes carried by each phage indicated at the bottom. (B) Altered expression of phage virulence genes at different growth stages (mid-log phase [LP] and stationary phase [SP]) in the invasive AP53/CovS− isolate compared to an isogenic noninvasive strain complemented with the covS gene, AP53/CovS+. speK-slaA from the novel phage ΦAP53.2 exhibited increased expression in the invasive isolates, while speC-spd1 from ΦAP53.3 showed the opposite trend. The expression of spd3 from ΦAP53.4 was not significantly changed. The expression profile was determined by qRT-PCR, and the expression levels are expressed as ratios relative to that for the AP53/CovS− strain at mid-log phase.
A novel prophage with missing virulence genes may confer survival advantages.
Phages ΦAP53.1 and ΦAP53.5 do not carry any virulence genes. Therefore, we identified the gene functions of the prophages from AP53 in order to identify alterations of gene contents in phages that may influence the virulence capacity of their lysogenic host strains. By comparison with data in protein databases using BLASTP, we were able to assign modularized gene functions to all five phages. As illustrated in Fig. 7, the phages from AP53 exhibited gene organizations typical of tailed temperate phages that function in lysogeny control, DNA replication, regulation, DNA packaging, head morphogenesis, head-tail joining, tail morphogenesis, host cell lysis, and phage-related virulence. Specifically, phage ΦAP53.5 contains only two modules, viz., lysogeny control and DNA replication, with a total length of 14.8 kb. This phage was reported to be a remnant widely present in GAS strains (36) and suffered large DNA deletions via interactions with lysogenic hosts during evolution. This phage remnant does not appear to be capable of contributing to the pathogenesis of AP53. However, ΦAP53.1 presents a full prophage, with all the functional modules except for the virulence cassette. The genetic profile of ΦAP53.1, the presence of the full-length phage modules, and the absence of virulence genes are in contrast to the concept of prophage-lysogen interactions, where the acquisition of phages enhances the fitness of the bacteria by providing the lysogens with additional virulence determinants while at the same time not substantially increasing the genomic burden. A possible explanation is that ΦAP53.1 was more harmful than beneficial to this strain, and selective recombination events occurred following the acquisition of the phage by deletion of the toxin genes. This idea is supported by comparisons showing that the gene sequences among the homologs of ΦAP53.1 are highly mosaic, and the homology terminates abruptly in the regions of host lysis and virulence (see Fig. S3A in the supplemental material). Furthermore, a holin gene in the host lysis module was interrupted by an early stop codon at amino acid 91. The overall genomic properties of the novel phage ΦAP53.1 demonstrate evidence for an ongoing loss of lytic functions from phages, conferring to the bacterium potential survival advantages as a whole.
FIG 7.

Gene maps and modularized functions of the five prophages present in the genome of AP53. Genes with known functions are indicated by directional orange arrows, and putative genes are indicated by directional gray arrows. The clustered genes with modular functions are grouped with brackets and color-coded. Phage ΦAP53.5 is a remnant with only two modules, viz., lysogeny control and DNA replication, and ΦAP53.1 represents a full prophage but lost the virulence cassette at the right terminus. A transposon gene (tra) is inserted in ΦAP53.5 between the modules of lysogeny control and DNA replication. ΦAP53.3 also suffered from DNA deletion of the module of DNA packaging and head morphogenesis. The module encompassed by the dotted bracket is hypothetical. The virulence genes harbored by ΦAP53.2, ΦAP53.3, and ΦAP53.4 are shown above the module of virulence. Sequence comparisons with the closest homologs of ΦAP53.1, ΦAP53.3, and ΦAP53.5 are shown for demonstration of gene content changes. The similarities are represented with a grayscale gradient.
Evolutionary implications of ΦAP53.2 for disease development.
In order to obtain insights into the evolutionary patterns of the phages and their implications for GAS virulence, we collected all the homologous phages from other previously reported GAS genomes and carried out pairwise comparisons. Although frequent recombination events made rigorous assessments of phage evolution difficult, we discovered that the five phages fall into distinct homology clusters that share rare intercluster similarities (see Fig. S4 in the supplemental material). Only sporadic similarities were detected in the segments of tail fiber genes, e.g., hyaluronidase. Surprisingly, inspection of the sequence alignments revealed that the phages within the same clusters contained conserved regions in the module of head and tail morphogenesis, although they were divergent in other regions, most significantly in DNA replication, host lysis, and virulence (see Fig. S3 in the supplemental material). We therefore narrowed our focus on the evolutionary patterns of phages within each cluster.
We first extracted the conserved regions in the module of head or tail morphogenesis for each group of phages and then performed multiple-sequence comparisons within the same groups, each of which included 8 to 23 members (see Table S3 in the supplemental material). The phylogenetic network representation of the phages differentiates ΦAP53.2-like phages from the others (Fig. 8). The three groups of ΦAP53.5-, ΦAP53.3-, and ΦAP53.4-like phages exhibited similar evolutionary patterns, where the phage members within the groups were split into two distant branches (Fig. 8A to C). Each branch is composed mainly of phages integrated at the same chromosomal locations (see Table S3 in the supplemental material). In contrast, the group of ΦAP53.2-like phages diverged in multiple branches, and the phages from different branches may have integrated at the same chromosomal locations (Fig. 8D). Close examination of sequence comparison profiles revealed that the two-branch divergence within the ΦAP53.5-, ΦAP53.3-, and ΦAP53.4-like phage groups was essentially induced by segmental sequence deletion or replacement. Exclusion of the related segments in multiple-sequence comparisons annihilated the two-branch structures. This resulted in branching structures similar to those of ΦAP53.2-like phages (Fig. 8). Furthermore, we estimated the SNP rates for each phage group after eliminating the divergent segments and found that the SNP rate for the ΦAP53.2-like phage group is significantly higher than that for the other three phage groups (175 SNPs/kb versus an average of 69 SNPs/kb; P value of ∼10−11, as determined by a t test).
FIG 8.

Phylogenetic network representation of phages from the ΦAP53.2-, ΦAP53.3-, ΦAP53.4-, and ΦAP53.5-like groups. The phylogeny structures were built based on multiple-sequence alignments of conserved regions in the gene modules of head and tail morphogenesis (top) in comparison with those of regions excluding sequences of segmental changes (bottom). (A to C) Phage members within the ΦAP53.5-like (A), ΦAP53.3-like (B), and ΦAP53.4-like (C) groups are split into two distant branches. The phages falling into different branches are color-coded with the phage nodes, and the phages from AP53 are highlighted in blue. (D) In contrast, the group of ΦAP53.2-like phages diverged into multiple branches. Phages with the same integration sites are shaded with the same color. The two-branch divergence in the ΦAP53.3-, ΦAP53.4-, and ΦAP535-like phage groups disappeared when the sequence comparisons were performed for the regions with excluded sequences of segmental changes.
It was further noted that the ΦAP53.2-like phage was not present in any skin-tropic GAS strains other than AP53. The recently reported genomes of skin-tropic isolates STAB1101, STAB1102, NGAS327, and NGAS638 (37) were not found to share any phage relationships with AP53, except for the remnant ΦAP53.5. This finding, coupled with the isolation of AP53 early in 1967 (38), suggests that AP53 is probably among the ancient isolates carrying the ΦAP53.2-like phage, which was transmitted to pharyngeal strains in occasional events and afterwards to a wider population of “throat” specialist strains.
The phylogenetic relationships of the group of ΦAP53.1-like phages were similar to those of the ΦAP53.3-, ΦAP53.4-, and ΦAP53.5-like phages, in that two explicit branches are formed (see Fig. S5 in the supplemental material). However, only four of the eight phage members, namely, ΦATCC 19615.2, ΦMGAS8232.1, ΦSTAB906.6, and ΦSSI-1.6, carry virulence genes. Analysis of their sequences identified diverse chromosomal locations for integration (see Table S3 in the supplemental material), reinforcing our view that ΦAP53.1 is potentially in the process of evolving into a new phage with enhanced lytic properties.
DISCUSSION
Group A Streptococcus pyogenes is recognized as the causative pathogen for a wide range of disease genotypes due to the high level of genetic variation found in its many strains, which confers to the organism multifaceted survival strategies and virulence capacities. The genetic factors for these variations include point mutations in essential regulatory genes; various combinations of virulence factors, including those found in incorporated phages; susceptibility to horizontal transfer of gene contents; and large segmental chromosomal changes and rearrangements. In this study, we present a genomic study of a serotype M53 GAS strain, AP53, which was obtained half a century ago from a low-virulence skin isolate (38). After many passages, this strain became highly virulent, as was the goal of the work during that time, but this strain has nonetheless remained very similar to its pattern D emm53-less virulent counterpart, Alab49, with respect to various genetic markers, i.e., emm pattern, FCT type, and SNPs. The gene content, organization, and individual sequences of AP53 are highly similar (>90%) to those of Alab49. Thus, a comparison of these two strains is highly valuable and should shed light on the genetic factors that led to the enhanced virulence of AP53 as well as reveal genetic mechanisms for the skin tropicity of these two strains. We found that there are indeed small yet distinct genomic changes between AP53 and Alab49, and a few other newly sequenced skin isolates, suggesting that the combination of these small changes at the genomic level likely constitute part of the driving force of the enhanced virulence and persistent survival of the invasive forms of the skin-professional class of GAS during infection.
The genome of AP53 differed from that of Alab49 by >30 point mutations, which are likely strain specific. However, a nucleotide deletion in AP53, not found in Alab49, was detected in the sensor kinase gene (covS) of the two-component regulatory system CovRS in AP53, a change that inactivated the sensor function of CovS (CovS−). Our work, and those of others, demonstrated that inactivated CovS constitutes one of the most important universal contributors to the invasiveness of strains such as AP53. Inactivation of CovS has been shown to switch the pathogenicity of nearly all GAS strains studied from low virulence to invasive by regulating the transcription of diverse virulence genes. A mouse survival study with the AP53/CovS− mutant strain along with the isogenic strain containing complemented covS (AP53/CovS+) showed that the lethality of the bacteria was reversed from noninvasive to invasive solely through switching of AP53/CovS+ to AP53/CovS−. This switch abrogated the expression of the extracellular protease speB and elevated the expression of the protective hyaluronic acid capsule in the AP53/CovS− strain (15), consistent with a survival mechanism for the bacteria that allows persistent infection to occur. These examples represent explicit manifestations of CovS playing a phenotype-switching role, converting a benign isolate to a hypervirulent one, and the frameshift deletion in covS, a gene hot spot for many different inactivating mutations, is a key factor that renders AP53 a highly invasive and lethal isolate. Such CovS inactivations, but not their reversal, occur during the course of infection in several GAS strains as the bacteria adapt to new environments (39). The triggers for this important inactivation of CovS, especially during the course of infection, are not fully understood.
While AP53 shares almost all of its essential virulence genes with Alab49, a potentially important difference between AP53 and Alab49 in the collagen-like binding protein SclB was detected. In this case, AP53 contains a different number of repeating pentanucleotides in the N-terminal signal sequence than Alab49 (and other GAS strains). The number of repeat units could determine the proper translation of sclB, since depending on their number, the initiator GTG codon may be out of frame with the ORF of SclB, as is the case in many strains of GAS. The finding that the sclB genes from the strains with emm pattern D are all fully functionally translatable logically implicates the role of a human collagen mimetic, sclB, in skin adherence. Repeat-sequence-controlled translation may have become an evolutionary mechanism of GAS to adapt to various infectious conditions. Another collagen-like protein, SclA, is also encoded by essentially all GAS genomes and is not under highly controlled translation, at least via the slipped-strand mispairing mechanism (33), as is the case with SclB. It is possible that SclA is a basic collagen-binding protein, while SclB plays a role in flexible adaptation to pressures during infection. However, the exact relationships between SclA and SclB are yet to be determined.
The most apparent deviations of the genome of AP53 from that of Alab49 are the acquisition of two novel phages (ΦAP53.1 and ΦAP53.2) and the loss of another phage (ΦAlab49.1). Phage ΦAP53.2 carries two adjacent virulence factors, speK-slaA, which showed increased expression in the AP53/CovS− strain relative to that in the AP53/CovS+ strain. SpeK is an exotoxin that has been shown to stimulate human T-cell proliferation (40). The phage toxin SlaA, a phospholipase A2 protein, catalyzes the hydrolysis of phospholipids in the cell membrane, with the consequent destruction of the membrane surface. This step releases arachidonic acids, which can mediate proinflammatory cascades in eukaryotic hosts (41). SlaA was identified predominantly in clones causing invasive infections or pharyngeal epidemiology (42). The increased production of immunoreactive SlaA and SpeK in a serotype M3 strain, MGAS315, causing STSS, was previously detected in in vitro cocultures with human pharyngeal epithelial cells (35). Here, we present the first report of elevated expression levels of slaA and the adjacent gene speK in a skin-tropic invasive GAS strain. This implies that SlaA may also be involved in host-pathogen interactions in the skin locale. Considering that speK-slaA are the only phage-borne virulence genes upregulated in the AP53/CovS− strain, we hypothesize that the acquisition of phage ΦAP53.2, carrying speK-slaA, may represent an important evolutionary event undergone by AP53 for enhanced virulence and fitness in adaption to host niches. Our phylogenetic analysis of the ΦAP53.2 homologs revealed that ΦAP53.2-like phages were rapidly evolving, active in lytic cycles, and therefore likely to have been transmitted frequently among GAS isolates from invasive infections or epidemic outbreaks. Notably, the isolation history of GAS strains provides clues that ΦAP53.2 is among the connecting points between the ancestors and their progenies, which are continuously disseminating geographically and causing disease.
However, not all of the derived phages conferred selective advantages to the bacteria. Phage ΦAP53.1 was not found to carry any virulence factors. The homologs of ΦAP53.1 in other GAS strains were characterized by a high frequency of missing virulence cassettes and diverse integration in chromosomal locations. These features lead us to propose that ΦAP53.1 is probably a relatively new phage and still in the process of adaptation to host defenses. The toxin genes may have been deleted by the host in order to reduce the harm from exogenous DNA content.
Taken together, the data presented here are consistent with the hypothesis that two-branch splitting in the ΦAP53.3-, ΦAP53.4-, and ΦAP53.5-like phage groups occurred in a single event of segmental DNA changes and that the phages were disseminated within each branch, most probably via transduction in the lysogenic cycle. However, the phages from the ΦAP53.2-like group were disseminated via a complex evolutionary pathway, probably involving a mixture of lysogenic and lytic cycles, and subsequently underwent rapid point mutations for better adaptation. The high genetic diversity of the ΦAP53.2-like phages suggests a greater diversifying selective capacity for survival and fitness in versatile environmental niches. Meanwhile, it is noteworthy that the evolutionary profile obtained from the sequence analysis of the limited number of known phage elements in this study may represent only a portion of the whole evolutionary history of phage populations, most of which have not yet been identified.
The influence of phage elements in enhancing fitness and pathogenesis may not be limited to the acquisition of virulence genes but could also result from a genetic switch mechanism via regulation of gene expression. A mutator phenotype was found in a serotype M1 GAS strain, SF370, where a defective phage, SpyClM1, may exist as lysogens or episomes depending on the growth status (36, 43). The switching status of the phage altered the global transcription profile, probably by controlling the regulation of the DNA mismatch repair (MMR) operon, thus facilitating alternative adaptation for survival of the bacteria in host cells (44). In this regard, it is possible that the phages in the strain studied here also have evolved to influence virulence at the gene regulation level in addition to the genetic level.
In summary, the data presented here demonstrate that hypervirulent GAS strain AP53 may have undergone several evolutionary adaptations, resulting in changes in multiple genetic factors which ultimately contributed to the enhanced adaptation and pathogenesis of AP53. In addition to genetic factors, it should also be noted that epigenetic factors may also play important roles in fitness and pathogenesis. The influence of DNA adenine methylation on virulence has been demonstrated for various pathogens, including Vibrio cholerae (45), Salmonella enterica (46), Yersinia pestis (47), and Mycobacterium tuberculosis (48). The methylation of DNA at specific sites may exert influences through the regulation of chromosome replication, transcriptional regulation, and mismatch repair in response to various environmental stimuli (49, 50). The specific combination of genetic factors, regulatory processes, and epigenetic machineries, coupled with environmental conditions, may ultimately generate adapted GAS genotypes that determine the disease phenotypes of the strains. Future studies with integrated genetic and epigenetic approaches will result in a better understanding of the mechanisms underlying the pathogenesis of bacteria in diverse host environments.
Supplementary Material
ACKNOWLEDGMENT
This work was supported in part by NIH grant HL013423.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01019-15.
REFERENCES
- 1.Lancefield RC. 1928. The antigenic complex of Streptococcus haemolyticus. I. Demonstration of a type-specific substance in extracts of Streptococcus haemolyticus. J Exp Med 47:91–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carapetis JR, Steer AC, Mulholland EK, Weber M. 2005. The global burden of group A streptococcal diseases. Lancet Infect Dis 5:685–694. doi: 10.1016/S1473-3099(05)70267-X. [DOI] [PubMed] [Google Scholar]
- 3.Schwartz B, Facklam RR, Breiman RF. 1990. Changing epidemiology of group A streptococcal infection in the USA. Lancet 336:1167–1171. doi: 10.1016/0140-6736(90)92777-F. [DOI] [PubMed] [Google Scholar]
- 4.Aziz RK, Kansal R, Aronow BJ, Taylor WL, Rowe SL, Kubal M, Chhatwal GS, Walker MJ, Kotb M. 2010. Microevolution of group A streptococci in vivo: capturing regulatory networks engaged in sociomicrobiology, niche adaptation, and hypervirulence. PLoS One 5:e9798. doi: 10.1371/journal.pone.0009798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walker MJ, Barnett TC, McArthur JD, Cole JN, Gillen CM, Henningham A, Sriprakash KS, Sanderson-Smith ML, Nizet V. 2014. Disease manifestations and pathogenic mechanisms of group A Streptococcus. Clin Microbiol Rev 27:264–301. doi: 10.1128/CMR.00101-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ribardo DA, McIver KS. 2006. Defining the Mga regulon: comparative transcriptome analysis reveals both direct and indirect regulation by Mga in the group A streptococcus. Mol Microbiol 62:491–508. doi: 10.1111/j.1365-2958.2006.05381.x. [DOI] [PubMed] [Google Scholar]
- 7.Bessen DE, Sotir CM, Readdy TL, Hollingshead SK. 1996. Genetic correlates of throat and skin isolates of group A streptococci. J Infect Dis 173:896–900. doi: 10.1093/infdis/173.4.896. [DOI] [PubMed] [Google Scholar]
- 8.McGregor KF, Spratt BG, Kalia A, Bennett A, Bilek N, Beall B, Bessen DE. 2004. Multilocus sequence typing of Streptococcus pyogenes representing most known emm types and distinctions among subpopulation genetic structures. J Bacteriol 186:4285–4294. doi: 10.1128/JB.186.13.4285-4294.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bessen DE, Lizano S. 2010. Tissue tropisms in group A streptococcal infections. Future Microbiol 5:623–638. doi: 10.2217/fmb.10.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barocchi MA, Ries J, Zogaj X, Hemsley C, Albiger B, Kanth A, Dahlberg S, Fernebro J, Moschioni M, Masignani V, Hultenby K, Taddei AR, Beiter K, Wartha F, von Euler A, Covacci A, Holden DW, Normark S, Rappuoli R, Henriques-Normark B. 2006. A pneumococcal pilus influences virulence and host inflammatory responses. Proc Natl Acad Sci U S A 103:2857–2862. doi: 10.1073/pnas.0511017103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kreikemeyer B, Gamez G, Margarit I, Giard JC, Hammerschmidt S, Hartke A, Podbielski A. 2011. Genomic organization, structure, regulation and pathogenic role of pilus constituents in major pathogenic streptococci and enterococci. Int J Med Microbiol 301:240–251. doi: 10.1016/j.ijmm.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 12.Falugi F, Zingaretti C, Pinto V, Mariani M, Amodeo L, Manetti AG, Capo S, Musser JM, Orefici G, Margarit I, Telford JL, Grandi G, Mora M. 2008. Sequence variation in group A Streptococcus pili and association of pilus backbone types with Lancefield T serotypes. J Infect Dis 198:1834–1841. doi: 10.1086/593176. [DOI] [PubMed] [Google Scholar]
- 13.Lizano S, Luo F, Bessen DE. 2007. Role of streptococcal T antigens in superficial skin infection. J Bacteriol 189:1426–1434. doi: 10.1128/JB.01179-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Steer AC, Law I, Matatolu L, Beall BW, Carapetis JR. 2009. Global emm type distribution of group A streptococci: systematic review and implications for vaccine development. Lancet Infect Dis 9:611–616. doi: 10.1016/S1473-3099(09)70178-1. [DOI] [PubMed] [Google Scholar]
- 15.Liang Z, Zhang Y, Agrahari G, Chandrahas V, Glinton K, Donahue DL, Balsara RD, Ploplis VA, Castellino FJ. 2013. A natural inactivating mutation in the CovS component of the CovRS regulatory operon in a pattern D streptococcal pyogenes strain influences virulence-associated genes. J Biol Chem 288:6561–6573. doi: 10.1074/jbc.M112.442657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Delcher AL, Bratke KA, Powers EC, Salzberg SL. 2007. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23:673–679. doi: 10.1093/bioinformatics/btm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lagesen K, Hallin P, Rodland EA, Staerfeldt HH, Rognes T, Ussery DW. 2007. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 35:3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aziz RK, Kotb M. 2008. Rise and persistence of global M1T1 clone of Streptococcus pyogenes. Emerg Infect Dis 14:1511–1517. doi: 10.3201/eid1410.071660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tatusova T, Ciufo S, Fedorov B, O'Neill K, Tolstoy I. 2014. RefSeq microbial genomes database: new representation and annotation strategy. Nucleic Acids Res 42:D553–D559. doi: 10.1093/nar/gkt1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA. 2011. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. doi: 10.1186/1471-2164-12-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carver TJ, Rutherford KM, Berriman M, Rajandream MA, Barrell BG, Parkhill J. 2005. ACT: the Artemis Comparison Tool. Bioinformatics 21:3422–3423. doi: 10.1093/bioinformatics/bti553. [DOI] [PubMed] [Google Scholar]
- 23.Nusbaum C, Ohsumi TK, Gomez J, Aquadro J, Victor TC, Warren RM, Hung DT, Birren BW, Lander ES, Jaffe DB. 2009. Sensitive, specific polymorphism discovery in bacteria using massively parallel sequencing. Nat Methods 6:67–69. doi: 10.1038/nmeth.1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huson DH, Bryant D. 2006. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23:254–267. [DOI] [PubMed] [Google Scholar]
- 26.Sun H, Ringdahl U, Homeister JW, Fay WP, Engelberg NC, Yang AY, Rozek LS, Wang X, Sjobring U, Ginsburg D. 2004. Plasminogen is a critical host pathogenicity factor for group A streptococcal infection. Science 305:1283–1286. doi: 10.1126/science.1101245. [DOI] [PubMed] [Google Scholar]
- 27.Kratovac Z, Manoharan A, Luo F, Lizano S, Bessen DE. 2007. Population genetics and linkage analysis of loci within the FCT region of Streptococcus pyogenes. J Bacteriol 189:1299–1310. doi: 10.1128/JB.01301-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kralovich KR, Li L, Hembrough TA, Webb DJ, Karns LR, Gonias SL. 1998. Characterization of the binding sites for plasminogen and tissue-type plasminogen activator in cytokeratin 8 and cytokeratin 18. J Protein Chem 17:845–854. doi: 10.1023/A:1020738620817. [DOI] [PubMed] [Google Scholar]
- 29.Selander RK, Caugant DA, Ochman H, Musser JM, Gilmour MN, Whittam TS. 1986. Methods of multilocus enzyme electrophoresis for bacterial population genetics and systematics. Appl Environ Microbiol 51:873–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Enright MC, Spratt BG. 1998. A multilocus sequence typing scheme for Streptococcus pneumoniae: identification of clones associated with serious invasive disease. Microbiology 144:3049–3060. doi: 10.1099/00221287-144-11-3049. [DOI] [PubMed] [Google Scholar]
- 31.Lukomski S, Nakashima K, Abdi I, Cipriano VJ, Ireland RM, Reid SD, Adams GG, Musser JM. 2000. Identification and characterization of the scl gene encoding a group A Streptococcus extracellular protein virulence factor with similarity to human collagen. Infect Immun 68:6542–6553. doi: 10.1128/IAI.68.12.6542-6553.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lukomski S, Nakashima K, Abdi I, Cipriano VJ, Shelvin BJ, Graviss EA, Musser JM. 2001. Identification and characterization of a second extracellular collagen-like protein made by group A Streptococcus: control of production at the level of translation. Infect Immun 69:1729–1738. doi: 10.1128/IAI.69.3.1729-1738.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rasmussen M, Bjorck L. 2001. Unique regulation of SclB—a novel collagen-like surface protein of Streptococcus pyogenes. Mol Microbiol 40:1427–1438. doi: 10.1046/j.1365-2958.2001.02493.x. [DOI] [PubMed] [Google Scholar]
- 34.Whatmore AM. 2001. Streptococcus pyogenes sclB encodes a putative hypervariable surface protein with a collagen-like repetitive structure. Microbiology 147:419–429. doi: 10.1099/00221287-147-2-419. [DOI] [PubMed] [Google Scholar]
- 35.Banks DJ, Lei B, Musser JM. 2003. Prophage induction and expression of prophage-encoded virulence factors in group A Streptococcus serotype M3 strain MGAS315. Infect Immun 71:7079–7086. doi: 10.1128/IAI.71.12.7079-7086.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scott J, Thompson-Mayberry P, Lahmamsi S, King CJ, McShan WM. 2008. Phage-associated mutator phenotype in group A streptococcus. J Bacteriol 190:6290–6301. doi: 10.1128/JB.01569-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Athey TB, Teatero S, Sieswerda LE, Gubbay JB, Marchand-Austin A, Li A, Wasserscheid J, Dewar K, McGeer A, Williams D, Fittipaldi N. 21 October 2015. High incidence of invasive group A Streptococcus disease caused by strains of uncommon emm types in Thunder Bay, Ontario, Canada. J Clin Microbiol doi: 10.1128/JCM.02201-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Svensson MD, Sjobring U, Bessen DE. 1999. Selective distribution of a high-affinity plasminogen-binding site among group A streptococci associated with impetigo. Infect Immun 67:3915–3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mayfield JA, Liang Z, Agrahari G, Lee SW, Donahue DL, Ploplis VA, Castellino FJ. 2014. Mutations in the control of virulence sensor gene from Streptococcus pyogenes after infection in mice lead to clonal bacterial variants with altered gene regulatory activity and virulence. PLoS One 9:e100698. doi: 10.1371/journal.pone.0100698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Beres SB, Sylva GL, Barbian KD, Lei B, Hoff JS, Mammarella ND, Liu MY, Smoot JC, Porcella SF, Parkins LD, Campbell DS, Smith TM, McCormick JK, Leung DY, Schlievert PM, Musser JM. 2002. Genome sequence of a serotype M3 strain of group A Streptococcus: phage-encoded toxins, the high-virulence phenotype, and clone emergence. Proc Natl Acad Sci U S A 99:10078–10083. doi: 10.1073/pnas.152298499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murakami M, Kudo I. 2002. Phospholipase A2. J Biochem 131:285–292. doi: 10.1093/oxfordjournals.jbchem.a003101. [DOI] [PubMed] [Google Scholar]
- 42.Nagiec MJ, Lei B, Parker SK, Vasil ML, Matsumoto M, Ireland RM, Beres SB, Hoe NP, Musser JM. 2004. Analysis of a novel prophage-encoded group A Streptococcus extracellular phospholipase A(2). J Biol Chem 279:45909–45918. doi: 10.1074/jbc.M405434200. [DOI] [PubMed] [Google Scholar]
- 43.Nguyen SV, McShan WM. 2014. Chromosomal islands of and related streptococci: molecular switches for survival and virulence. Front Cell Infect Microbiol 4:109. doi: 10.3389/fcimb.2014.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hendrickson C, Euler CW, Nguyen SV, Rahman M, McCullor KA, King CJ, Fischetti VA, McShan WM. 2015. Elimination of chromosomal island SpyCIM1 from Streptococcus pyogenes strain SF370 reverses the mutator phenotype and alters global transcription. PLoS One 10:e0145884. doi: 10.1371/journal.pone.0145884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Julio SM, Heithoff DM, Provenzano D, Klose KE, Sinsheimer RL, Low DA, Mahan MJ. 2001. DNA adenine methylase is essential for viability and plays a role in the pathogenesis of Yersinia pseudotuberculosis and Vibrio cholerae. Infect Immun 69:7610–7615. doi: 10.1128/IAI.69.12.7610-7615.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heithoff DM, Sinsheimer RL, Low DA, Mahan MJ. 1999. An essential role for DNA adenine methylation in bacterial virulence. Science 284:967–970. doi: 10.1126/science.284.5416.967. [DOI] [PubMed] [Google Scholar]
- 47.Robinson VL, Oyston PC, Titball RW. 2005. A dam mutant of Yersinia pestis is attenuated and induces protection against plague. FEMS Microbiol Lett 252:251–256. doi: 10.1016/j.femsle.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 48.Shell SS, Prestwich EG, Baek SH, Shah RR, Sassetti CM, Dedon PC, Fortune SM. 2013. DNA methylation impacts gene expression and ensures hypoxic survival of Mycobacterium tuberculosis. PLoS Pathog 9:e1003419. doi: 10.1371/journal.ppat.1003419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Low DA, Weyand NJ, Mahan MJ. 2001. Roles of DNA adenine methylation in regulating bacterial gene expression and virulence. Infect Immun 69:7197–7204. doi: 10.1128/IAI.69.12.7197-7204.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heusipp G, Falker S, Schmidt MA. 2007. DNA adenine methylation and bacterial pathogenesis. Int J Med Microbiol 297:1–7. doi: 10.1016/j.ijmm.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 51.Fittipaldi N, Beres SB, Olsen RJ, Kapur V, Shea PR, Watkins ME, Cantu CC, Laucirica DR, Jenkins L, Flores AR, Lovgren M, Ardanuy C, Linares J, Low DE, Tyrrell GJ, Musser JM. 2012. Full-genome dissection of an epidemic of severe invasive disease caused by a hypervirulent, recently emerged clone of group A Streptococcus. Am J Pathol 180:1522–1534. doi: 10.1016/j.ajpath.2011.12.037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.