

ABSTRACT
The herpes simplex virus 1 (HSV-1) Us8A gene overlaps the gene that encodes glycoprotein E (gE). Previous studies have investigated the roles of Us8A in HSV-1 infection using null mutations in Us8A and gE; therefore, the role of Us8A remains to be elucidated. In this study, we investigated the function of Us8A and its phosphorylation at serine 61 (Ser-61), which we recently identified as a phosphorylation site by mass spectrometry-based phosphoproteomic analysis of HSV-1-infected cells, in HSV-1 pathogenesis. We observed that (i) the phosphorylation of Us8A Ser-61 in infected cells was dependent on the activity of the virus-encoded Us3 protein kinase; (ii) the Us8A null mutant virus exhibited a 10-fold increase in the 50% lethal dose for virulence in the central nervous system (CNS) of mice following intracranial infection compared with a repaired virus; (iii) replacement of Ser-61 with alanine (S61A) in Us8A had little effect on virulence in the CNS of mice following intracranial infection, whereas it significantly reduced the mortality of mice following ocular infection to levels similar to the Us8A null mutant virus; (iv) the Us8A S61A mutation also significantly reduced viral yields in mice following ocular infection, mainly in the trigeminal ganglia and brains; and (v) a phosphomimetic mutation at Us8A Ser-61 restored wild-type viral yields and virulence. Collectively, these results indicate that Us8A is a novel HSV-1 virulence factor and suggest that the Us3-mediated phosphorylation of Us8A Ser-61 regulates Us8A function for viral invasion into the CNS from peripheral sites.
IMPORTANCE The DNA genomes of viruses within the subfamily Alphaherpesvirinae are divided into unique long (UL) and unique short (Us) regions. Us regions contain alphaherpesvirus-specific genes. Recently, high-throughput sequencing of ocular isolates of HSV-1 showed that Us8A was the most highly conserved of 13 herpes simplex virus 1 (HSV-1) genes mapped to the Us region, suggesting Us8A may have an important role in the HSV-1 life cycle. However, the specific role of Us8A in HSV-1 infection remains to be elucidated. Here, we show that Us8A is a virulence factor for HSV-1 infection in mice, and the function of Us8A for viral invasion into the central nervous system from peripheral sites is regulated by Us3-mediated phosphorylation of the protein at Ser-61. This is the first study to report the significance of Us8A and its regulation in HSV-1 infection.
INTRODUCTION
Mammalian herpesviruses belong to the family Herpesviridae and are classified into three subfamilies: Alphaherpesvirinae, Betaherpesvirinae, and Gammaherpesvirinae (1). The DNA genomes of viruses within the Alphaherpesvirinae are divided into unique long (UL) and unique short (Us) regions, each of which is flanked by long inverted repeats (1). Although many of the genes within the UL regions are conserved throughout the virus family, the Us regions contain alphaherpesvirus-specific genes that lack detectable homologues in other subfamilies (1). Moreover, the gene content of the Us region varies considerably even between different alphaherpesviruses, the best characterized of which is the alphaherpesvirus Us region from herpes simplex virus 1 (HSV-1) (1).
Of the 13 HSV-1 genes that map to the Us region by DNA sequence analyses combined with transcriptional studies, all but one gene, encoding glycoprotein D (gD), have been reported to be dispensable for viral replication in cell culture (2–10). It is assumed that the nonessential proteins encoded by these genes enable the virus to evade host immunity or to replicate in a variety of cells under different conditions, and consequently, the virus can replicate, spread, and survive in vivo (1, 11). In support of this hypothesis, it has been reported that ICP47, Us3 protein kinase, or glycoprotein J (gJ) can regulate cytotoxic T lymphocyte (CTL)-induced cytotoxicity (12–16), and heterodimeric glycoproteins, glycoprotein E and glycoprotein I (gE/gI), are involved in species-specific binding of immunoglobulin G as Fc receptors (17–20). Furthermore, recombinant viruses lacking ICP22 or Us3 show significantly impaired cell-type-dependent viral replication in cell culture, as well as reduced virulence in mouse models, establishment of latency, and reactivation from latency (21–26).
Illumina high-throughput sequencing analysis of seven ocular isolates of HSV-1 demonstrated that the Us8A gene, the subject of this study, was the most highly conserved among the 13 HSV-1 genes that mapped to the Us region (27), suggesting it might have important roles in HSV-1 replication and/or pathogenesis in vivo. In agreement with this hypothesis, the 50% lethal dose (LD50) in mice following intracranial infection with recombinant HSV-1 containing a deletion spanning the Us8 gene encoding gE and part of the Us8A gene was nearly 100-fold higher than that of the parent HSV-1 (23). However, the specific role of Us8A in HSV-1 pathogenesis remains largely unclear because the Us8A gene overlaps the Us8 gene and the construction of a recombinant HSV-1 in which only the Us8A gene has been deleted has not been reported.
The Us8A gene is predicted to encode a type II membrane protein, and its product was reported to be phosphorylated in HSV-1-infected cells (28, 29). Recently, we reported two large-scale phosphoproteomic analyses of HSV-1-infected human carcinoma HEp-2 cells and human neuroblastoma SK-N-SH cells (30, 31). Both studies identified the serine at position 61 (Ser-61) in Us8A as a physiological phosphorylation site in HSV-1-infected cells. However, there is a lack of information on the biological significance of Us8A Ser-61 phosphorylation for viral replication and/or pathogenesis. In this study, we investigated the specific effects of Us8A and Ser-61 phosphorylation on HSV-1 infection, especially HSV-1 replication and pathogenesis in vivo.
MATERIALS AND METHODS
Cells and viruses.
Simian kidney epithelial Vero cells and rabbit skin cells, as well as the HSV-1 wild-type strain HSV-1(F), were described previously (32, 33). Recombinant virus YK511, encoding an enzymatically inactive Us3 mutant in which lysine at Us3 residue 220 was replaced with methionine (Us3K220M), and recombinant virus YK513, in which the Us3K220M mutation in YK511 was repaired (Us3K220M-repair), were described previously (34).
Plasmids.
pGEM-1NC-EGFP/BAC, used to generate recombinant viruses in the two-step Red-mediated mutagenesis procedures described below, was constructed as follows. pBS-1C was constructed by amplifying a 1-kb sequence containing UL51, its upstream region, and part of UL52 from the HSV-1(F) genome by PCR and was then cloned into the NotI and SalI sites of pBluescript II KS(+) (Stratagene). pBS-1NC was constructed by amplifying a 1-kb sequence containing genes within UL50 and its downstream region from the HSV-1(F) genome by PCR and cloned into the EcoRI and NotI sites of pBS-1C. A NotI fragment of pBS246-GFP-BAC containing the bacterial artificial chromosome (BAC) sequence and an enhanced green fluorescent protein (EGFP) expression cassette flanked by loxP sites (25) was cloned into a NotI site of pBS-1NC to construct pBS-1NC-EGFP/BAC.
pBS-CREin-Kan, used to generate recombinant viruses in the two-step Red-mediated mutagenesis procedures described below, was constructed as follows. pBS-CREin was constructed by amplifying the Cre-encoding gene, containing a synthetic intron that prevented expression of the functional protein in Escherichia coli, by PCR from pCREin (35) (a gift from G. A. Smith, Northwestern University Feinberg School of Medicine, Chicago, IL, USA) and cloned into the EcoRI and SpeI sites of pBluescript II KS(+) (Stratagene). pBS-CREin-Kan was constructed by amplifying the pEPkan-S domain (36, 37), encoding the I-SceI site, and a kanamycin resistance gene by PCR from pEPkan-S using the primers shown in Table 1 and cloned into the BamHI site of pBS-CREin.
TABLE 1.
Oligonucleotide sequences for the construction of plasmids and recombinant viruses

pMAL-Us8A-P3 and pMAL-Us8A-P4, used to generate a fusion protein of maltose binding protein (MBP) and a domain of Us8A, were constructed by cloning the domains of Us8A encoded by Us8A codons 20 to 94 and 20 to 71, respectively, amplified by PCR from pYEbac102 (32), into pMAL-c (New England BioLabs) in frame with the MBP.
Construction of recombinant virus YK5000.
To construct recombinant virus YK5000, in which the BAC sequence and EGFP expression cassette, flanked by loxP sites, were inserted into the intergenic region between the HSV-1 UL50 and UL51 genes (Fig. 1), rabbit skin cells were cotransfected with pBS-1NC-EGFP/BAC and HSV-1(F) viral DNA using the calcium phosphate precipitation technique as described previously (32). At 3 days posttransfection, the transfected cells were harvested, freeze-thawed, and sonicated. The cell lysates were diluted and inoculated onto Vero cells, and plaques were screened for fluorescence with an inverted fluorescence microscope (Olympus IX71) (38). Recombinant viruses were plaque purified three times on Vero cells as described previously (32, 38).
FIG 1.

Strategy to construct a self-excisable HSV-1(F) BAC clone. Shown is a schematic diagram of the genome structure of HSV-1(F), the intergenic regions of recombinant viruses with or without the BAC sequence and the EGFP expression cassette insertions, and E. coli containing HSV-1(F) BAC. Line 1, wild-type HSV-1(F) genome; line 2, intergenic region between HSV-1(F) UL50 and UL51 genes; lines 3 and 7, schematic diagrams of recombinant viruses YK5000 and YK5002, respectively; lines 4 and 5, schematic diagrams of E. coli plasmid pYEbac5001 contained in DH10B and GS1783, respectively; line 6, schematic diagram of E. coli plasmid pYEbac5002 contained in GS1783.
Construction of an E. coli strain harboring HSV-1(F) BAC.
YK5000 circular viral DNA was isolated from infected Vero cells by the Hirt method, as described previously (32). The resulting circular viral DNA was electroporated into E. coli DH10B (Invitrogen), and the transformed bacteria were grown on LB agar plates containing 12.5 μg of chloramphenicol/ml as described previously (32). Chloramphenicol-resistant colonies were screened by PCR with appropriate primers, which led to the identification of E. coli YEbac5001, an E. coli DH10B strain harboring the HSV-1 BAC plasmid (pYEbac5001) (Fig. 1).
Construction of a self-excisable HSV-1(F) BAC.
For the two-step Red-mediated mutagenesis procedures described below, pYEbac5001 was isolated by alkaline lysis and further purified by centrifugation in cesium chloride gradients as previously described (32). The isolated pYEbac5001 was electroporated into E. coli strain GS1783 (36, 37), and the transformed bacteria were grown on LB agar plates containing 12.5 μg of chloramphenicol/ml as described previously (32). To construct E. coli strain GS1783 harboring a self-excisable HSV-1 BAC plasmid (pYEbac5002) (Fig. 1), the EGFP sequence of pYEbac5001 was replaced with the Cre-encoding gene (35) by the two-step Red-mediated mutagenesis procedure using E. coli strain GS1783 containing pYEbac5001 as described previously (34, 36, 37), except using the primers listed in Table 1 and pBS-CREin-KanS.
Recombinant viruses YK5003 (ΔUs8A), encoding Us8A with threonine substituted for methionine-1 and methionine-20 (M1T/M20T) (Fig. 2); YK5005 (Us8A-S61A), encoding Us8A with alanine substituted for serine at residue Ser-61 (Fig. 2); and YK5007, (Us8A-S61E) encoding Us8A with glutamic acid substituted for serine at residue Ser-61 (Fig. 2), were generated by the two-step Red-mediated mutagenesis procedure using E. coli strain GS1783 containing pYEbac5002 as described previously (34, 36, 37), except using the primers listed in Table 1. Recombinant viruses YK5004 (ΔUs8A-repair), YK5006 (Us8A-SA-repair), and YK5008 (Us8A-SE-repair), in which the Us8A-M1T/M20T, Us8A-S61A, and UsA8-S61E mutations in YK5003, YK5005, and YK5007, respectively (Fig. 2), were repaired, were generated as described previously (34, 36, 37), except using the primers listed in Table 1.
FIG 2.
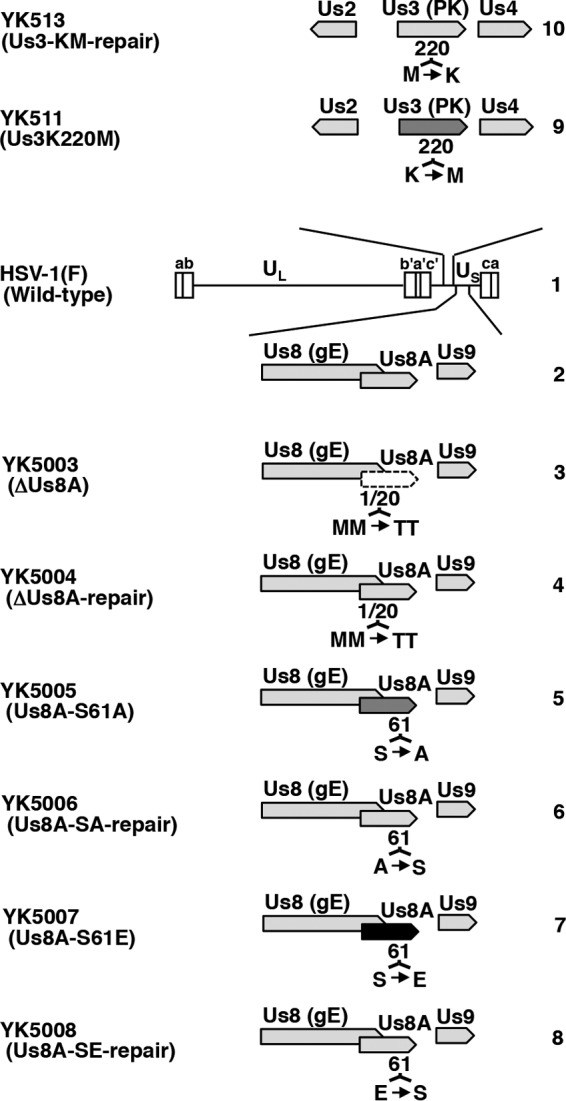
Schematic diagrams of the genome structures of HSV-1(F) and recombinant viruses used in this study. Line 1, wild-type HSV-1(F) genome; line 2, domain carrying the Us8 (gE), Us8A, and Us9 open reading frames; lines 3 to 8, recombinant viruses with mutations in the Us8A gene; lines 9 and 10, recombinant viruses with a mutation in the Us3 gene.
Phosphatase treatment.
Lysates of Vero cells that were mock infected or infected with wild-type HSV-1(F) at a multiplicity of infection (MOI) of 10 for 18 h were treated with alkaline phosphatase (CIP) (New England BioLabs) as described previously (39, 40).
Production and purification of MBP in E. coli.
MBP fusion proteins MBP-Us8A-P3, MBP-Us8A-P4, MBP-UL34, and MBP-Us9 were expressed in E. coli that had been transformed with pMAL-Us8A-P3, pMAL-Us8A-P4, pMAL-UL34 (39), and pMAL-Us9 (39), respectively, and purified using amylose resin (New England BioLabs) as described previously (41).
Purification of GST fusion proteins from baculovirus-infected cells.
Glutathione S-transferase (GST)-Us3 and GST-Us3K220M proteins were purified using glutathione-Sepharose resin (GE Healthcare Life Sciences) from the lysates of Sf9 cells infected with Bac-GST-Us3 and Bac-GST-Us3K220M, respectively, as described previously (39).
In vitro kinase assays.
MBP fusion proteins were captured on amylose beads (New England BioLabs) and used as substrates for in vitro kinase assays with purified GST-Us3 and GST-Us3K220M as described previously (31, 34, 39, 42, 43).
Antibodies.
To generate mouse polyclonal antibodies to HSV-1 Us8A, BALB/c mice were immunized once with purified MBP-Us8A-P3 with TiterMax Gold (TiterMax USA, Inc.). Sera from immunized mice were used as a source of anti-Us8A mouse polyclonal antibodies. To generate rabbit polyclonal antibodies to HSV-1 Us9, rabbits were immunized with purified MBP-Us9, following the standard protocol at MBL (Nagoya, Japan). Rabbit polyclonal antibodies to UL34 were described previously (40, 44). Mouse monoclonal antibodies to ICP27 (8.F.1378), gE (9H3), and glycoprotein B (gB) (H1817) were purchased from Virusys. Mouse monoclonal antibodies to ICP0 (5H7), ICP8 (10A3), and β-actin (AC15) were purchased from Santa Cruz Biotechnology, Millipore, and Sigma, respectively. Goat polyclonal antibodies to HSV-1 thymidine kinase (vTK) (vL-20) were purchased from Santa Cruz Biotechnology.
Immunoblotting.
Immunoblotting was performed as described previously (45). The amount of protein present in immunoblot bands was quantified using the ImageQuant LAS 4000 system with ImageQuant TL7.0 analysis software (GE Healthcare Life Sciences).
Animal studies.
Female ICR mice were purchased from Charles River. For intracranial infection, 3-week-old mice were infected intracranially with 10-fold serial dilutions of the indicated viruses as described previously (25, 32). The mice were monitored daily, and mortality from 1 to 14 days postinfection was attributed to the infected virus. The LD50 values were calculated by the Behrens-Karber method (32). For ocular infection, 5-week-old mice were infected with 1 × 105 PFU/eye of each of the indicated viruses as described previously (21, 24). The scoring scale for the severity of herpes stromal keratitis (HSK) was described previously (21, 24). Infected mice were euthanized at the indicated time points after infection, and eyes, trigeminal ganglia, and/or brains were removed, sonicated in 0.5 ml medium 199 (Sigma) containing 1% fetal calf serum and antibiotics, and frozen at −80°C as described previously (26). The frozen samples were later thawed and centrifuged, after which viral titers in the supernatants were determined by standard plaque assays on Vero cells. All animal experiments were carried out in accordance with the Guidelines for Proper Conduct of Animal Experiments, Science Council of Japan. The protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of the Institute of Medical Science, University of Tokyo (IACUC protocol approval number 19-26).
Statistical analysis.
Differences in the relative amounts of phosphorylated Us8A were statistically analyzed using analysis of variance (ANOVA) and Tukey's test. Differences in viral yields and HSK scores were statistically analyzed using a two-tailed Student t test. Differences in mortality of infected mice were statistically analyzed by the log rank test. A P value of <0.05 was considered statistically significant.
RESULTS
Construction and characterization of recombinant viruses.
To examine the effects of Us8A and its phosphorylation at Ser-61, we constructed a series of Us8A mutant viruses (shown in Fig. 2) using a self-excisable HSV-1(F) BAC clone generated in this study. The HSV-1(F) BAC clone, designated pYEbac5002, harbors the BAC sequences and expression cassette for a gene encoding Cre flanked by loxP sites, which were inserted into the intergenic region between HSV-1 UL50 and UL51 (Fig. 1). Recombinant viruses reconstituted from pYEbac5002 and its derivatives were expected to excise the BAC sequences through the functional Cre enzyme in infected cells as described previously (35). This newly generated HSV-1(F) BAC (pYEbac5002) was characterized as follows. (i) The pattern of BamHI digestion of the viral DNA of YK5002 (ΔBAC) reconstituted from pYEbac5002 was similar to that of wild-type HSV-1(F) viral DNA; however, the BamHI-digested YK5002 (ΔBAC) viral DNA had a 5.3-kbp fragment (Fig. 3A, fragment a) and was missing a 5.2-kbp fragment (Fig. 3A, fragment b), caused by the loxP insertion, compared to BamHI-digested HSV-1(F) DNA (Fig. 1). Furthermore, sequence analysis of the intergenic region between the YK5002 (ΔBAC) UL50 and UL51 genes showed that the expected Cre-mediated excision had occurred and only one loxP site remained in the region (data not shown). (ii) The maintenance of OriL and OriS in YK5002 (ΔBAC) was confirmed by a method described previously (32) (Fig. 3B), because the HSV-1 Ori sequences were reported to be unstable in bacteria (46). (iii) Expression levels of ICP0, ICP27, ICP8, vTK, gB, and UL34 from Vero cells infected at an MOI of 10 with YK5002 (ΔBAC) at 18 h postinfection were similar to those of wild-type HSV-1 (F) (Fig. 3C). (iv) The growth curves for YK5002 (ΔBAC) were similar to those of wild-type HSV-1(F) in Vero cells infected at MOIs of 10 and 0.01 (Fig. 3D and E). (v) The LD50 value of YK5002 (ΔBAC) for intracranial infection in mice was similar to that of wild-type HSV-1(F) (Table 2). These results indicate that, for the properties examined in these studies, the YK5002 (ΔBAC) virus reconstituted from pYEbac5002 retained the wild-type expression level of viral proteins and replication kinetics in cell culture and maintained a wild-type HSV-1(F) level of virulence in mice following intracranial infection.
FIG 3.
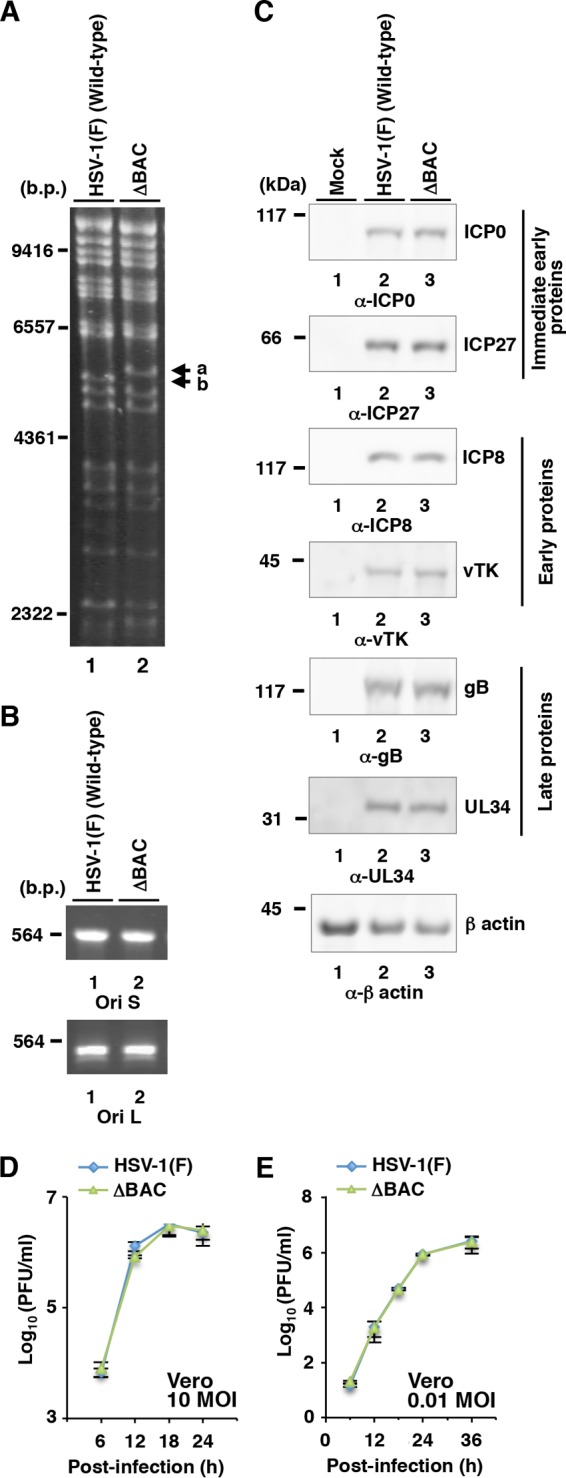
Viral genomes, expression of viral proteins, and growth curves from cells infected with HSV-1(F) and YK5002 (ΔBAC). (A) Agarose gel electrophoresis of BamHI-digested viral DNAs of HSV-1(F) (lane 1) and recombinant virus YK5002 (ΔBAC) (lane 2). Fragment a (5.3 kb) and fragment b (5.2 kb) were detected in the YK5002 and HSV-1(F) genomes, respectively, as a result of insertion of the loxP sequence. (B) Agarose gel electrophoresis of PCR products containing OriS (top) and OriL (bottom) of wild-type HSV-1(F) (lanes 1) and YK5002 (ΔBAC) (lanes 2). (C) Vero cells were mock infected (lanes 1) or infected with wild-type HSV-1(F) (lanes 2) or YK5002 (ΔBAC) (lanes 3) at an MOI of 10, harvested at 18 h postinfection, and then analyzed by immunoblotting with the indicated antibodies. Molecular size (A and B) and mass (C) markers are shown on the left. (D and E) Vero cells were infected at an MOI of 10 (D) or 0.01 (E) with wild-type HSV-1(F) or YK5002 (ΔBAC). Total virus from the cell culture supernatants and in fected cells was harvested at the indicated times and assayed on Vero cells. Each value represents the mean ± standard error of the mean (SEM) of the results of three independent experiments.
TABLE 2.
LD50 values of wild-type HSV-1(F) and YK5002 (ΔBAC) in mice following intracranial infectiona
| Virus | LD50 (PFU)b |
|---|---|
| HSV-1(F) (wild type) | 31.6 |
| YK5002 (ΔBAC) | 46.4 |
Three-week-old female ICR mice were infected intracranially with serial 10-fold dilutions of each virus in groups of six per dilution and monitored for 14 days postinfection.
The LD50 values were determined by the Behrens-Karber method.
Using pYEbac5002, we constructed a Us8A null mutant YK5003 (ΔUs8A) virus in which the initiation methionine codon of Us8A (Met-1) and the second methionine (Met-20) were replaced with threonines (M1T/M20T); a recombinant YK5005 (Us8A-S61A) virus encoding a mutant Us8A in which Ser-61 was replaced with alanine; and their repaired viruses, YK5004 (ΔUs8A-repair) and YK5006 (Us8A-SA-repair), respectively (Fig. 2). These recombinant viruses were characterized as follows: (i) Vero cells infected with wild-type HSV-1(F), YK5005 (Us8A-S61A), YK5004 (ΔUs8A-repair), or YK5006 (Us8A-SA-repair) expressed Us8A, whereas those infected with YK5003 (ΔUs8A) did not (Fig. 4A and D), indicating that the M1T/M20T mutation in Us8A inactivated expression of the Us8A gene; (ii) the null mutation in Us8A had no effect on the expression of the overlapping and/or neighboring Us8 (gE) and Us9 genes (Fig. 4B), indicating that the silent mutations in the wobble base of Us8(gE) for an asparagine at position 503 and an aspartic acid at position 522, caused by the M1T/M20T mutation in Us8A, had no effect on the expression of the Us8 (gE) and Us9 genes; and (iii) the growth curves of recombinant viruses YK5003 (ΔUs8A) and YK5005 (Us8A-S61A) were similar to those of the repaired viruses [YK5004 (ΔUs8A-repair) and YK5006 (Us8A-SA-repair)] and wild-type HSV-1(F) in Vero cells infected at MOIs of 10 and 0.01 (Fig. 5). These results were in agreement with a previous report that the double-null mutation of gE and Us8A had no effect on viral replication in Vero cells (23).
FIG 4.

Us3-mediated phosphorylation of Us8A Ser-61 in infected cells. (A and B) Vero cells were mock infected (lanes 1) or infected with wild-type HSV-1(F) (lanes 2), YK5003 (ΔUs8A) (lanes 3), or YK5004 (ΔUs8A-repair) (lanes 4) at an MOI of 10; harvested at 18 h postinfection; and then analyzed by immunoblotting with the indicated antibodies. (C) The infected Vero cell lysates were untreated (lane 1) or treated with CIP (lane 2) and then analyzed as described for panel A. (D) Vero cells were mock infected (lanes 1) or infected with wild-type HSV-1(F) (lanes 2), YK5005 (Us8A-S61A) (lanes 3), or YK5006 (Us8A-SA-repair) (lanes 4) at an MOI of 10; harvested at 18 h postinfection; and then analyzed as described for panel A. (E) Vero cells were mock infected (lanes 1) or infected with wild-type HSV-1(F) (lanes 2), YK511 (Us3K220M) (lanes 3), or YK513 (Us3K220M-repair) (lanes 4) at an MOI of 10; harvested at 18 h postinfection; and then analyzed as described for panel A. Molecular mass markers are shown on the left. (F) Quantitation of the amount of the slower-migrating Us8A band relative to that of the faster-migrating Us8A band shown in panel E (top, lanes 2, 3, and 4). Each value represents the mean and SEM of the results of three independent experiments and is expressed relative to the mean value of wild-type HSV-1(F)-infected cells, which was normalized to 100. The statistical significance according to ANOVA and Tukey's test is indicated. n.s., not significant.
FIG 5.
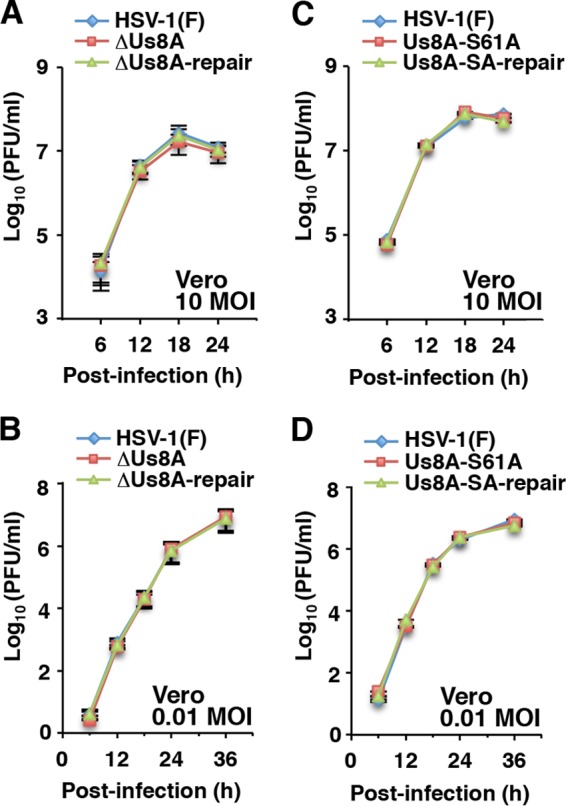
Effects of Us8A null or S61A mutations on growth curves for HSV-1-infected cells. Vero cells were infected at an MOI of 10 (A and C) or 0.01 (B and D) with wild-type HSV-1(F), YK5003 (ΔUs8A), or YK5004 (ΔUs8A-repair) (A and B) or wild-type HSV-1(F), YK5005 (Us8A-S61A), or YK5006 (Us8A-SA-repair) (C and D). Total virus from cell culture supernatants and infected cells was harvested at the indicated times and assayed on Vero cells. Each value represents the mean ± SEM of the results of three independent experiments.
Phosphorylation of Us8A at Ser-61 in HSV-1-infected cells.
Notably, Us8A proteins from Vero cells infected with wild-type HSV-1(F), YK5004 (ΔUs8A-repair), and YK5005 (Us8A-SA-repair) were detected as two bands with mobility differences in denaturing gels (Fig. 4A, top, lanes 2 and 4, and D, top, lanes 2 and 4). After the phosphatase treatment of lysates from wild-type HSV-1(F)-infected cells, the slower-migrating Us8A band disappeared (Fig. 4C, lane 2). Similarly, the disappearance of the slower-migrating band after phosphatase treatment was also observed for Us8A from Vero cells infected with YK5005 (Us8A-S61A) (Fig. 4D, top, lane 3).
It has been reported that Us8A from cells infected with recombinant HSV-1 carrying Us8A tagged with a 23-amino-acid epitope from the human cytomegalovirus (HCMV)-encoded glycoprotein B was detected in a denaturing gel as doublet bands, in agreement with the results shown in Fig. 4A, while the tagged Us8A from an in vitro transcription and translation system was detected as a single band (28). These observations suggested that phosphorylation of Us8A Ser-61 depended on another viral protein(s) and/or cellular protein(s) induced by HSV-1 infection. Interestingly, the amino acid sequence around Us8A Ser-61 resembles the consensus sequence recognized by the HSV-1-encoded Us3 protein kinase (47–49). These observations led us to examine whether Us3 was involved in the phosphorylation of Us8A Ser-61 in HSV-1-infected cells. As shown in Fig. 4E, a significant reduction of the slower-migrating band of Us8A was observed with Us8A from Vero cells infected with the Us3 kinase-dead mutant virus YK511 (Us3K220M) compared to Us8A from Vero cells infected with wild-type HSV-1(F) or YK513 (Us3K220M-repair) (Fig. 4E and F).
Collectively, these results indicated that the slower-migrating band was Us8A phosphorylated at Ser-61 and the faster-migrating band was Us8A lacking phosphorylation at Ser-61. In addition, these results indicated that Us3 mediated the phosphorylation of Us8A Ser-61 in infected cells. Of note, there was a low level of Us8A Ser-61 phosphorylation in the absence of Us3 kinase activity in infected cells (Fig. 4E, top, lane 3), indicating that a protein kinase other than Us3, likely a cellular protein kinase, also mediated the phosphorylation of Us8A Ser-61 in infected cells.
Us3-mediated phosphorylation of Us8A in vitro.
To examine whether HSV-1 Us3 can directly phosphorylate Us8A in vitro, we generated and purified chimeric proteins consisting of MBP fused to peptides encoded by Us8A codons 20 to 94 (MBP-Us8A-P3) and codons 20 to 71 (MBP-Us8A-P4) (Fig. 6A). The MBP fusion proteins were captured on amylose beads and used as substrates for in vitro kinase assays with purified wild-type GST-Us3 and a kinase-negative mutant, GST-Us3K220M (31, 34, 39, 42, 43). As shown in Fig. 6C, MBP-Us8A-P3 and MBP-Us8A-P4 were not labeled with [γ-32P]ATP in kinase assays using GST-Us3 (Fig. 6C, lanes 1 and 3), while MBP-UL34 was efficiently labeled (Fig. 6C, lane 5), as demonstrated in our previous reports (34, 39). When the kinase-negative mutant GST-Us3K220M was used, none of the MBP fusion proteins were labeled (Fig. 6C, lanes 2, 4, and 6). The presence of each MBP fusion protein and the radiolabeled MBP-UL34 band was verified by Coomassie brilliant blue (CBB) staining (Fig. 6B). These data indicate that Us3 cannot directly phosphorylate Us8A in vitro. This suggests an additional cellular and/or viral component(s) is required for the Us3-mediated phosphorylation of Us8A in HSV-1-infected cells.
FIG 6.
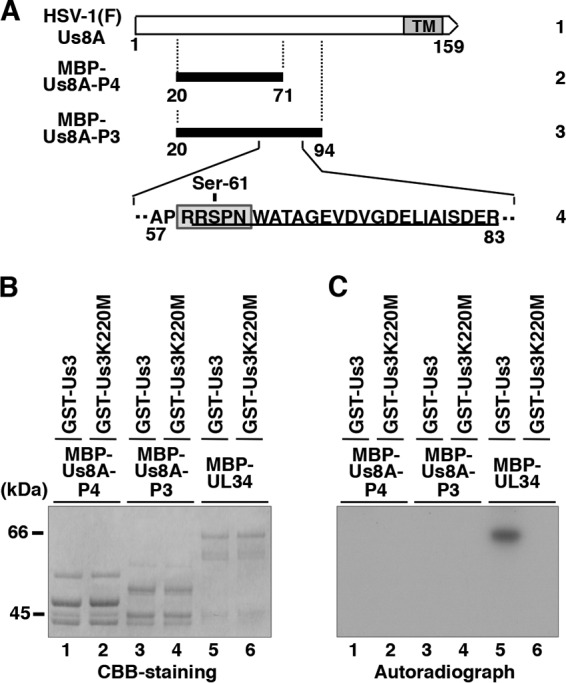
Us3-mediated phosphorylation of Us8A in vitro. (A) Schematic diagram of Us8A. Line 1, structure of the Us8A open reading frame. The shaded area represents a predicted transmembrane domain. Line 2, domain of the Us8A gene encoding Us8A residues 20 to 71, which were used in these studies to generate the MBP-Us8A-P4 fusion protein. Line 3, amino acid sequence of Us8A residues 20 to 94. Line 4, domain of the Us8A gene encoding Us8A residues 57 to 83. The amino acid sequence similar to the consensus sequence recognized by Us3 is boxed. A phosphopeptide detected in our previous studies (30, 31) is underlined. (B) Purified MBP-Us8A-P4 (lanes 1 and 2), MBP-Us8A-P3 (lanes 3 and 4), and MBP-UL34 (lanes 5 and 6) were incubated in kinase buffer containing [γ-32P]ATP and purified GST-Us3 (lanes 1, 3, and 5) or GST-Us3K220M (lanes 2, 4, and 6); separated on an SDS-PAGE gel; and stained with CBB. Molecular mass markers are shown on the left. (C) Autoradiograph of the gel in panel B. Molecular mass markers are shown on the left.
Effect of Us8A and its phosphorylation at Ser-61 on HSV-1 pathogenicity in mice following intracranial infection.
Simple experimental murine models of HSV-1 infection induced by the direct intracranial injection of HSV-1 are used to study the effect of viral replication on destruction of the central nervous system (CNS). In contrast, multiple aspects of HSV-1 infection, including pathogenic manifestations at peripheral sites of infection and the capacity to invade the CNS from peripheral sites (neuroinvasiveness) and the subsequent induction of CNS damage by viral replication, can be studied in mice following their peripheral (e.g., ocular or vaginal) infection. Therefore, to clarify the roles of Us8A and Us3-mediated phosphorylation of Us8A Ser-61 on HSV-1 infection in vivo, mice were infected intracranially with Us8A mutant viruses or wild-type HSV-1. Three-week-old female ICR mice were infected intracranially with 10-fold dilutions of wild-type HSV-1(F), YK5003 (ΔUs8A), YK5005 (Us8A-S61A), YK5004 (ΔUs8A-repair), or YK5006 (Us8A-SA-repair), and mortality was monitored for 14 days postinfection. As shown in Table 3, the LD50 of YK5003 (ΔUs8A) was approximately 10-fold higher than that of YK5004 (ΔUs8A-repair). This result is in agreement with previous reports that a deletion spanning gE and part of Us8A significantly reduced mortality in mice following intracranial infection (23). In contrast, the LD50 of YK5005 (Us8A-S61A) was comparable to that of YK5006 (Us8A-SA-repair) (Table 3). These results suggested that Us8A, but not the Us3-mediated phosphorylation of Us8A Ser-61, is required for efficient virulence in mice following intracranial infection.
TABLE 3.
LD50 values of recombinant viruses carrying mutations in Us8A and their repaired viruses in mice following intracranial infectiona
| Virus | LD50 (PFU)b |
|---|---|
| YK5003 (ΔUs8A) | 464 |
| YK5004 (ΔUs8A-repair) | 46.4 |
| YK5005 (Us8A-S61A) | 46.4 |
| YK5006 (Us8A-SA-repair) | 68.1 |
Three-week-old female ICR mice were infected intracranially with serial 10-fold dilutions of each virus in groups of six per dilution and monitored for 14 days postinfection.
The LD50 values were determined by the Behrens-Karber method.
Effects of Us8A and its phosphorylation at Ser-61 on HSV-1 pathogenesis in mice following ocular infection.
Next, 5-week-old female ICR mice were infected ocularly with 1 × 105 PFU/eye of YK5003 (ΔUs8A), YK5004 (ΔUs8A-repair), YK5005 (Us8A-S61A), or YK5006 (Us8A-SA-repair). The survival of the infected mice was monitored for 21 days postinfection, and the development of HSK in their eyes was determined at 6 days postinfection. In agreement with the virulence analysis in mice following intracranial infection (Table 3), the null mutation in Us8A significantly reduced the development of HSK, and survival was significantly longer for mice infected with YK5003 (ΔUs8A) than for mice infected with YK5004 (ΔUs8A-repair) (Fig. 7A and C). These results indicate that Us8A is required for both efficient virulence and the development of HSK in mice following ocular infection.
FIG 7.
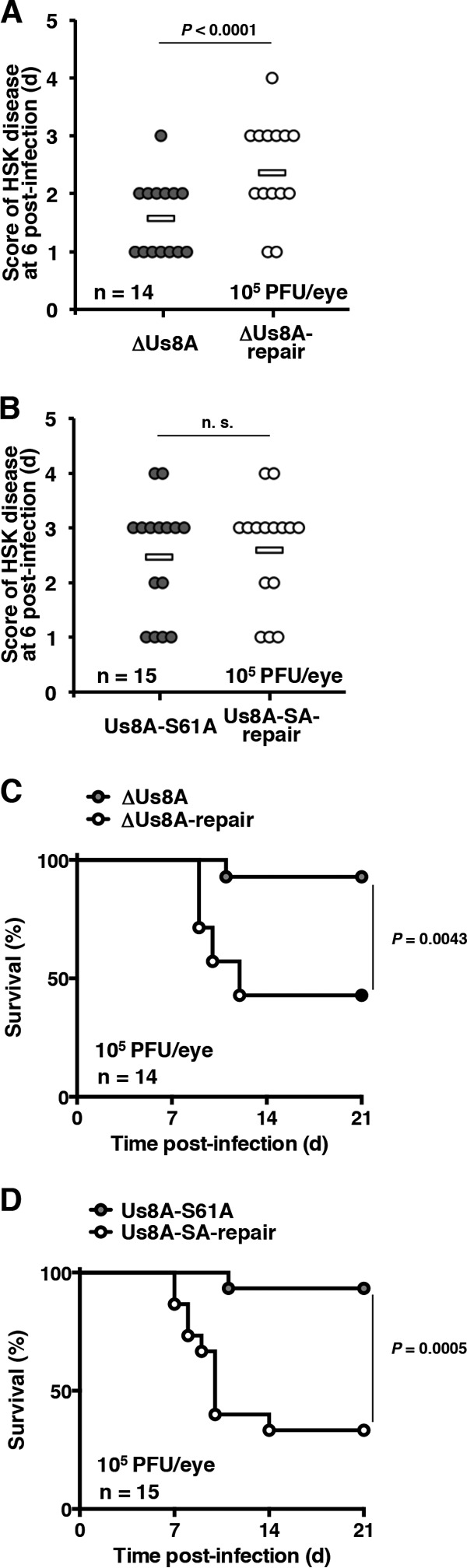
Effects of Us8A null and S61A mutations on the development of HSK and mortality of ocularly infected mice. (A and B) Fourteen (A) or 15 (B) 5-week-old female ICR mice infected ocularly with 1 × 105 PFU/eye of YK5003 (ΔUs8A) (A), YK5004 (ΔUs8A-repair) (A), YK5005 (Us8A-S61A) (B), or YK5006 (Us8A-SA-repair) (B) were scored for HSK at 6 days postinfection. Each data point represents the HSK score from one mouse. The horizontal bars represent the mean for each group. The statistical significance according to a two-tailed Student t test is indicated. n.s., not significant. (C and D) Survival of mice in the experiment described for panels A and B was monitored for 21 days postinfection. The statistical significance according to a log rank test is shown.
In contrast, the Us8A S61A mutation had no significant effect on the development of HSK, whereas the mutation significantly reduced mortality in mice at levels similar to those for the null mutation (Fig. 7B and 8D). To investigate further the linkage between the phosphorylation of Us8A Ser-61 and the mortality of ocularly infected mice, we constructed a recombinant virus, YK5007 (Us8A-S61E), encoding a phosphomimetic mutation at Us8A Ser-61, and the repaired virus YK5008 (Us8A-SE-repair). These recombinant viruses were characterized as follows: (i) Us8A from Vero cells infected with YK5007 (Us8A-S61E) migrated as a single band with intermediate mobility on denaturing gels (Fig. 8A, top, lane 3), suggesting that a negatively charged amino acid at Us8A position 61, either phosphorylated Ser-61 or an S61E substitution, affected the electrophoretic mobility of Us8A on denaturing gels, and (ii) the growth curves of these recombinant viruses were similar to that of wild-type HSV-1(F) in Vero cells infected at MOIs of 10 and 0.01 (Fig. 8B and C).
FIG 8.
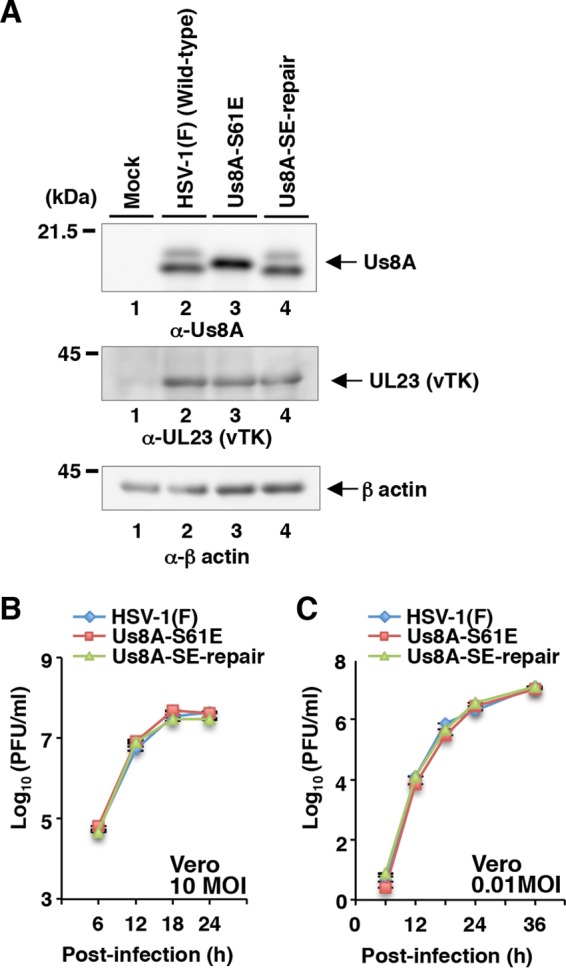
Effects of the Us8A S61E mutation on expression of Us8A and growth of HSV-1 in infected cells. (A) Vero cells were mock infected (lanes 1) or infected with wild-type HSV-1(F) (lanes 2), YK5007 (Us8A-S61E) (lanes 3), or YK5008 (ΔUs8A-SE-repair) (lanes 4) at an MOI of 10; harvested at 18 h postinfection; and then analyzed by immunoblotting with the indicated antibodies. Molecular mass markers are shown on the left. (B and C) Vero cells were infected at an MOI of 10 (B) or 0.01 (C) with wild-type HSV-1(F), YK5007 (Us8A-S61E), or YK5008 (ΔUs8A-SE-repair). Total virus from the cell culture supernatants and infected cells was harvested at the indicated times and assayed on Vero cells. Each value represents the mean ± SEM of the results of three independent experiments.
The recombinant viruses were then tested in ocularly infected mice. As observed with YK5005 (Us8A-S61A) (Fig. 7B), following ocular infection, the development of HSK in the eyes of mice infected with YK5007 (Us8A-S61E) was similar to that of mice infected with YK5008 (Us8A-SE-repair) (Fig. 9A). In addition, the survival of mice infected with YK5007 (Us8A-S61E) was similar to that of mice infected with YK5008 (Us8A-SE-repair) (Fig. 9B). These results suggested that a negatively charged amino acid at position 61 of Us8A, either phosphorylated Ser-61 or an S61E substitution, was required for wild-type mortality of mice following ocular infection. Taken together, these results indicate that Us8A Ser-61 and its phosphorylation are required for efficient viral virulence in mice following ocular infection but not for the pathogenic effects of HSV-1 in eyes.
FIG 9.
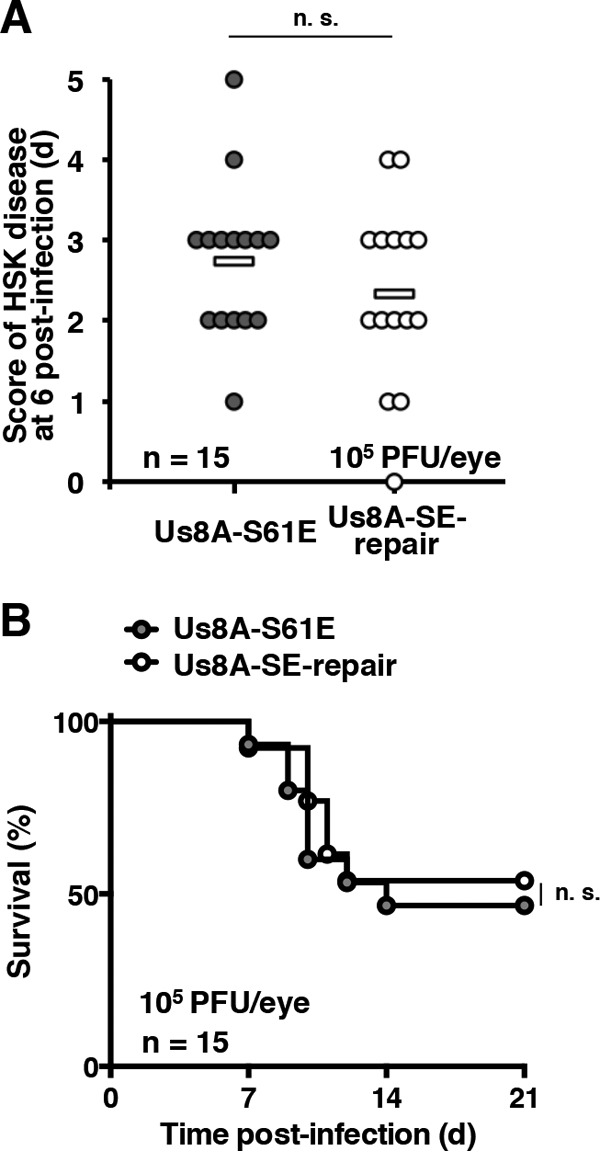
Effects of Us8A S61E mutation on the development of HSK and mortality in ocularly infected mice. (A) Fifteen 5-week-old female ICR mice infected ocularly with 1 × 105 PFU/eye of YK5007 (Us8A-S61E) or YK5008 (ΔUs8A-SE-repair) were scored for HSK at 6 days postinfection. Each data point represents the HSK score from one mouse. The horizontal bars represent the mean for each group. n.s., not significant. (B) Survival of mice in the experiment described for panel A was monitored for 21 days postinfection.
Effect of Us8A Ser-61 phosphorylation on viral replication in the eyes, trigeminal ganglia, and brains of mice following ocular infection.
In the murine model of HSK, when sufficient quantities of virus and/or a virulent strain of HSV-1 infects the eye, HSV-1 replicates in the epithelial layer of the cornea (1) and then infects nerve endings and is transported through neural axons toward the nerve cell bodies of the trigeminal ganglia, where the virus multiplies (1). Then, the virus returns from the trigeminal ganglia to the eye and can invade the CNS, causing encephalitis and death (1). To investigate the anatomical location of the restriction caused by blocking the phosphorylation of Us8A Ser-61, kinetic analysis of viral replication in various mouse tissues was performed. Five-week-old female ICR mice were infected ocularly with 1 × 105 PFU/eye of YK5005 (Us8A-S61A), YK5006 (Us8A-SA-repair), YK5007 (Us8A-S61E), or YK5008 (Us8A-SE-repair), and the yields of the indicated viruses in the eyes, trigeminal ganglia, and brains of the mice were measured.
The results were as follows. (i) As reported previously (26), viral yields in the eyes of mice infected with YK5006 (Us8A-SA-repair) or YK5008 (Us8A-SE-repair) peaked at 1 day postinfection and declined thereafter. Viral yields in the trigeminal ganglia of mice infected with each of these viruses reached a plateau at 3 and 4 days postinfection, and those in the brains were detectable from 3 days postinfection and increased at 4 days postinfection (Fig. 10). (ii) In the eyes of mice infected with YK5005 (Us8A-S61A) at 1 to 3 days postinfection, viral yields were similar to those of mice infected with YK5006 (Us8A-SA-repair). In contrast, at 4 days postinfection, when the virus is thought to return to the eyes from the trigeminal ganglia, viral yields in the eyes of mice infected with YK5005 (Us8A-S61A) were significantly (4.58-fold) lower than in YK5006 (Us8A-SA-repair)-infected mice (Fig. 10A). (iii) in the trigeminal ganglia of mice infected with YK5005 (Us8A-S61A), viral yields were comparable to those of mice infected with YK5006 (Us8A-SA-repair) at 1 and 2 days postinfection, whereas at 3 and 4 days postinfection, viral yields were significantly (3.07- and 10.4-fold, respectively) lower than those of mice infected with YK5006 (Us8A-SA-repair) (Fig. 10B). (iv) consistent with the viral yields from the trigeminal ganglia of mice, the viral yields from the brains of mice infected with YK5005 (Us8A-S61A) were significantly (2.62- and 4.63-fold) lower than those from mice infected with YK5006 (Us8A-SA-repair) at 3 and 4 days postinfection, respectively (Fig. 10C). (v) the phosphomimetic mutation at position 61 of Us8A restored wild-type viral yields in the eyes, trigeminal ganglia, and brains of mice following ocular infection (Fig. 10D to F).
FIG 10.

Effects of Us8A S61A or S61E mutations on viral yields in the eyes, trigeminal ganglia (TG), and brains of ocularly infected mice. Fifteen 5-week-old female ICR mice were infected ocularly with 1 × 105 PFU/eye of YK5005 (Us8A-S61A), YK5006 (Us8A-SA-repair), YK5007 (Us8A-S61E), or YK5008 (ΔUs8A-SE-repair). The eyes, trigeminal ganglia, and brains were recovered at 1, 2, 3, or 4 days postinfection, and viral yields were assayed on Vero cells. The detection limit of viral yields from trigeminal ganglia and brains were 2.5 PFU/ml and 25 PFU/ml, respectively. Because the mean weight of one trigeminal ganglion or one brain was 0.007 or 0.464 g, respectively, the limit of detection was about 356 or 53.9 PFU/ml/g. The dashed lines indicate the limit of detection. n.d., not detected. Each data point represents the viral yield from one mouse. The horizontal bars represent the mean for each group. The statistical significance according to a two-tailed Student t test is indicated. n.s., not significant.
Collectively, these results suggest that in this murine model of HSK, (i) the phosphorylation of Us8A Ser-61 has little effect on viral replication in the eyes and trigeminal ganglia early in infection (at 1 to 3 days and 1 and 2 days postinfection, respectively) but is required for efficient viral replication in these tissues later in infection (at 4 days and at 3 and 4 days postinfection, respectively) and (ii) this phosphorylation is required for efficient viral replication in the brains from the time the virus become detectable (at 3 and 4 days postinfection).
DISCUSSION
In this study, we present data showing that Us8A is required for efficient virulence in mice following both intracranial and ocular infection, thereby clarifying that Us8A is a novel HSV-1 virulence factor. We also demonstrate that the phosphorylation of a physiological phosphorylation site (Ser-61) in Us8A in infected cells is mediated by Us3 and the regulation of Us8A by Ser-61 phosphorylation is required for efficient virulence in mice following ocular infection. To our knowledge, this is the first report directly indicating the biological significance of Us8A alone and its regulation during the HSV-1 life cycle.
Another striking observation in this study was that the phosphorylation of Us8A at Ser-61 was required for efficient virulence in mice following ocular infection, but not intracranial infection. As described above, virulence in mice following intracranial infection appears to depend on the ability of HSV-1 to replicate in the CNS (1), and therefore, our results suggest that the phosphorylation of Us8A Ser-61 has no effect on viral replication in the CNS. In contrast, virulence in mice following ocular infection appears to result from the sum of the multiple capacities of HSV-1 to replicate in the eye, trigeminal ganglia, and brain; to gain access to the trigeminal ganglia from the eye; and to invade the CNS from the trigeminal ganglia (1). In this study, we also showed that (i) the mutation in Us8A that precludes phosphorylation at Ser-61 had no effect on viral replication in the eyes of ocularly infected mice at 1 to 3 days postinfection, during which time viral replication in the trigeminal ganglia reached a plateau; (ii) the mutation had no effect on viral replication in the trigeminal ganglia at 1 and 2 days postinfection but significantly reduced viral replication at 3 and 4 days postinfection; and (iii) the mutation in Us8A that mimics constitutive phosphorylation at Ser-61 restored wild-type viral replication in the trigeminal ganglia. Collectively, this series of observations suggests that the phosphorylation of Us8A Ser-61 plays no obvious role in viral replication in the eyes in these early stages of infection or in the viral transport from the eyes to the trigeminal ganglia but is required for efficient viral replication in the trigeminal ganglia. In support of this, the mutation that precludes the phosphorylation of Us8A Ser-61 also significantly reduced viral replication in the eyes of ocularly infected mice at 4 days postinfection, when the virus is thought to return from the trigeminal ganglia, and in the brains at 3 and 4 days postinfection. Furthermore, the phosphomimetic mutation at Us8A Ser-61 restored wild-type viral replication in the eyes at 4 days postinfection and in the brains at 3 and 4 days postinfection. The reduced viral replication in the trigeminal ganglia caused by the preclusion of Us8A Ser-61 phosphorylation may lead to reduced viral yields in the eyes at 4 days postinfection and in the brains at 3 and 4 days postinfection. Taken together with the data in this study showing that Us3 is a major kinase responsible for mediating the phosphorylation of Us8A Ser-61 in infected cells, it seems likely that the Us3 phosphorylation of Us8A Ser-61 regulates viral neuroinvasiveness, especially the viral capacity to replicate in the trigeminal ganglia, which is one of the critical aspects for viral invasion of the CNS from peripheral sites. In support of this hypothesis, it has been reported that Us3 is critical for viral neuroinvasiveness in mice following ocular infection (26). Us8A may be one of the targets of Us3 for the regulation of viral neuroinvasiveness. In addition, Negatsch et al. previously reported that glutamine replacement at the stop codon of Us8A in the HSV-1 strain KOS led to an extension of the predicted protein from 159 to 190 amino acids compared to HSV-1 strains 17 (50), H129, McKrae, and F (Fig. 11A). Interestingly, it has been reported that HSV-1 KOS exhibits phenotypes similar to that of a recombinant HSV-1(F) strain carrying a mutation to preclude phosphorylation of Us8A Ser-61. Thus, HSV-1 KOS grows in cell cultures and is virulent in mice following intracranial infection, similar to other HSV-1 strains (17, F, H129, and McKrae), whereas HSV-1 KOS is several orders of magnitude less neuroinvasive than the other HSV-1 strains (51–54). These observations, along with our results, raise the interesting possibility that the C-terminal 31-amino-acid extension of Us8A from HSV-1 KOS may affect the function of Us8A to regulate viral neuroinvasiveness, similar to the S61A mutation in HSV-1(F) Us8A shown in this study, and may be one of the polymorphic variations seen in HSV-1 KOS involved in the attenuation of its viral neuroinvasiveness.
FIG 11.

Sequence alignment of the Us8A protein homologues from HSV-1(F), HSV-1(17), HSV-1(KOS), HSV-2(HG52), and HSV-2(SD90e) strains. Shown are the stop codon (asterisks) (A) and Us8A Ser-61 (B) of HSV-1(F) and the corresponding residues of other HSV-1 and HSV-2 strains. The residues conserved in the sequences are shaded.
In general, identification of the physiological substrate of a viral protein kinase and its phosphorylation sites requires the demonstration that the substrate and its phosphorylation sites are specifically and directly phosphorylated by the kinase in vitro, and that the phosphorylation of the substrate is altered in cells infected with a mutant virus lacking the protein kinase activity. In this study, we showed that a kinase-dead mutation in Us3 significantly reduced the phosphorylation of Us8A Ser-61 in infected cells. However, we could not detect the direct phosphorylation of Us8A by Us3 in vitro. One explanation for this might be that a protein kinase(s) other than Us3 phosphorylates Us8A Ser-61 in infected cells and this kinase is induced by the presence of Us3 kinase activity. In support of this hypothesis, it has been reported that Us3 activates cellular protein kinase A (PKA) in infected cells (55), and in silico analysis of the Us8A amino acid sequence (using GPS 3.0 kinase-specific phosphorylation site prediction [56, 57; http://gps.biocuckoo.org/online.php]) predicted that Ser-61 of Us8A could be phosphorylated by PKA. Protein kinases other than PKA that have regulation and substrate specificity similar to PKA in HSV-1-infected cells and/or PKA might directly phosphorylate Us8A Ser-61. However, there is a possibility that Us3 directly phosphorylates Us8 at Ser-61 in HSV-1-infected cells. In the in vitro kinase assay used in this study, the kinase and substrate, Us3 and Us8A, were tagged with GST and MBP, respectively, and this tagging may induce conformational changes of the proteins, which may prevent conformation-dependent phosphorylation of Us8A by Us3.
Us8A Ser-61 is conserved among HSV-1 strains, but not HSV-2 strains (Fig. 11B). This suggests that the Us3-mediated phosphorylation of Us8A Ser-61 and its relevance to the regulation of viral neuroinvasiveness, clarified in this study, are specific to HSV-1. In agreement with this, it has been reported that HSV-1 Us3 and HSV-2 Us3 differ in their contributions to viral neuroinvasiveness in mice (26, 58). Recently, it has been reported that the replacement of HSV-1 Us3 with HSV-2 Us3 compensated for the phosphorylation of HSV-1 gB Thr-887, which is not conserved in HSV-2 gB, leading to the effect of its phosphorylation and the downregulation of the cell surface expression of gB in infected cells (59). These observations suggest that phosphorylation sites in viral proteins may have evolved to enable viral kinases to acquire new functions in viral protein regulation. HSV-1 Us8A Ser-61 may be such a phosphorylation site, similar to HSV-1 gB Thr-887.
We used another HSV-1-BAC clone (pYEbac102) (32) to generate and analyze HSV-1 mutants in previous studies. In the course of the studies, we realized that the excision of the BAC sequence from the HSV-1 genome was required for efficient viral neuroinvasiveness in mice following peripheral infection (data not shown). Although the BAC sequence can be excised from pYEbac102, it requires coinfection of cells with a recombinant virus reconstituted from pYEbac102 and adenovirus expressing Cre recombinase, followed by plaque purification (32). In this study, analyses of HSV-1 neuroinvasiveness using HSV-1 Us8A mutants were necessary, and therefore, we generated a new self-excisable HSV-1-BAC clone (pYEbac5002). Using pYEbac5002, the excision of the BAC sequence from the HSV-1 genome was much easier than that using pYEbac102, as described previously (35). Since we have not determined the entire sequence of pYEbac5002, there is a possibility that this new HSV-1-BAC clone may have a small deletion(s) that has no obvious effect on viral replication in cell cultures and neurovirulence in mice following intracranial infection.
Thus far, information on HSV-1 factors regulating neuroinvasiveness has been limited because it is difficult to investigate the efficiency of HSV-1 transport from peripheral sites to the CNS in vivo. Therefore, previous studies have identified HSV-1 factors involved in neuroinvasiveness based on the fact that (i) a mutation in the corresponding HSV-1 factor has no effect on virulence in mice following intracranial infection and (ii) the mutation significantly reduced virulence in mice following peripheral infection, and these factors include only gD, ICP0, Us9, and gB (60–64). Therefore, the identification of Us8A as a novel HSV-1 factor for neuroinvasiveness in this study provides insight into the mechanisms by which HSV-1 regulates viral neuroinvasiveness and contributes to our understanding of HSV-1 pathogenesis.
ACKNOWLEDGMENTS
We thank Gregory A. Smith for providing pCREin.
Funding for this work was provided by the Funding Program for Next Generation World-Leading Researchers from the Japan Society for the Promotion of Science (JSPS) to Yasushi Kawaguchi; Grants for Scientific Research from JSPS to Yasushi Kawaguchi, Akihisa Kato, and Jun Arii; Grants for Scientific Research on Innovative Areas from the Ministry of Education, Culture, Science, Sports and Technology (MEXT) of Japan to Yasushi Kawaguchi and Akihisa Kato; a contract research fund for the Japan Initiative for Global Research Network on Infectious Diseases (J-GRID) program from MEXT and the Japan Agency for Medical Research and Development (AMED) to Yasushi Kawaguchi; grants from the Takeda Science Foundation to Yasushi Kawaguchi, Akihisa Kato, and Jun Arii; a grant from the Mitsubishi Foundation to Yasushi Kawaguchi; and grants from the Naito Foundation, the Kao Foundation for Arts and Science, and the Life Science Foundation of Japan to Akihisa Kato.
REFERENCES
- 1.Roizman B, Knipe DM, Whitley RJ. 2013. Herpes simplex viruses, p 1823–1897. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott-Williams &Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Post LE, Roizman B. 1981. A generalized technique for deletion of specific genes in large genomes: alpha gene 22 of herpes simplex virus 1 is not essential for growth. Cell 25:227–232. doi: 10.1016/0092-8674(81)90247-6. [DOI] [PubMed] [Google Scholar]
- 3.Mavromara-Nazos P, Ackermann M, Roizman B. 1986. Construction and properties of a viable herpes simplex virus 1 recombinant lacking coding sequences of the alpha 47 gene. J Virol 60:807–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weber PC, Levine M, Glorioso JC. 1987. Rapid identification of nonessential genes of herpes simplex virus type 1 by Tn5 mutagenesis. Science 236:576–579. doi: 10.1126/science.3033824. [DOI] [PubMed] [Google Scholar]
- 5.Longnecker R, Chatterjee S, Whitley RJ, Roizman B. 1987. Identification of a herpes simplex virus 1 glycoprotein gene within a gene cluster dispensable for growth in cell culture. Proc Natl Acad Sci U S A 84:4303–4307. doi: 10.1073/pnas.84.12.4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neidhardt H, Schroder CH, Kaerner HC. 1987. Herpes simplex virus type 1 glycoprotein E is not indispensable for viral infectivity. J Virol 61:600–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson DC, Ligas MW. 1988. Herpes simplex viruses lacking glycoprotein D are unable to inhibit virus penetration: quantitative evidence for virus-specific cell surface receptors. J Virol 62:4605–4612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Longnecker R, Roizman B. 1986. Generation of an inverting herpes simplex virus 1 mutant lacking the L-S junction A sequences, an origin of DNA synthesis, and several genes including those specifying glycoprotein E and the alpha 47 gene. J Virol 58:583–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Longnecker R, Roizman B. 1987. Clustering of genes dispensable for growth in culture in the S component of the HSV-1 genome. Science 236:573–576. doi: 10.1126/science.3033823. [DOI] [PubMed] [Google Scholar]
- 10.Ligas MW, Johnson DC. 1988. A herpes simplex virus mutant in which glycoprotein D sequences are replaced by beta-galactosidase sequences binds to but is unable to penetrate into cells. J Virol 62:1486–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nishiyama Y. 2004. Herpes simplex virus gene products: the accessories reflect her lifestyle well. Rev Med Virol 14:33–46. doi: 10.1002/rmv.409. [DOI] [PubMed] [Google Scholar]
- 12.Fruh K, Ahn K, Djaballah H, Sempe P, van Endert PM, Tampe R, Peterson PA, Yang Y. 1995. A viral inhibitor of peptide transporters for antigen presentation. Nature 375:415–418. doi: 10.1038/375415a0. [DOI] [PubMed] [Google Scholar]
- 13.York IA, Roop C, Andrews DW, Riddell SR, Graham FL, Johnson DC. 1994. A cytosolic herpes simplex virus protein inhibits antigen presentation to CD8+ T lymphocytes. Cell 77:525–535. doi: 10.1016/0092-8674(94)90215-1. [DOI] [PubMed] [Google Scholar]
- 14.Hill A, Jugovic P, York I, Russ G, Bennink J, Yewdell J, Ploegh H, Johnson D. 1995. Herpes simplex virus turns off the TAP to evade host immunity. Nature 375:411–415. doi: 10.1038/375411a0. [DOI] [PubMed] [Google Scholar]
- 15.Aubert M, Krantz EM, Jerome KR. 2006. Herpes simplex virus genes Us3, Us5, and Us12 differentially regulate cytotoxic T lymphocyte-induced cytotoxicity. Viral Immunol 19:391–408. doi: 10.1089/vim.2006.19.391. [DOI] [PubMed] [Google Scholar]
- 16.Imai T, Koyanagi N, Ogawa R, Shindo K, Suenaga T, Sato A, Arii J, Kato A, Kiyono H, Arase H, Kawaguchi Y. 2013. Us3 kinase encoded by herpes simplex virus 1 mediates downregulation of cell surface major histocompatibility complex class I and evasion of CD8+ T cells. PLoS One 8:e72050. doi: 10.1371/journal.pone.0072050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frank I, Friedman HM. 1989. A novel function of the herpes simplex virus type 1 Fc receptor: participation in bipolar bridging of antiviral immunoglobulin G. J Virol 63:4479–4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson DC, Frame MC, Ligas MW, Cross AM, Stow ND. 1988. Herpes simplex virus immunoglobulin G Fc receptor activity depends on a complex of two viral glycoproteins, gE and gI. J Virol 62:1347–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hanke T, Graham FL, Lulitanond V, Johnson DC. 1990. Herpes simplex virus IgG Fc receptors induced using recombinant adenovirus vectors expressing glycoproteins E and I. Virology 177:437–444. doi: 10.1016/0042-6822(90)90507-N. [DOI] [PubMed] [Google Scholar]
- 20.Bell S, Cranage M, Borysiewicz L, Minson T. 1990. Induction of immunoglobulin G Fc receptors by recombinant vaccinia viruses expressing glycoproteins E and I of herpes simplex virus type 1. J Virol 64:2181–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sagou K, Imai T, Sagara H, Uema M, Kawaguchi Y. 2009. Regulation of the catalytic activity of herpes simplex virus 1 protein kinase Us3 by autophosphorylation and its role in pathogenesis. J Virol 83:5773–5783. doi: 10.1128/JVI.00103-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sears AE, Halliburton IW, Meignier B, Silver S, Roizman B. 1985. Herpes simplex virus 1 mutant deleted in the alpha 22 gene: growth and gene expression in permissive and restrictive cells and establishment of latency in mice. J Virol 55:338–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meignier B, Longnecker R, Mavromara-Nazos P, Sears AE, Roizman B. 1988. Virulence of and establishment of latency by genetically engineered deletion mutants of herpes simplex virus 1. Virology 162:251–254. doi: 10.1016/0042-6822(88)90417-5. [DOI] [PubMed] [Google Scholar]
- 24.Kato A, Shindo K, Maruzuru Y, Kawaguchi Y. 2014. Phosphorylation of a herpes simplex virus 1 dUTPase by a viral protein kinase, Us3, dictates viral pathogenicity in the central nervous system but not at the periphery. J Virol 88:2775–2785. doi: 10.1128/JVI.03300-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morimoto T, Arii J, Tanaka M, Sata T, Akashi H, Yamada M, Nishiyama Y, Uema M, Kawaguchi Y. 2009. Differences in the regulatory and functional effects of the Us3 protein kinase activities of herpes simplex virus 1 and 2. J Virol 83:11624–11634. doi: 10.1128/JVI.00993-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koyanagi N, Imai T, Arii J, Kato A, Kawaguchi Y. 2014. Role of herpes simplex virus 1 Us3 in viral neuroinvasiveness. Microbiol Immunol 58:31–37. doi: 10.1111/1348-0421.12108. [DOI] [PubMed] [Google Scholar]
- 27.Kolb AW, Adams M, Cabot EL, Craven M, Brandt CR. 2011. Multiplex sequencing of seven ocular herpes simplex virus type-1 genomes: phylogeny, sequence variability, and SNP distribution. Invest Ophthalmol Vis Sci 52:9061–9073. doi: 10.1167/iovs.11-7812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Georgopoulou U, Michaelidou A, Roizman B, Mavromara-Nazos P. 1993. Identification of a new transcriptional unit that yields a gene product within the unique sequences of the short component of the herpes simplex virus 1 genome. J Virol 67:3961–3968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Georgopoulou U, Kakkanas A, Miriagou V, Michaelidou A, Mavromara P. 1995. Characterization of the US8.5 protein of herpes simplex virus. Arch Virol 140:2227–2241. doi: 10.1007/BF01323242. [DOI] [PubMed] [Google Scholar]
- 30.Kobayashi R, Kato A, Oda S, Koyanagi N, Oyama M, Kozuka-Hata H, Arii J, Kawaguchi Y. 2015. Function of the herpes simplex virus 1 small capsid protein VP26 is regulated by phosphorylation at a specific site. J Virol 89:6141–6147. doi: 10.1128/JVI.00547-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kato A, Tsuda S, Liu Z, Kozuka-Hata H, Oyama M, Kawaguchi Y. 2014. Herpes simplex virus 1 protein kinase Us3 phosphorylates viral dUTPase and regulates its catalytic activity in infected cells. J Virol 88:655–666. doi: 10.1128/JVI.02710-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tanaka M, Kagawa H, Yamanashi Y, Sata T, Kawaguchi Y. 2003. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J Virol 77:1382–1391. doi: 10.1128/JVI.77.2.1382-1391.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ejercito PM, Kieff ED, Roizman B. 1968. Characterization of herpes simplex virus strains differing in their effects on social behaviour of infected cells. J Gen Virol 2:357–364. doi: 10.1099/0022-1317-2-3-357. [DOI] [PubMed] [Google Scholar]
- 34.Kato A, Tanaka M, Yamamoto M, Asai R, Sata T, Nishiyama Y, Kawaguchi Y. 2008. Identification of a physiological phosphorylation site of the herpes simplex virus 1-encoded protein kinase Us3 which regulates its optimal catalytic activity in vitro and influences its function in infected cells. J Virol 82:6172–6189. doi: 10.1128/JVI.00044-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith GA, Enquist LW. 2000. A self-recombining bacterial artificial chromosome and its application for analysis of herpesvirus pathogenesis. Proc Natl Acad Sci U S A 97:4873–4878. doi: 10.1073/pnas.080502497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tischer BK, Smith GA, Osterrieder N. 2010. En passant mutagenesis: a two step markerless red recombination system. Methods Mol Biol 634:421–430. doi: 10.1007/978-1-60761-652-8_30. [DOI] [PubMed] [Google Scholar]
- 37.Tischer BK, von Einem J, Kaufer B, Osterrieder N. 2006. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197. doi: 10.2144/000112096. [DOI] [PubMed] [Google Scholar]
- 38.Sugimoto K, Uema M, Sagara H, Tanaka M, Sata T, Hashimoto Y, Kawaguchi Y. 2008. Simultaneous tracking of capsid, tegument, and envelope protein localization in living cells infected with triply fluorescent herpes simplex virus 1. J Virol 82:5198–5211. doi: 10.1128/JVI.02681-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kato A, Yamamoto M, Ohno T, Kodaira H, Nishiyama Y, Kawaguchi Y. 2005. Identification of proteins phosphorylated directly by the Us3 protein kinase encoded by herpes simplex virus 1. J Virol 79:9325–9331. doi: 10.1128/JVI.79.14.9325-9331.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kato A, Yamamoto M, Ohno T, Tanaka M, Sata T, Nishiyama Y, Kawaguchi Y. 2006. Herpes simplex virus 1-encoded protein kinase UL13 phosphorylates viral Us3 protein kinase and regulates nuclear localization of viral envelopment factors UL34 and UL31. J Virol 80:1476–1486. doi: 10.1128/JVI.80.3.1476-1486.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kawaguchi Y, Kato K, Tanaka M, Kanamori M, Nishiyama Y, Yamanashi Y. 2003. Conserved protein kinases encoded by herpesviruses and cellular protein kinase cdc2 target the same phosphorylation site in eukaryotic elongation factor 1delta. J Virol 77:2359–2368. doi: 10.1128/JVI.77.4.2359-2368.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kato A, Arii J, Shiratori I, Akashi H, Arase H, Kawaguchi Y. 2009. Herpes simplex virus 1 protein kinase Us3 phosphorylates viral envelope glycoprotein B and regulates its expression on the cell surface. J Virol 83:250–261. doi: 10.1128/JVI.01451-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kato A, Liu Z, Minowa A, Imai T, Tanaka M, Sugimoto K, Nishiyama Y, Arii J, Kawaguchi Y. 2011. Herpes simplex virus 1 protein kinase Us3 and major tegument protein UL47 reciprocally regulate their subcellular localization in infected cells. J Virol 85:9599–9613. doi: 10.1128/JVI.00845-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shiba C, Daikoku T, Goshima F, Takakuwa H, Yamauchi Y, Koiwai O, Nishiyama Y. 2000. The UL34 gene product of herpes simplex virus type 2 is a tail-anchored type II membrane protein that is significant for virus envelopment. J Gen Virol 81:2397–2405. doi: 10.1099/0022-1317-81-10-2397. [DOI] [PubMed] [Google Scholar]
- 45.Kawaguchi Y, Van Sant C, Roizman B. 1997. Herpes simplex virus 1 alpha regulatory protein ICP0 interacts with and stabilizes the cell cycle regulator cyclin D3. J Virol 71:7328–7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cunningham C, Davison AJ. 1993. A cosmid-based system for constructing mutants of herpes simplex virus type 1. Virology 197:116–124. doi: 10.1006/viro.1993.1572. [DOI] [PubMed] [Google Scholar]
- 47.Leader DP. 1993. Viral protein kinases and protein phosphatases. Pharmacol Ther 59:343–389. doi: 10.1016/0163-7258(93)90075-O. [DOI] [PubMed] [Google Scholar]
- 48.Leader DP, Deana AD, Marchiori F, Purves FC, Pinna LA. 1991. Further definition of the substrate specificity of the alpha-herpesvirus protein kinase and comparison with protein kinases A and C. Biochim Biophys Acta 1091:426–431. doi: 10.1016/0167-4889(91)90210-O. [DOI] [PubMed] [Google Scholar]
- 49.Purves FC, Deana AD, Marchiori F, Leader DP, Pinna LA. 1986. The substrate specificity of the protein kinase induced in cells infected with herpesviruses: studies with synthetic substrates indicate structural requirements distinct from other protein kinases. Biochim Biophys Acta 889:208–215. doi: 10.1016/0167-4889(86)90106-0. [DOI] [PubMed] [Google Scholar]
- 50.Negatsch A, Mettenleiter TC, Fuchs W. 2011. Herpes simplex virus type 1 strain KOS carries a defective US9 and a mutated US8A gene. J Gen Virol 92:167–172. doi: 10.1099/vir.0.026484-0. [DOI] [PubMed] [Google Scholar]
- 51.Strelow LI, Laycock KA, Jun PY, Rader KA, Brady RH, Miller JK, Pepose JS, Leib DA. 1994. A structural and functional comparison of the latency-associated transcript promoters of herpes simplex virus type 1 strains KOS and McKrae. J Gen Virol 75:2475–2480. doi: 10.1099/0022-1317-75-9-2475. [DOI] [PubMed] [Google Scholar]
- 52.Thompson RL, Cook ML, Devi-Rao GB, Wagner EK, Stevens JG. 1986. Functional and molecular analyses of the avirulent wild-type herpes simplex virus type 1 strain KOS. J Virol 58:203–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dix RD, McKendall RR, Baringer JR. 1983. Comparative neurovirulence of herpes simplex virus type 1 strains after peripheral or intracerebral inoculation of BALB/c mice. Infect Immun 40:103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang H, Davido DJ, Morrison LA. 2013. HSV-1 strain McKrae is more neuroinvasive than HSV-1 KOS after corneal or vaginal inoculation in mice. Virus Res 173:436–440. doi: 10.1016/j.virusres.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Benetti L, Roizman B. 2004. Herpes simplex virus protein kinase US3 activates and functionally overlaps protein kinase A to block apoptosis. Proc Natl Acad Sci U S A 101:9411–9416. doi: 10.1073/pnas.0403160101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang D, Harry GJ, Blackshear PJ, Zeldin DC. 2008. G-protein pathway suppressor 2 (GPS2) interacts with the regulatory factor X4 variant 3 (RFX4_v3) and functions as a transcriptional co-activator. J Biol Chem 283:8580–8590. doi: 10.1074/jbc.M708209200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xue Y, Zhou F, Zhu M, Ahmed K, Chen G, Yao X. 2005. GPS: a comprehensive www server for phosphorylation sites prediction. Nucleic Acids Res 33:W184–W187. doi: 10.1093/nar/gki393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nishiyama Y, Yamada Y, Kurachi R, Daikoku T. 1992. Construction of a US3 lacZ insertion mutant of herpes simplex virus type 2 and characterization of its phenotype in vitro and in vivo. Virology 190:256–268. doi: 10.1016/0042-6822(92)91212-D. [DOI] [PubMed] [Google Scholar]
- 59.Shindo K, Kato A, Koyanagi N, Sagara H, Arii J, Kawaguchi Y. 2015. Characterization of a herpes simplex virus 1 (HSV-1) chimera in which the Us3 protein kinase gene is replaced with the HSV-2 Us3 gene. J Virol 90:457–473. doi: 10.1128/JVI.02376-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nishiyama Y, Kurachi R, Daikoku T, Umene K. 1993. The US 9, 10, 11, and 12 genes of herpes simplex virus type 1 are of no importance for its neurovirulence and latency in mice. Virology 194:419–423. doi: 10.1006/viro.1993.1279. [DOI] [PubMed] [Google Scholar]
- 61.Izumi KM, Stevens JG. 1990. Molecular and biological characterization of a herpes simplex virus type 1 (HSV-1) neuroinvasiveness gene. J Exp Med 172:487–496. doi: 10.1084/jem.172.2.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Arii J, Wang J, Morimoto T, Suenaga T, Akashi H, Arase H, Kawaguchi Y. 2010. A single-amino-acid substitution in herpes simplex virus 1 envelope glycoprotein B at a site required for binding to the paired immunoglobulin-like type 2 receptor alpha (PILRalpha) abrogates PILRalpha-dependent viral entry and reduces pathogenesis. J Virol 84:10773–10783. doi: 10.1128/JVI.01166-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Van Sant C, Kawaguchi Y, Roizman B. 1999. A single amino acid substitution in the cyclin D binding domain of the infected cell protein no. 0 abrogates the neuroinvasiveness of herpes simplex virus without affecting its ability to replicate. Proc Natl Acad Sci U S A 96:8184–8189. doi: 10.1073/pnas.96.14.8184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Polcicova K, Biswas PS, Banerjee K, Wisner TW, Rouse BT, Johnson DC. 2005. Herpes keratitis in the absence of anterograde transport of virus from sensory ganglia to the cornea. Proc Natl Acad Sci U S A 102:11462–11467. doi: 10.1073/pnas.0503230102. [DOI] [PMC free article] [PubMed] [Google Scholar]