

ABSTRACT
Due to enzootic infections in poultry and persistent human infections in China, influenza A (H7N9) virus has remained a public health threat. The Yangtze River Delta region, which is located in eastern China, is well recognized as the original source for H7N9 outbreaks. Based on the evolutionary analysis of H7N9 viruses from all three outbreak waves since 2013, we identified the Pearl River Delta region as an additional H7N9 outbreak source. H7N9 viruses are repeatedly introduced from these two sources to the other areas, and the persistent circulation of H7N9 viruses occurs in poultry, causing continuous outbreak waves. Poultry movements may contribute to the geographic expansion of the virus. In addition, the AnH1 genotype, which was predominant during wave 1, was replaced by JS537, JS18828, and AnH1887 genotypes during waves 2 and 3. The establishment of a new source and the continuous evolution of the virus hamper the elimination of H7N9 viruses, thus posing a long-term threat of H7N9 infection in humans. Therefore, both surveillance of H7N9 viruses in humans and poultry and supervision of poultry movements should be strengthened.
IMPORTANCE Since its occurrence in humans in eastern China in spring 2013, the avian H7N9 viruses have been demonstrating the continuing pandemic threat posed by the current influenza ecosystem in China. As the viruses are silently circulated in poultry, with potentially severe outcomes in humans, H7N9 virus activity in humans in China is very important to understand. In this study, we identified a newly emerged H7N9 outbreak source in the Pearl River Delta region. Both sources in the Yangtze River Delta region and the Pearl River Delta region have been established and found to be responsible for the H7N9 outbreaks in mainland China.
INTRODUCTION
Since the first emergence of influenza A (H7N9) virus in Eastern China (1), the novel virus has caused three outbreak waves in humans. The first outbreak emerged in late February 2013 and decreased rapidly after the middle of April 2013. The second wave began in October 2013, peaked in January 2014, and decreased in late February 2014. The number of H7N9 cases increased again in late 2014 and peaked in January 2015 during the third wave (Fig. 1A). By the end of September 2015, a total of 17 provinces or municipalities had been affected in Mainland China (Fig. 1), and 656 H7N9 cases had been reported, 268 of which were fatal. Imported cases were documented in Hong Kong SAR, in Taiwan, China, in Malaysia, and in Canada. Since October 2015, over 60 additional laboratory-confirmed cases of human infection with avian influenza A (H7N9) virus have been notified, indicating the beginning of the fourth outbreak wave.
FIG 1.

H7N9 case distribution from wave 1 to wave 3 in mainland China. From 9 Feb 2013 to 21 August 2015, a total of 656 H7N9 cases have been reported. (A) Epicurve of human H7N9 cases by date of illness onset. The cases were accumulated weekly. Colors indicate the H7N9-affected provinces. (B to D) H7N9 case distribution from the emergence of H7N9 to September 2013 (wave 1) (B), from October 2013 to July 2014 (wave 2) (C), and from October 2014 to August 2015 (wave 3) (D). H7N9-affected provinces are highlighted in light green. Solid violet circles vary from smallest to largest, indicating that the case number of H7N9 patients increased from 1 to >100.
Previous studies on the H7N9 virus have elucidated the potential origin and source of the virus (2–6). Dynamic reassortment among H7N9 and H9N2 viruses has been reported to increase the diversity of the virus (3–5, 7, 8). Frequent poultry movement and the silent spread of H7N9 viruses in poultry might have contributed to the H7N9 virus being enzootic in China (5). Several mammal-adapted mutations detected in H7N9 viruses have enhanced the threat of the novel virus in humans. It is important to monitor the virus evolution for pandemic preparedness. Hence, we conducted a panoramic molecular epidemiology study to analyze the three waves of H7N9 viruses.
MATERIALS AND METHODS
Virus isolation and subtyping.
Fifty-three human H7N9 and avian origin H7N9 viruses have been previously sequenced (4). Between May 2013 and May 2015, a total of 232 H7N9 viruses were isolated from H7N9-positive human swabs (n = 189) or environmental samples (n = 43) confirmed by real-time PCR results in this study.
Virus isolation was conducted in a biosafety level 3 facility by inoculating 0.2-ml original samples allantoically into 9- to 11-day specific-pathogen-free (SPF) embryonated chicken eggs. After incubation at 37°C for 48 to 72 h, the allantoic fluids were harvested and tested for hemagglutinin (HA) agglutination by turkey red blood cells (TRBC). RNA was extracted for each virus using the RNeasy minikit (Qiagen, Hilden, Germany), and the subtype was further confirmed by sequencing.
Genome sequencing.
Based on the real-time reverse transcription (RT)-PCR results, H7N9-positive RNAs were selected for Sanger sequencing. Gene segments were amplified using the Qiagen OneStep RT-PCR kit. A total of 48 primer pairs were used to generate PCR amplicons among 378 and 1,123 bp in length for full-genome sequencing. Primer sequences are available from the authors on request. Amplified PCR products were purified using ExoSAP-IT reagent (USB, Cleveland, OH, USA). Complete genome sequencing was performed with an ABI 3730XL automatic DNA analyzer (Applied Biosystems, Foster City, CA, USA) using the ABI BigDye Terminator v3.1 cycle sequencing kit (Applied Biosystems), according to the manufacturer's recommendations.
Sequence alignment and phylogenetic analysis.
The nucleotide sequences generated were assembled using Lasergene (Dnastar, Inc.) and combined with publicly available sequences of influenza A virus available in Global Initiative on Sharing All Influenza Data (GISAID) databases (http://www.gisaid.org). Reference sequences, which were phylogenetically related to the H7N9 viruses and had full-length sequences, were included with our sequences for phylogenic analysis.
A maximum likelihood phylogenetic tree for nucleotide sequences of each gene of selected influenza viruses was constructed under the GTRCAT model (a computational workaround for the general time-reversible model of nucleotide substitution under the Gamma model of rate heterogeneity) with 1,000 bootstrap replicates, using RAxML (9) via CIPRES Science Gateway (10). According to the bootstrap value and branch length, we classified each viral gene into clades. Combinations of the clades of all six internal genes of an isolate were defined as one genotype.
For BEAST analysis, virus sequences without sampling date information were excluded and the missing dates were set as the middle of month. According to the virus isolation locations, all sequences were classified into three regions as the Yangtze River Delta, the Pearl River Delta, and other regions.
The time-resolved Bayesian phylogenetic tree of HA gene was estimated by BEAST v1.8.2. The GTR + Gamma + Invariant substitution model was selected using jModelTest-2.1.4. The uncorrelated log-normal relaxed-clock model was employed to account for lineage-specific rate heterogeneity. The coalescent Bayesian sky grid model was used. Phylogeographic patterns were estimated using a symmetric continuous-time Markov chain (CTMC) model for discrete state reconstructions. The prior ucld.mean was set as uniform distribution. Markov chain Monte Carlo (MCMC) was run 3 times, each with 50 million steps sampled every 5,000 steps with burn-in of 5 million steps and samples combined. The effective sample size (ESS) of all priors was greater than 200. The migration rates were calculated using posterior analysis of coalescent trees (PACT) from Github.
Nucleotide sequence accession numbers.
Full-genome sequences of 232 H7N9 viruses in our study have been deposited into GISAID with the accession numbers EPI626978 to EPI628832.
RESULTS
Evolutionary relationships of hemagglutinin genes of H7N9 viruses during waves 1 to 3.
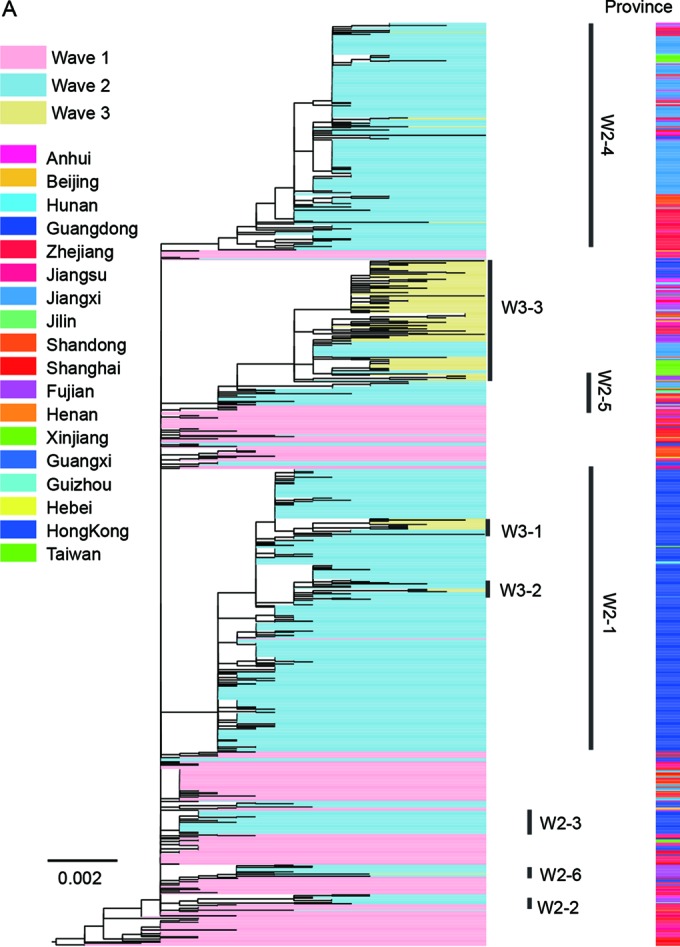
Based on both RAxML and BEAST analyses of the H7 hemagglutinin (HA) genes (Fig. 2 and 3), all viruses from all affected regions in wave 1 clustered into one group and originated exclusively from the Yangtze River Delta region. This result further supported that the Yangtze River Delta region was the source responsible for the wave 1 H7N9 outbreak (4). In contrast to wave 1, the HA gene of wave 2 viruses clustered into 6 clades (W2-1, W2-2, W2-3, W2-4, W2-5, and W2-6 [Fig. 2A]). All outgroup viruses of these six clades were detected from wave 1, indicating that all H7N9 viruses of wave 2 originated exclusively from wave 1. In each clade, H7N9 viruses of human and avian origins shared numerous similarities and clustered together (Fig. 2D), further verifying the avian-to-human transmission of influenza A (H7N9) viruses.
FIG 2.

Phylogenetic trees of HA genes of influenza A (H7N9) viruses from wave 1 to wave 3 estimated using RAxML. Trees are presented in a circular format with the same topology in all panels. (A) H7N9 viruses collected during the first wave are highlighted with the background shading in pink, and those during the second and third waves are in light blue and light yellow, respectively. (B) H7N9 viruses isolated from the Yangtze River Delta regions, the Pearl River Delta regions, and other regions are shown in red, blue, and green, respectively. (C) Different colors indicate the provinces from which the corresponding H7N9 viruses came. (D) Black or gray denote the human or nonhuman H7N9 viruses, respectively. Reference viruses representing each subclade are listed in Table 7.
FIG 3.

Bayesian maximum clade credibility (MCC) phylogeny of H7 gene sequences of wave 1 to wave 3. All viruses isolated in the Yangtze River Delta region, the Pearl River Delta region, and other regions are highlighted in red, blue, and green, respectively. (A) BEAST tree of the H7N9 viruses estimated using the HA gene sequences. (B and C) Representative clades showing virus expansion from the Pearl River Delta region to other regions. (D and E) Representative clades showing virus expansion from the Yangtze River Delta region to the Pearl River Delta region. (F) Representative clades showing virus expansion from the Yangtze River Delta region to the Pearl River Delta region or other regions. (G) Virus migration rate among the Yangtze River Delta region, the Pearl River Delta region, and other regions. Colors in the solid circles correspond to the regions shown in the phylogenetic tree. Thicker arrows indicate the higher migration rate between two indicated regions. The migration rates were calculated using PACT.
Clade W2-1 included primarily viruses isolated in the Pearl River Delta region, including Guangdong province and Hong Kong SAR, China (Fig. 2B and C). In agreement with a previous study (5), clade W2-1 includes a large number of H7N9 viruses of both human and avian origins, suggesting that influenza A (H7N9) viruses may have become established in poultry during the second wave (Fig. 2D). Thus, the establishment of H7N9 in poultry caused the large number of human infections in wave 2 in this area. As shown in Fig. 2B and C, clade W2-1 viruses originated from the Yangtze River region and persisted in Guangdong Province. Some viruses of this clade spread to the Guangxi and Hunan provinces, indicating that another potential source in the Pearl River Delta region readily emerged during wave 2.
Viruses of other clades of wave 2 (W2-2, W2-3, W2-4, W2-5, and W2-6) contain viruses detected in multiple provinces, especially W2-4 and W2-5 (Fig. 2C). Human isolates from Jilin and Xinjiang, which are geographically far away from eastern China, involved clades W2-4 and W2-5, respectively. Outgroup viruses of these two clades were detected exclusively in the Yangtze River region (Fig. 2B). This result indicated that viruses of these clades may have originated and spread from the Yangtze River region to other regions. In addition, several viruses in clade W2-4 were isolated from Guangdong (Fig. 2B and 3E), suggesting the repeated introduction of H7N9 viruses from the Yangtze River region to the Pearl River Delta region.
The majority of viruses from wave 3 were clustered into three clades (W3-1, W3-2, and W3-3) (Fig. 2). The viruses of wave 3 were descended exclusively from the viruses of wave 2. Among the viruses, W3-1 and W3-2 viruses circulated primarily in Guangdong and originated from viruses of W2-1, indicating that the virus historically circulating in Guangdong during wave 2 was persistent in this area during wave 3. More importantly, viruses isolated from Fujian Province clustered into clade W3-1 (Fig. 2C and 3C), further supporting the notion that the Pearl River Delta region was an additional source of H7N9 outbreaks. W3-3 viruses that originated from W2-5 constituted the primary circulated cluster during wave 3. It contained viruses circulated in the Yangtze River Delta region and in Guangdong, providing evidence on the introduction of genes from the Yangtze River Delta region to the Pearl River Delta region (Fig. 2 and 3). Besides, clade W3-3 viruses caused human infections both in the Yangtze River Delta region and in other provinces, including northwestern and southeastern China (Fig. 2). This result strongly supports the contention that the Yangtze River Delta region was an important source for H7N9 outbreaks.
Evolutionary relationships of NA gene and six internal genes of H7N9 viruses during waves 1 to 3.
As shown in Fig. 4A, the phylogenetic tree of the neuraminidase (NA) gene (N9) exhibited a topology similar to that of the HA tree, with the wave 2 viruses divided into 6 corresponding clades and wave 3 viruses separated into 3 clades. Of note, six human H7N9 viruses isolated from the Fujian province [A/Fujian/1/2015(H7N9) and others] in wave 3 had HA genes descended from viruses detected in Guangdong (clade W3-1) and the NA genes grouped into clade W3-3, indicating that the H7N9 viruses were transmitted from Guangdong to Fujian and generated new variants.
FIG 4.

Evolution of NA (A) and six internal genes (B) of influenza A (H7N9) viruses from wave 1 to wave 3. RAxML was used to estimate the phylogenetic relationships. In the phylogenetic tree, H7N9 viruses collected during the first wave are highlighted with the background shading in pink, and those during the second and third waves are in light blue and light yellow, respectively. Colors in the right side of panel A indicate the provinces from which the corresponding H7N9 viruses came.
Six internal genes of all H7N9 viruses studied were broadly classified into multiple clades. The PB2 and PB1 genes were divided into four major clades, and the NP and M genes were divided into three clades. For the PA and NS genes, two major clades were clustered (Fig. 4B).
Classification of H7N9 genotypes during waves 1 to 3.
Based on different clade combinations of 6 internal genes, at least 93 genotypes were identified (Fig. 5). Most of the genotypes (83/93) were circulated only transiently. During wave 1, AnH1, JS1, and SH7 genotype viruses were the predominant viruses, while in wave 2, JS537, which was detected only once during wave 1, rapidly became a dominant H7N9 virus genotype. In addition, another six genotypes (JS18828, AnH1887, GD1, GD10, GD2, and GD429) were cocirculated as well; however, only two AnH1 viruses and one JS1 virus were detected during the early stage of wave 2. During wave 3, four genotypes, including JS18828, AnH1887, JS537, and JS1, were persistent from wave 2. Another 4 genotypes (GD1, GD10, GD2, and GD429), which dominated in wave 2, on the other hand, were not detected during this period.
FIG 5.

Timeline of influenza A (H7N9) virus genotypes during three outbreak waves. Ten predominant genotypes (GD429, GD2, GD10, GD1, AnH1887, JS18828, JS537, SH7, JS1, and AnH1) are listed on the left. All 83 transient genotypes are on the bottom line. Symbols represent the corresponding genotypes of H7N9 viruses and their time of isolation.
Formation and establishment of two outbreak sources of H7N9 viruses.
The genotype geographical distribution data identified the Pearl River Delta region as the subsource of H7N9 viruses, which were generated from the Yangtze River Delta region (Fig. 6). Six dominant genotypes, including AnH1, JS1, SH7, JS537, JS18828, and AnH1887, were generated in the Yangtze River Delta region and responsible for the majority of cases in other affected regions. Genotypes AnH1, JS1, JS537, and JS18828 were introduced into the Pearl River Delta region. Genotypes GD1, GD10, GD2, and GD429, which were generated by reassortment with local H9N2 viruses (11), circulated mainly in Guangdong Province. Genotypes GD1 and GD10 further spread to Guangxi Province and genotype GD429 to Hunan Province. Because the AnH1 genotype disappeared rapidly after wave 2 and JS537 genotype viruses were detected widely and persistently during all three waves, we speculated that JS537 might have replaced AnH1 genotype and become the predominant circulated genotype.
FIG 6.

Genotype flow of the influenza A (H7N9) viruses. All H7N9-affected regions are highlighted in light yellow. Two H7N9 sources, the Yangtze River Delta and the Pearl River Delta regions, are highlighted with larger and smaller dotted black triangles, respectively. Dominant genotypes in each source are listed below the corresponding triangles. Virus spreads are indicated by solid arrows. The dotted arrows indicate that the imported H7N9 cases occurred in Taiwan, China, in Malaysia, or in Canada.
As shown in Fig. 1B to D, the geographic distribution of H7N9 cases further supported the two sources of H7N9 outbreaks. During wave 1, the majority of H7N9 cases were distributed in the Yangtze River Delta region, including Jiangsu, Shanghai, and Zhejiang Provinces (Fig. 1A and B and Table 1). The H7N9 virus from the Yangtze River Delta region was introduced into Guangdong Province and caused the first human infection in Huizhou City at the end of wave 1 (early August 2013 [12]) (Fig. 1B). During waves 2 and 3, the H7N9 viruses were persistent and continually reassorted their six internal genes with local H9N2 viruses circulating in poultry in the Pearl River region. The progeny H7N9 viruses were disseminated to the neighbor provinces, such as Guangxi, Hunan, and Fujian Provinces. Supported by the distribution and expansion of H7N9 viruses, human cases in Guangdong increased and the largest numbers of H7N9 cases were reported in the same province during wave 2, followed by Zhejiang and Jiangsu Provinces (Fig. 1C and Table 1). Moreover, the majority of H7N9 cases were distributed along the southeastern coastal line provinces in wave 3, covering both the Yangtze River Delta and Pearl River Delta regions (Fig. 1D and Table 1). The Yangtze River Delta region persistently acted as the main source during all three waves and was responsible for the H7N9 cases in most affected regions, limited not only to the neighboring provinces but also to the distant provinces, such as Xinjiang in northwestern China and Jilin in northern China (Fig. 1B to D).
TABLE 1.
Geographic distribution of influenza A (H7N9) cases during the three waves
| Region and province or city | No. of cases |
|||
|---|---|---|---|---|
| First wave | Second wave | Third wave | Total | |
| Yangtze River Delta | 136 | 61 | 22 | 219 |
| Zhejiang | 41 | 46 | 14 | 101 |
| Jiangsu | 48 | 8 | 8 | 64 |
| Shanghai | 47 | 7 | 54 | |
| Pearl River Delta | 16 | 297 | 28 | 341 |
| Guangdong | 16 | 286 | 25 | 327 |
| Hongkong | 11 | 3 | 14 | |
| Other regions | 44 | 191 | 63 | 298 |
| Jiangxi | 8 | 126 | 134 | |
| Fujian | 3 | 16 | 27 | 46 |
| Anhui | 5 | 12 | 11 | 28 |
| Shandong | 13 | 10 | 5 | 28 |
| Hunan | 4 | 9 | 5 | 18 |
| Beijing | 4 | 1 | 1 | 6 |
| Henan | 3 | 3 | ||
| Hebei | 1 | 1 | ||
| Guangxi | 3 | 1 | 4 | |
| Guizhou | 1 | 1 | 2 | |
| Xinjiang | 3 | 11 | 14 | |
| Jilin | 7 | 7 | ||
| Hubei | 1 | 1 | ||
We further calculated the migration rates among the Yangtze River Delta region, the Pearl River Delta region, and the other regions. As shown in Fig. 3G and in Table 2, the migration rate from Yangtze River Delta region to other regions was the highest (mean, 0.94), further supporting the Yangtze River Delta region as the major H7N9 outbreak source. Besides, the migration rate from the Pearl River Delta region to the other regions is 0.25, indicating this region as another potential H7N9 outbreak source.
TABLE 2.
Posterior estimate of migration rates between regionsa
| Statistic | Posterior migration rate mean (95% interval) |
|---|---|
| YRD to PRD | 0.26 (0.16, 0.38) |
| YRD to OR | 0.94 (0.71, 1.2) |
| PRD to YRD | 0.11 (0.05, 0.2) |
| PRD to OR | 0.25 (0.19, 0.34) |
| OR to YRD | 0.38 (0.19, 0.62) |
| OR to PRD | 0.22 (0.12, 0.35) |
The migration rate is calculated as migration events per lineage per year between two regions, using PACT. Abbreviations: YRD, Yangtze River Delta; PRD, Pearl River Delta; OR, other region.
Host adaption mutations and antigenic characterization during waves 1 to 3.
Given the wide distributions of the viruses and the frequent reassortment among H7N9 and H9N2 viruses, we further analyzed the amino acid mutations associated with mammalian adaptation and antiviral drug resistance. The results are summarized in Table 3.
TABLE 3.
Key amino acid substitutions of influenza A (H7N9) viruses during the three wavesa
| Gene | Function | Mutation | Amino acid | First wave | Second wave | Third wave | Nonhuman isolate |
|---|---|---|---|---|---|---|---|
| HA | Altered receptor specificity | A160S/T | A | 97 | 166 | 92 | 490 |
| S/T | 1 | 3 | 0 | 0 | |||
| Favor mammalian adaptation, Receptor binding site | G186V | G | 3 | 0 | 0 | 0 | |
| V | 95 | 170 | 84 | 487 | |||
| A | 0 | 0 | 8 | 3 | |||
| Q226L/I | Q | 5 | 1 | 0 | 20 | ||
| L | 87 | 168 | 84 | 460 | |||
| I | 5 | 0 | 7 | 5 | |||
| S | 0 | 1 | 1 | 1 | |||
| P | 1 | 0 | 0 | 0 | |||
| NA | Related to drug resistance | E119V | E | 91 | 163 | 92 | 488 |
| V | 1 | 3 | 0 | 0 | |||
| R292K | R | 86 | 160 | 92 | 488 | ||
| K | 6 | 6 | 0 | 0 | |||
| PB2 | Mammalian host adaptation | D256G | D | 91 | 160 | 90 | 476 |
| G | 1 | 0 | 1 | 0 | |||
| Enhance the 627K and 701N function | K526R | K | 84 | 159 | 89 | 474 | |
| R | 8 | 1 | 2 | 2 | |||
| Altered replication efficiency and virulence in mice | Q591K | Q | 86 | 152 | 87 | 475 | |
| K | 4 | 4 | 3 | 1 | |||
| L | 0 | 3 | 0 | 0 | |||
| Increased virulence in mice | E627K | E | 29 | 32 | 32 | 475 | |
| K | 62 | 122 | 52 | 1 | |||
| Q | 0 | 0 | 1 | 0 | |||
| V | 0 | 2 | 2 | 0 | |||
| Enhanced transmission in guinea pigs | D701N | D | 83 | 146 | 88 | 474 | |
| N | 8 | 14 | 3 | 1 | |||
| Species-associated signature positions | K702R | K | 92 | 157 | 91 | 473 | |
| R | 0 | 3 | 0 | 3 | |||
| PB1 | Increased transmission in ferret | I368V | I | 11 | 0 | 0 | 6 |
| V | 75 | 161 | 92 | 467 | |||
| Increased replication in mammalian cells | L598P | L | 85 | 162 | 91 | 473 | |
| Q | 0 | 0 | 1 | 0 | |||
| M | 1 | 0 | 0 | 0 | |||
| PB1-F2 | Increased pathogenicity in mice | 87–90 amino acids in length | 11AA | 1 | 0 | 0 | 0 |
| 25AA | 11 | 1 | 0 | 24 | |||
| 27AA | 0 | 0 | 1 | 0 | |||
| 34AA | 0 | 0 | 11 | 45 | |||
| 57AA | 0 | 17 | 0 | 35 | |||
| 76AA | 3 | 0 | 1 | 13 | |||
| 79AA | 0 | 0 | 1 | 0 | |||
| 90AA | 71 | 144 | 76 | 355 | |||
| 101AA | 0 | 0 | 1 | 0 | |||
| Altered virulence and antivirus response in mice | N66S | N | 86 | 162 | 92 | 473 | |
| S | 0 | 2 | 0 | 0 | |||
| PA | Species-associated signature positions | V100A | V | 9 | 55 | 35 | 261 |
| A | 77 | 87 | 55 | 202 | |||
| I | 0 | 21 | 2 | 2 | |||
| T | 0 | 0 | 0 | 1 | |||
| Increase the polymerase activity in mice | L336M | L | 86 | 129 | 90 | 368 | |
| M | 0 | 34 | 2 | 98 | |||
| Species-associated signature positions | K356R | K | 0 | 35 | 2 | 99 | |
| R | 86 | 128 | 90 | 367 | |||
| S409N | S | 6 | 12 | 7 | 60 | ||
| N | 80 | 151 | 85 | 406 | |||
| NS1 | Altered virulence in mice | D92E | D | 89 | 164 | 92 | 476 |
| E | 2 | 0 | 0 | 0 | |||
| Altered antiviral response in host | N205S | N | 0 | 3 | 0 | 6 | |
| S | 91 | 161 | 90 | 464 | |||
| C | 0 | 0 | 0 | 5 | |||
| R | 0 | 0 | 2 | 1 | |||
| G210R | G | 91 | 164 | 92 | 472 | ||
| R | 0 | 0 | 0 | 4 |
Values indicate the number of isolates with the given mutation. Identical molecular markers that exist in all influenza A (H7N9) virus isolates and have been reported to have functions in other influenza virus subtypes are not listed in this table. They include the following: PB2 (89V and 357H, indicating increased pathology in mice), PB1 (473V, indicating increased replication in mammalian cells), PA (36A, reported to be related to increased replication), M1 (30D and 215A, related to increased virulence in mice), M2 (31N, indicating reduced sensitivity to amantadine), NS1 (42S, indicating increased virulence in mice; PDZ motif deletion, indicating decreased virulence in mice). Mixed residues are not included for analysis. The HA gene was under the H3 numbering system and the NA gene under the N2 numbering system. Other internal genes were numbered from the start codon (M).
For the HA gene, most viruses contained G186V and Q226L/I substitutions (H3 numbering), increasing the human receptor binding of the viruses (13–16). During the three waves, a reduced number of human H7N9 viruses retained the residue with 186G or 226Q. The majority of human origin H7N9 viruses of each wave acquired PB2-E627K, which increased viral pathogenicity in mice (17–19). Of note, almost all of the human- or avian-origin H7N9 viruses possessed the I368V mutation in PB1 protein, which would increase the H5N1 viral transmissibility in ferrets (20). A total of 5.4% (19/352) of the H7N9 viruses possessed substitutions associated with drug resistance in the NA protein, indicating the sensitivity of neuraminidase inhibitors. More specifically, a total of 4, 1, and 14 human-origin H7N9 viruses possessed either E119V, A246T, or R292K substitutions in NA protein, respectively (21). None of the mutations occurred in avian-origin H7N9 viruses. All H7N9 viruses were resistant to adamantine due to the S31N mutation in M2 protein (1, 16, 22).
Hemagglutinin inhibition assays were conducted on the selected viruses representing each subclade of all three waves according to standard protocols using 0.5% turkey red blood cells (23, 24). The results showed that all H7N9 viruses reacted well with the antisera of the vaccine virus A/Anhui/1/2013 (Tables 4, 5, and 6). No antigenic differences were observed.
TABLE 4.
Hemagglutination inhibition reactions of influenza H7N9 viruses in experiments conducted on 15 February 2016a
| Isolate no. | Virus description | Titer |
Passage history | Collection date (yr-mo-day) | Phylogenetic clade | ||
|---|---|---|---|---|---|---|---|
| HA | AH1 | SH2 | |||||
| 1 | A/Anhui/1/2013 (reference) | 256 | 160 | 320 | E2 | 2013-03-20 | W1 |
| 2 | A/Shanghai/2/2013 (reference) | 64 | 160 | 320 | E2 | 2013-03-05 | W1 |
| 3 | A/Guangdong/24997/2013 | 1,024 | 320 | 320 | E1 | 2013-11-30 | W2-1 |
| 4 | A/Guangdong/24999/2013 | 128 | 160 | 320 | E1 | 2013-12-16 | W2-1 |
| 5 | A/Guangdong/02125/2014 | 128 | 160 | 320 | E1 | 2014-01-08 | W2-1 |
| 6 | A/Guangdong-Guangzhou/XN00509/2014 | 512 | 320 | 320 | E1 | 2014-02-10 | W2-1 |
| 7 | A/Guangdong-Guangzhou/XN08763/2014 | 256 | 160 | 160 | E1 | 2014-02-21 | W2-1 |
| 8 | A/Shandong/02/2014 | 512 | 160 | 320 | E1 | 2014-03-04 | W2-3 |
| 9 | A/Jiangsu/09041/2014 | 512 | 160 | 320 | E1 | 2014-01-20 | W2-3 |
| 10 | A/Guangdong/15SF017/2015 | 64 | 160 | 320 | E1 | 2015-01-15 | W3-2 |
| 11 | A/Jiangsu/19758/2015 | 512 | 160 | 160 | E1 | 2015-01-23 | W3-3 |
| 12 | A/Jiangsu/18828/2014 | 256 | 80 | 80 | E1 | 2014-12-31 | W3-3 |
| 13 | A/Anhui/01887/2014 | 64 | 80 | 80 | E1 | 2014-05-20 | W3-3 |
| 14 | A/Anhui/33228/2015 | 128 | 160 | 160 | E1 | 2015-04-08 | W3-3 |
| 15 | A/Beijing/40610/2015 | 64 | 80 | 80 | E1 | 2015-05-26 | W3-3 |
| 16 | A/Anhui/40095/2015 | 64 | 80 | 160 | E1 | 2015-05-23 | W3-3 |
| 17 | A/Guangdong/15SF010/2015 | 512 | 160 | 320 | E1 | 2015-01-06 | W3-1 |
| 18 | A/Environment/Guangdong/22351/2015 | 1,024 | 80 | 160 | E1 | 2015-01-11 | W3-1 |
| 19 | A/Environment/Guangdong/21123/2015 | 256 | 160 | 320 | E1 | 2015-01-20 | W3-1 |
TABLE 5.
Hemagglutination inhibition reactions of influenza H7N9 viruses in experiments conducted on 15 August 2014
| Isolate no. | Virus description | Titer |
Passage history | Collection date (yr-mo-day) | Phylogenetic clade | ||
|---|---|---|---|---|---|---|---|
| HA | AH1 | SH2 | |||||
| 1 | A/Anhui/1/2013 (reference) | 256 | 160 | 640 | E2 | 2013-03-20 | W1 |
| 2 | A/Shanghai/2/2013 (reference) | 512 | 160 | 640 | E1 | 2013-03-05 | W1 |
| 3 | A/Fujian/16/2014 | 512 | 160 | 1,280 | E1 | 2014-03-28 | W2-6 |
| 4 | A/Zhejiang/24/2014 | 1,024 | 320 | 1,280 | E1 | 2014-01-24 | W2-4 |
| 5 | A/Zhejiang/23/2014 | 512 | 320 | 1,280 | E1 | 2014-02-09 | W2-4 |
| 6 | A/Zhejiang/22/2014 | 512 | 80 | 640 | E1 | 2014-02-08 | W2-4 |
| 7 | A/Zhejiang/21/2014 | 128 | 160 | 1,280 | E1 | 2014-02-10 | W2-4 |
| 8 | A/Zhejiang/20/2014 | 128 | 160 | 1,280 | E1 | 2014-02-03 | W2-4 |
| 9 | A/Zhejiang/17/2014 | 4,096 | 80 | 640 | E1 | 2014-02-03 | W2-4 |
| 10 | A/Zhejiang/16/2014 | 4,096 | 160 | 320 | E1 | 2014-02-03 | W2-4 |
| 11 | A/Zhejiang/15/2014 | 256 | 320 | 1,280 | E1 | 2014-01-25 | W2-4 |
| 12 | A/Zhejiang/14/2014 | 512 | 160 | 640 | E1 | 2014-01-14 | W2-4 |
| 13 | A/Zhejiang/13/2014 | 512 | 160 | 640 | E1 | 2014-01-27 | W2-3 |
| 14 | A/Fujian/15/2014 | 64 | 160 | 640 | E1 | 2014-02-26 | W2-2 |
| 15 | A/Jiangxi/27569/2014 | 512 | 160 | 1,280 | E1 | 2014/4/21 | W2-4 |
| 16 | A/Hunan/26937/2014 | 256 | 160 | 640 | E1 | 2014/4/19 | W2-1 |
| 17 | A/Hunan/26938/2014 | 256 | 80 | 640 | E1 | 2014/4/19 | W2-1 |
| 18 | A/Environment/Xinjiang/73033/2014 | 128 | 80 | 80 | E1 | 2014/7/25 | W2-5 |
TABLE 6.
Hemagglutination inhibition reactions of influenza H7N9 viruses in experiments conducted on 17 June 2013
| Isolate no. | Virus description | Titer |
Passage history | Collection date (yr-mo-day) | Phylogenetic clade | ||
|---|---|---|---|---|---|---|---|
| HA | AH1 | SH2 | |||||
| 1 | A/Anhui/1/2013 (reference) | 16 | 320 | 640 | E1 | 2013-03-20 | W1 |
| 2 | A/Shanghai/2/2013 (reference) | 256 | 320 | 640 | E1 | 2013-03-05 | W1 |
| 3 | A/Shanghai/1/2013 | 256 | 160 | 320 | E2 | 2013-02-26 | W1 |
| 4 | A/Shanghai/3/2013 | 256 | 320 | 640 | E1 | 2013-02-27 | W1 |
| 5 | A/Shanghai/4/2013 | 512 | 320 | 640 | E2 | 2013-03-09 | W1 |
| 6 | A/Jiangsu/01/2013 | 64 | 320 | 640 | E2 | 2013-03-30 | W1 |
| 7 | A/Zhejiang/01/2013 | 512 | 320 | 640 | E1 | 2013-03-25 | W1 |
| 8 | A/Shanghai/05/2013 | 1,024 | 80 | 160 | E1 | 2013-04-02 | W1 |
| 9 | A/Shanghai/07/2013 | 128 | 640 | 1,280 | C2 | 2013-03-18 | W1 |
| 10 | A/Jiangsu/02/2013 | 64 | 320 | 1,280 | E1 | 2013-03-31 | W1 |
| 11 | A/Jiangsu/03/2013 | 1,024 | 320 | 640 | E1 | 2013-04-06 | W1 |
| 12 | A/Jiangsu/04/2013 | 512 | 160 | 320 | E1 | 2013-04-05 | W1 |
| 13 | A/Beijing/01-A/2013 | 512 | 320 | 640 | E1 | 2013-04-12 | W1 |
| 14 | A/Shanghai/06-A/2013 | 256 | 320 | 640 | E1 | 2013-04-10 | W1 |
| 15 | A/Shanghai/10/2013 | 512 | 640 | 1,280 | E1 | 2013-04-09 | W1 |
| 16 | A/Shanghai/11/2013 | 128 | 320 | 640 | E1 | 2013-04-10 | W1 |
| 17 | A/Shanghai/12/2013 | 256 | 640 | 1,280 | E1 | 2013-04-10 | W1 |
| 18 | A/Shanghai/13/2013 | 32 | 160 | 320 | E1 | 2013-04-10 | W1 |
| 19 | A/Shanghai/14/2013 | 1,024 | 640 | 1,280 | E1 | 2013-04-10 | W1 |
| 20 | A/Shanghai/17/2013 | 512 | 160 | 640 | E1 | 2013-04-03 | W1 |
| 21 | A/Shanghai/8/2013 | 32 | 320 | 1,280 | E1 | 2013-04-07 | W1 |
| 22 | A/Shanghai/9/2013 | 256 | 320 | 640 | E1 | 2013-04-08 | W1 |
| 23 | A/Zhejiang/02/2013 | 128 | 320 | 640 | E1 | 2013-04-03 | W1 |
| 24 | A/Shanghai/15/2013 | 128 | 320 | 640 | E2 | 2013-04-09 | W1 |
| 25 | A/Shanghai/16/2013 | 512 | 640 | 1,280 | E2 | 2013-04-10 | W1 |
| 26 | A/Henan/01/2013 | 12 | 160 | 320 | E1 | 2013-04-16 | W1 |
| 27 | A/Shandong/01/2013 | 256 | 320 | 640 | E1 | 2013-04-21 | W1 |
| 28 | A/Fujian/01/2013 | 512 | 320 | 640 | E1 | 2013-04-23 | W1 |
| 29 | A/Jiangxi/01/2013 | 128 | 320 | 640 | E1 | 2013-04-24 | W1 |
| 30 | A/Hunan/01/2013 | 256 | 160 | 640 | E1 | 2013-04-24 | W1 |
| 31 | A/Hunan/02/2013 | 128 | 320 | 640 | E1 | 2013-04-25 | W1 |
| 32 | A/Jiangsu/05/2013 | 512 | 160 | 640 | E1 | 2013-04-08 | W1 |
| 33 | A/Jiangsu/06/2013 | 128 | 320 | 640 | E1 | 2013-04-10 | W1 |
| 34 | A/Jiangsu/07/2013 | 512 | 320 | 640 | E1 | 2013-04-03 | W1 |
| 35 | A/Jiangsu/08/2013 | 512 | 640 | 1,280 | E1 | 2013-04-11 | W1 |
| 36 | A/Jiangsu/09/2013 | 512 | 320 | 640 | E1 | 2013-04-09 | W1 |
| 37 | A/Anhui/02/2013 | 512 | 320 | 1,280 | E1 | 2013-04-14 | W1 |
| 38 | A/Anhui/03/2013 | 64 | 640 | 1,280 | E1 | 2013-04-21 | W1 |
| 42 | A/Chicken/Anhui-Chuzhou/01/2013 | 512 | 320 | 1,280 | E1 | 2013-03-29 | W1 |
| 43 | A/Environment/Shandong/1/2013 | 128 | 320 | 640 | E1 | 2013-04-27 | W1 |
TABLE 7.
Reference viruses representing each subclade on the HA phylogenetic tree
| GISAID accession no. | Isolate name | Subclade |
|---|---|---|
| EPI447598 | A/Shanghai/05/2013 | W1 |
| EPI447629 | A/Anhui/02/2013 | W1 |
| EPI447614 | A/Zhejiang/02/2013 | W1 |
| EPI439507 | A/Anhui/1/2013 | W1 |
| EPI447599 | A/Shanghai/07/2013 | W1 |
| EPI447596 | A/Jiangsu/01/2013 | W1 |
| EPI627705 | A/Guangdong-Guangzhou/XN08763/2014 | W2-1 |
| EPI628512 | A/Guangdong/24999/2013 | W2-1 |
| EPI627649 | A/Guangdong-Guangzhou/XN08423/2014 | W2-1 |
| EPI627745 | A/Guangdong/0010/2014 | W2-1 |
| EPI628520 | A/Guangdong/24997/2013 | W2-1 |
| EPI628096 | A/Guangdong/0012/2014 | W2-1 |
| EPI628440 | A/Environment/Guangdong/02621/2013 | W2-1 |
| EPI627928 | A/Fujian/15/2014 | W2-2 |
| EPI628056 | A/Jiangsu/09387/2014 | W2-2 |
| EPI628032 | A/Anhui/09186/2014 | W2-2 |
| EPI628248 | A/Jiangsu/09041/2014 | W2-3 |
| EPI627593 | A/Zhejiang/13/2014 | W2-3 |
| EPI576623 | A/Environment/Shanghai/PD-JZ-01/2014 | W2-3 |
| EPI627808 | A/Shandong/01/2014 | W2-4 |
| EPI627537 | A/Zhejiang/23/2014 | W2-4 |
| EPI627808 | A/Shandong/01/2014 | W2-4 |
| EPI552399 | A/Zhejiang/17/2014 | W2-4 |
| EPI628312 | A/Zhejiang/07803/2014 | W2-4 |
| EPI627441 | A/Anhui/01887/2014 | W2-5 |
| EPI627912 | A/Environment/Xinjiang/73033/2014 | W2-5 |
| EPI628416 | A/Environment/Jiangsu/03137/2013 | W2-5 |
| EPI628224 | A/Fujian/6/2014 | W2-6 |
| EPI628272 | A/Fujian/3/2014 | W2-6 |
| EPI628160 | A/Fujian/13/2014 | W2-6 |
| EPI566044 | A/Guangdong/15SF010/2015 | W3-1 |
| EPI628712 | A/Environment/Guangdong/22351/2015 | W3-1 |
| EPI627489 | A/Fujian/19/2014 | W3-1 |
| EPI559417 | A/Hong Kong/2550/2015 | W3-1 |
| EPI566036 | A/Guangdong/15SF018/2015 | W3-1 |
| EPI627209 | A/Guangdong/15SF002/2015 | W3-2 |
| EPI628648 | A/Environment/Guangdong/21123/2015 | W3-2 |
| EPI627057 | A/Guangdong/15SF051/2015 | W3-2 |
| EPI628728 | A/Anhui/33228/2015 | W3-3 |
| EPI627385 | A/Guangdong/02497/2014 | W3-3 |
| EPI627097 | A/Jiangsu/19758/2015 | W3-3 |
| EPI628568 | A/Anhui/40095/2015 | W3-3 |
| EPI627017 | A/Jiangsu/18828/2014 | W3-3 |
| EPI628544 | A/Beijing/40610/2015 | W3-3 |
| EPI566052 | A/Xinjiang/05916/2014 | W3-3 |
DISCUSSION
Due to the asymptomatic infections of H7N9 viruses in poultry (25–27), humans acted as the sentinels for the presence of these viruses in avian infections. After three outbreak waves, the fourth epidemic is ongoing. In view of influenza pandemic preparedness, H7N9 activities of the past three waves in humans in China are very important to understand. Thus, genomic, epidemic, and antigenic analyses of the three waves of H7N9 viruses were conducted in this study.
The results have shown that two H7N9 sources have been established in China (Fig. 6). The larger one is the Yangtze River Delta region, which is well recognized as the original source of the H7N9 outbreaks, and the other is the Pearl River Delta region, where viruses also originated from the Yangtze River Delta region. Multiple introductions of the viruses from the Yangtze Delta region into the Pearl River region and the spread of the virus from these two sources to other regions resulted in the virus expansion and human infections.
Several contributions may facilitate the Pearl River Delta region as the subsource of H7N9 outbreaks. The extensive and semi-intensive poultry production systems and live poultry markets in the Pearl River Delta region resulted in the fact that multiple species of poultry shared the same environment (28). A large diversity of avian influenza subtypes, including H5N1 and H9N2 subtype viruses, have been identified from both wild and domestic birds in the Pearl River Delta region (29–31). These may benefit the reassortment of H7N9 viruses with local avian influenza viruses (11, 32) and enhance the opportunity for the genesis and transmission of different genotypes of H7N9 viruses. Moreover, poultry movements could increase opportunities for the spread of the virus from this contaminated region to other regions.
Our results have shown that the H7N9 viruses generated in the Yangtze River Delta or Pearl River Delta region could spread to other regions in Mainland China, including Xinjiang and Jilin, which were distant from these sources but in which human infections still occurred. The poultry trade or movement of poultry may contribute to the geographic expansion of H7N9 viruses, thereby increasing the risk of the H7N9 viruses crossing the border and spreading to other regions or countries. Regional expansion of H7N9 viruses increased the possibilities of reassortment among H7N9 and other subtype viruses. Furthermore, the accumulation proportion of the mammal-adapted substitutions, such as substitutions occurring in position 186 or 226 in HA protein, could increase the likelihood of the viral interspecies transmission. Therefore, these two regions should be monitored to control any H7N9 outbreaks, and the following should be encouraged: relocation of the poultry productions systems away from live poultry markets, raising of the biosecurity level in farms and markets, closure of live poultry markets, active surveillance in humans and avian species, and supervision of poultry movement from affected regions. In addition, regular serosurveys of high-risk personnel (e.g., live poultry markets workers) as well as the general public could be conducted to monitor in a timely fashion potential infections in humans.
ACKNOWLEDGMENTS
This study was supported by the National Mega-projects for Infectious Diseases (2014ZX10004002 and 2013ZX10004218-004, to Y. Shu).
We acknowledge the authors and laboratories for sharing the H7N9 sequences in GISAID's EpiFlu Database. The contents of this article are solely the responsibility of the authors and do not necessarily represent the views of China CDC and other organizations.
REFERENCES
- 1.Gao R, Cao B, Hu Y, Feng Z, Wang D, Hu W, Chen J, Jie Z, Qiu H, Xu K, Xu X, Lu H, Zhu W, Gao Z, Xiang N, Shen Y, He Z, Gu Y, Zhang Z, Yang Y, Zhao X, Zhou L, Li X, Zou S, Zhang Y, Li X, Yang L, Guo J, Dong J, Li Q, Dong L, Zhu Y, Bai T, Wang S, Hao P, Yang W, Zhang Y, Han J, Yu H, Li D, Gao GF, Wu G, Wang Y, Yuan Z, Shu Y. 2013. Human infection with a novel avian-origin influenza A (H7N9) virus. N Engl J Med 368:1888–1897. doi: 10.1056/NEJMoa1304459. [DOI] [PubMed] [Google Scholar]
- 2.Zhu W, Shu Y. 2015. Genetic tuning of avian influenza A (H7N9) virus promotes viral fitness within different species. Microbes Infect 17:118–122. doi: 10.1016/j.micinf.2014.11.010. [DOI] [PubMed] [Google Scholar]
- 3.Wu A, Su C, Wang D, Peng Y, Liu M, Hua S, Li T, Gao GF, Tang H, Chen J, Liu X, Shu Y, Peng D, Jiang T. 2013. Sequential reassortments underlie diverse influenza H7N9 genotypes in China. Cell Host Microbe 14:446–452. doi: 10.1016/j.chom.2013.09.001. [DOI] [PubMed] [Google Scholar]
- 4.Wang D, Yang L, Gao R, Zhang X, Tan Y, Wu A, Zhu W, Zhou J, Zou S, Li X, Sun Y, Zhang Y, Liu Y, Liu T, Xiong Y, Xu J, Chen L, Weng Y, Qi X, Guo J, Li X, Dong J, Huang W, Zhang Y, Dong L, Zhao X, Liu L, Lu J, Lan Y, Wei H, Xin L, Chen Y, Xu C, Chen T, Zhu Y, Jiang T, Feng Z, Yang W, Wang Y, Zhu H, Guan Y, Gao GF, Li D, Han J, Wang S, Wu G, Shu Y. 2014. Genetic tuning of the novel avian influenza A(H7N9) virus during interspecies transmission, China, 2013. Euro Surveill http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=20836. [DOI] [PubMed]
- 5.Lam TT, Zhou B, Wang J, Chai Y, Shen Y, Chen X, Ma C, Hong W, Chen Y, Zhang Y, Duan L, Chen P, Jiang J, Zhang Y, Li L, Poon LL, Webby RJ, Smith DK, Leung GM, Peiris JS, Holmes EC, Guan Y, Zhu H. 2015. Dissemination, divergence and establishment of H7N9 influenza viruses in China. Nature 522:102–105. doi: 10.1038/nature14348. [DOI] [PubMed] [Google Scholar]
- 6.Lam TT, Wang J, Shen Y, Zhou B, Duan L, Cheung CL, Ma C, Lycett SJ, Leung CY, Chen X, Li L, Hong W, Chai Y, Zhou L, Liang H, Ou Z, Liu Y, Farooqui A, Kelvin DJ, Poon LL, Smith DK, Pybus OG, Leung GM, Shu Y, Webster RG, Webby RJ, Peiris JS, Rambaut A, Zhu H, Guan Y. 2013. The genesis and source of the H7N9 influenza viruses causing human infections in China. Nature 502:241–244. doi: 10.1038/nature12515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang L, Zhang Z, Weng Z. 2013. Rapid reassortment of internal genes in avian influenza A(H7N9) virus. Clin Infect Dis 57:1059–1061. doi: 10.1093/cid/cit414. [DOI] [PubMed] [Google Scholar]
- 8.Cui L, Liu D, Shi W, Pan J, Qi X, Li X, Guo X, Zhou M, Li W, Li J, Haywood J, Xiao H, Yu X, Pu X, Wu Y, Yu H, Zhao K, Zhu Y, Wu B, Jin T, Shi Z, Tang F, Zhu F, Sun Q, Wu L, Yang R, Yan J, Lei F, Zhu B, Liu W, Ma J, Wang H, Gao GF. 2014. Dynamic reassortments and genetic heterogeneity of the human-infecting influenza A (H7N9) virus. Nat Commun 5:3142. doi: 10.1038/ncomms4142. [DOI] [PubMed] [Google Scholar]
- 9.Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miller MA, Pfeiffer W, Schwartz T. 2010. Creating the CIPRES Science Gateway for inference of large phylogenetic trees, p 1–8. In Proceedings of the Gateway Computing Environments Workshop (GCE), 14 November 2010, New Orleans, LA. [Google Scholar]
- 11.Lu J, Wu J, Zeng X, Guan D, Zou L, Yi L, Liang L, Ni H, Kang M, Zhang X, Zhong H, He X, Monagin C, Lin J, Ke C. 2014. Continuing reassortment leads to the genetic diversity of influenza virus H7N9 in Guangdong, China. J Virol 88:8297–8306. doi: 10.1128/JVI.00630-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang ZF, He JF, Li XB, Guan WD, Ke CW, Wu SG, Pan SH, Li RF, Kang M, Wu J, Lin JY, Ding GY, Huang JC, Pan WQ, Zhou R, Lin YP, Chen RC, Li YM, Chen L, Xiao WL, Zhang YH, Zhong NS. 2014. Epidemiological and viral genome characteristics of the first human H7N9 influenza infection in Guangdong Province, China. J Thorac Dis 6:1785–1793. doi: 10.3978/j.issn.2072-1439.2014.12.09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shi Y, Zhang W, Wang F, Qi J, Wu Y, Song H, Gao F, Bi Y, Zhang Y, Fan Z, Qin C, Sun H, Liu J, Haywood J, Liu W, Gong W, Wang D, Shu Y, Wang Y, Yan J, Gao GF. 2013. Structures and receptor binding of hemagglutinins from human-infecting H7N9 influenza viruses. Science 342:243–247. doi: 10.1126/science.1242917. [DOI] [PubMed] [Google Scholar]
- 14.Tharakaraman K, Jayaraman A, Raman R, Viswanathan K, Stebbins NW, Johnson D, Shriver Z, Sasisekharan V, Sasisekharan R. 2013. Glycan receptor binding of the influenza A virus H7N9 hemagglutinin. Cell 153:1486–1493. doi: 10.1016/j.cell.2013.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiong X, Martin SR, Haire LF, Wharton SA, Daniels RS, Bennett MS, McCauley JW, Collins PJ, Walker PA, Skehel JJ, Gamblin SJ. 2013. Receptor binding by an H7N9 influenza virus from humans. Nature 499:496–499. doi: 10.1038/nature12372. [DOI] [PubMed] [Google Scholar]
- 16.Zhou J, Wang D, Gao R, Zhao B, Song J, Qi X, Zhang Y, Shi Y, Yang L, Zhu W, Bai T, Qin K, Lan Y, Zou S, Guo J, Dong J, Dong L, Zhang Y, Wei H, Li X, Lu J, Liu L, Zhao X, Li X, Huang W, Wen L, Bo H, Xin L, Chen Y, Xu C, Pei Y, Yang Y, Zhang X, Wang S, Feng Z, Han J, Yang W, Gao GF, Wu G, Li D, Wang Y, Shu Y. 2013. Biological features of novel avian influenza A (H7N9) virus. Nature 499:500–503. doi: 10.1038/nature12379. [DOI] [PubMed] [Google Scholar]
- 17.Zhang H, Li X, Guo J, Li L, Chang C, Li Y, Bian C, Xu K, Chen H, Sun B. 2014. The PB2 E627K mutation contributes to the high polymerase activity and enhanced replication of H7N9 influenza virus. J Gen Virol 95:779–786. doi: 10.1099/vir.0.061721-0. [DOI] [PubMed] [Google Scholar]
- 18.Mok CK, Lee HH, Lestra M, Nicholls JM, Chan MC, Sia SF, Zhu H, Poon LL, Guan Y, Peiris JS. 2014. Amino acid substitutions in polymerase basic protein 2 gene contribute to the pathogenicity of the novel A/H7N9 influenza virus in mammalian hosts. J Virol 88:3568–3576. doi: 10.1128/JVI.02740-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu W, Li L, Yan Z, Gan T, Li L, Chen R, Chen R, Zheng Z, Hong W, Wang J, Smith DK, Guan Y, Zhu H, Shu Y. 2015. Dual E627K and D701N mutations in the PB2 protein of A(H7N9) influenza virus increased its virulence in mammalian models. Sci Rep 5:14170. doi: 10.1038/srep14170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herfst S, Schrauwen EJ, Linster M, Chutinimitkul S, de Wit E, Munster VJ, Sorrell EM, Bestebroer TM, Burke DF, Smith DJ, Rimmelzwaan GF, Osterhaus AD, Fouchier RA. 2012. Airborne transmission of influenza A/H5N1 virus between ferrets. Science 336:1534–1541. doi: 10.1126/science.1213362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Song MS, Marathe BM, Kumar G, Wong SS, Rubrum A, Zanin M, Choi YK, Webster RG, Govorkova EA, Webby RJ. 2015. Unique determinants of neuraminidase inhibitor resistance among N3, N7, and N9 avian influenza viruses. J Virol 89:10891–10900. doi: 10.1128/JVI.01514-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Y, Liang W, Yang S, Wu N, Gao H, Sheng J, Yao H, Wo J, Fang Q, Cui D, Li Y, Yao X, Zhang Y, Wu H, Zheng S, Diao H, Xia S, Zhang Y, Chan KH, Tsoi HW, Teng JL, Song W, Wang P, Lau SY, Zheng M, Chan JF, To KK, Chen H, Li L, Yuen KY. 2013. Human infections with the emerging avian influenza A H7N9 virus from wet market poultry: clinical analysis and characterisation of viral genome. Lancet 381:1916–1925. doi: 10.1016/S0140-6736(13)60903-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kendal APSJ, Pereira MS. 1982. World Health Organization Collaborating Centers from Reference and Research on Influenza: concepts and procedures for laboratory-based influenza surveillance, p B17–B35. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 24.WHO. WHO/CDS/CSR/NCS/2002.5. http://www.wpro.who.int/NR/rdonlyres/EFD2B9A7-2265-4AD0-BC98-97937B4FA83C/0/manualonanimalaidiagnosisandsurveillance.pdf Accessed 21 April 2009.
- 25.Pantin-Jackwood MJ, Miller PJ, Spackman E, Swayne DE, Susta L, Costa-Hurtado M, Suarez DL. 2014. Role of poultry in the spread of novel H7N9 influenza virus in China. J Virol 88:5381–5390. doi: 10.1128/JVI.03689-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zaraket H, Baranovich T, Kaplan BS, Carter R, Song MS, Paulson JC, Rehg JE, Bahl J, Crumpton JC, Seiler J, Edmonson M, Wu G, Karlsson E, Fabrizio T, Zhu H, Guan Y, Husain M, Schultz-Cherry S, Krauss S, McBride R, Webster RG, Govorkova EA, Zhang J, Russell CJ, Webby RJ. 2015. Mammalian adaptation of influenza A(H7N9) virus is limited by a narrow genetic bottleneck. Nat Commun 6:6553. doi: 10.1038/ncomms7553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kalthoff D, Bogs J, Grund C, Tauscher K, Teifke JP, Starick E, Harder T, Beer M. 2014. Avian influenza H7N9/13 and H7N7/13: a comparative virulence study in chickens, pigeons, and ferrets. J Virol 88:9153–9165. doi: 10.1128/JVI.01241-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gilbert M, Golding N, Zhou H, Wint GR, Robinson TP, Tatem AJ, Lai S, Zhou S, Jiang H, Guo D, Huang Z, Messina JP, Xiao X, Linard C, Van Boeckel TP, Martin V, Bhatt S, Gething PW, Farrar JJ, Hay SI, Yu H. 2014. Predicting the risk of avian influenza A H7N9 infection in live-poultry markets across Asia. Nat Commun 5:4116. doi: 10.1038/ncomms5116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Duan L, Zhu H, Wang J, Huang K, Cheung CL, Peiris JS, Chen H, Guan Y. 2011. Influenza virus surveillance in migratory ducks and sentinel ducks at Poyang Lake, China. Influenza Other Respir Viruses 5(Suppl 1):65–68. [PubMed] [Google Scholar]
- 30.Martin V, Pfeiffer DU, Zhou X, Xiao X, Prosser DJ, Guo F, Gilbert M. 2011. Spatial distribution and risk factors of highly pathogenic avian influenza (HPAI) H5N1 in China. PLoS Pathog 7:e1001308. doi: 10.1371/journal.ppat.1001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu X, Jin T, Cui Y, Pu X, Li J, Xu J, Liu G, Jia H, Liu D, Song S, Yu Y, Xie L, Huang R, Ding H, Kou Y, Zhou Y, Wang Y, Xu X, Yin Y, Wang J, Guo C, Yang X, Hu L, Wu X, Wang H, Liu J, Zhao G, Zhou J, Pan J, Gao GF, Yang R, Wang J. 2014. Influenza H7N9 and H9N2 viruses: coexistence in poultry linked to human H7N9 infection and genome characteristics. J Virol 88:3423–3431. doi: 10.1128/JVI.02059-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xie S, Jia W, Lin Y, Xing K, Ren X, Qi W, Liao M. 2015. Third wave of influenza A(H7N9) virus from poultry, Guangdong Province, China, 2014-2015. Emerg Infect Dis 21:1657–1660. doi: 10.3201/eid2109.150635. [DOI] [PMC free article] [PubMed] [Google Scholar]