

ABSTRACT
When multiple prion strains are inoculated into the same host, they can interfere with each other. Strains with long incubation periods can suppress conversion of strains with short incubation periods; however, nothing is known about the conversion of the long-incubation-period strain during strain interference. To investigate this, we inoculated hamsters in the sciatic nerve with long-incubation-period strain 139H prior to superinfection with the short-incubation-period hyper (HY) strain of transmissible mink encephalopathy (TME). First, we found that 139H is transported along the same neuroanatomical tracks as HY TME, adding to the growing body of evidence indicating that PrPSc favors retrograde transneuronal transport. In contrast to a previous report, we found that 139H interferes with HY TME infection, which is likely due to both strains targeting the same population of neurons following sciatic nerve inoculation. Under conditions where 139H blocked HY TME from causing disease, the strain-specific properties of PrPSc corresponded with the strain that caused disease, consistent with our previous findings. In the groups of animals where incubation periods were not altered, we found that the animals contained a mixture of 139H and HY TME PrPSc. This finding expands the definition of strain interference to include conditions where PrPSc formation is altered yet disease outcome is unaltered. Overall, these results contradict the premise that prion strains are static entities and instead suggest that strain mixtures are dynamic regardless of incubation period or clinical outcome of disease.
IMPORTANCE Prions can exist as a mixture of strains in naturally infected animals, where they are able to interfere with the conversion of each other and to extend incubation periods. Little is known, however, about the dynamics of strain conversion under conditions where incubation periods are not affected. We found that inoculation of the same animal with two strains can result in the alteration of conversion of both strains under conditions where the resulting disease was consistent with infection with only a single strain. These data challenge the idea that prion strains are static and suggests that strain mixtures are more dynamic than previously appreciated. This observation has significant implications for prion adaptation.
INTRODUCTION
Prion diseases are transmissible neurodegenerative diseases of animals, including humans, with no known effective treatment. Prion diseases of animals include scrapie of sheep and goats, chronic wasting disease of cervids, bovine spongiform encephalopathy of cattle, and transmissible mink encephalopathy (TME) of ranch-raised mink. Prion diseases of humans include kuru of the Fore people of Papua New Guinea, Creutzfeldt-Jakob disease (CJD), Gerstmann-Sträussler-Scheinker syndrome, and fatal familial insomnia (1–8). Human prion diseases are unique in biology in that they can have infectious, familial, or sporadic etiologies. Interestingly, brain material from all three etiologies is infectious and can transmit disease to animals.
The prion agent is comprised mainly, if not entirely, of PrPSc, the pathogenic conformation of the normal host protein PrPC (9–13). Formation of PrPSc occurs when existing PrPSc binds to PrPC, resulting in a conformational change of PrPC to PrPSc. Fragmentation of the growing PrPSc fibril results in formation of new free ends for PrPC binding, leading to exponential formation of PrPSc (14–16). In the absence of preexisting PrPSc, PrPC can spontaneously misfold into PrPSc; this process is enhanced by mutations of PrPC, providing a molecular basis for sporadic and familial forms of human prion disease (17).
Prion strains are operationally defined by differences in neuropathology that are observed under controlled experimental conditions. The PrP genotypes of the agent and host, titer of the agent, route of infection, and gender of the host can all influence the outcome of disease (18–25). When these agent and host parameters are controlled, strain-specific differences in disease phenotype such as incubation period, clinical signs, tissue tropism, host range, and neuropathology are observed.
Differences in the conformation of PrPSc may encode prion strain properties. Strain-specific differences in the biochemical properties of PrPSc, such as relative resistance to protease digestion, conformational stability, and relative α-helical and β-sheet contents, are observed (16, 26–29). Recent work suggests that host cofactors may also contribute to strain diversity (30–32). The relationship between the strain-specific biochemical properties of PrPSc and the observed differences in disease outcome are poorly understood.
More than one prion strain can be present in a single host in natural prion disease. Passage of sheep scrapie to wild-type or transgenic mice expressing ovine PrPC results in the isolation of distinct prion strains (33–35). This suggests that more than one strain of sheep scrapie is present in the inoculum. Interspecies transmission can also lead to the generation of new strains (36). More direct evidence for the coexistence of prion strains has been found in humans with CJD. Two major types of PrPSc, type 1 and type 2, have been identified in CJD and are characterized by migration of the unglycosylated PrPSc polypeptide at 21 and 19 kDa, respectively (37, 38). Transmission of type 1 or type 2 CJD to transgenic mice expressing human PrPC maintains the type-specific PrPSc properties, indicating that these are bona fide human prion strains (39). The cooccurrence of these PrPSc subtypes in natural cases of CJD has been identified based on PrPSc migration and by using anti-PrP antibodies that are type specific (40–44). While the relative percentage of CJD cases where type 1 and type 2 PrPSc coexist is controversial, it is clear that in natural prion disease a mixture of prion strains can occur.
Prion strains in the same host can interfere with each other. Strain interference was first observed in mice in which the long-incubation-period strain 22C was inoculated prior to superinfection with short-incubation-period strain 22A (45). As the interval between inoculation with the blocking strain 22C and superinfection with 22A increased, 22C was able to extend the incubation period of 22A or to completely block 22A from causing disease. Prion strain interference can also occur when the strains are inoculated at the same time, and in this instance the ratio of the strains coinfected determines the outcome of disease (46, 47). When two prion strains are targeted to infect the same population of neurons, the onset of conversion of each strain is a critical parameter of strain interference (48). Under conditions where the blocking strain extends the incubation period of the superinfecting strain or completely blocks the superinfecting strain from causing disease, the blocking strain can suppress conversion of the superinfecting strain (48). Nothing is known about the conversion of the blocking strain following coinfection or superinfection with a short-incubation-period strain, whether or not there is interference.
MATERIALS AND METHODS
Ethics statement.
All procedures involving animals were approved by the Creighton University Institutional Animal Care and Use Committee and comply with the Guide for the Care and Use of Laboratory Animals (49).
Animal inoculations.
Male Syrian golden hamsters (Harlan-Sprague-Dawley, Indianapolis, IN) were used. Animals were inoculated in the sciatic nerve with 2 μl of a 1% (wt/vol) brain homogenate from a hamster at the clinical stage of disease that was infected with the 139H strain of hamster-adapted scrapie or hyper (HY) strain of hamster-adapted transmissible mink encephalopathy (TME) as described previously (28, 48). Hamsters were observed three times per week for the onset of clinical signs, and the incubation period was calculated as the number of days between inoculation and the onset of clinical disease (28).
Tissue collection.
Prion-infected and age-matched mock-infected hamsters were anesthetized with isoflurane and transcardially perfused with 50 ml of 0.01 M Dulbecco's phosphate-buffered saline (DPBS) followed by 75 ml of McLean's paraformaldehyde-lysine-periodate (PLP) fixative (50) in preparation for immunohistochemical processing. Selected central nervous system (CNS) tissue was removed and immersed in PLP for 5 to 7 h at room temperature prior to paraffin processing. For Western blot analysis, animals were euthanized with CO2, followed by thoracotomy. Brain tissue was immediately collected, frozen on dry ice, and stored at −80°C.
IHC.
Immunohistochemistry (IHC) was performed as previously described (51). Briefly, 7-μm tissue sections were deparaffinized and incubated in 95% formic acid (Sigma-Aldrich, St. Louis, MO) for 20 min at room temperature. Endogenous peroxidase activity was blocked using 0.3% H2O2 in methanol for 20 min at room temperature. Nonspecific staining was blocked with 10% normal horse serum (Vector Laboratories, Burlingame, CA) in Tris-buffered saline for 30 min at room temperature. The sections were incubated with the anti-PrP monoclonal antibody 3F4 (1:600; Chemicon, Billerica, MA) at 4°C overnight. The sections were then incubated in biotinylated horse anti-mouse immunoglobulin G conjugate and subsequently incubated in ABC solution (Elite kit; Vector Laboratories, Burlingame, CA). The chromogen was visualized using 0.05% (wt/vol) 3,3′-diaminobenzidine (Sigma-Aldrich, St. Louis, MO) in Tris-buffered saline containing 0.0015% H2O2 and counterstained with hematoxylin (Richard Allen Scientific, Kalamazoo, MI). Light microscopy was performed using a Nikon i80 microscope (Nikon, Melville, NY), and images were captured and identically processed for white balance using Adobe Bridge CS6 (San Jose, CA). Tissue sections were analyzed at sampling intervals of no greater than 126 μm. The rate of PrPSc spread was determined as previously described (51). Briefly, the distance from the site of inoculation in the sciatic nerve to ventral motor neurons (VMNs), lateral vestibular nucleus, red nucleus, and hind limb motor cortex was measured in millimeters. The rate of PrPSc spread to each structure was calculated by dividing the distance in millimeters between the inoculation site and the specific CNS structure by the number of days postinfection (p.i.) when PrPSc immunoreactivity was first detected. Sections were stained for Nissl or with hematoxylin and eosin (H&E) as described previously (51, 52).
Western blot analysis.
Western blot detection of PrPSc from brain homogenate was performed as described previously (53). Briefly, brain homogenate (5%, wt/vol) was digested with proteinase K (PK) at a final concentration of 2 U/ml (Roche Diagnostics Corporation, Indianapolis, IN) at 37°C for 60 min. PK digestion was terminated by incubating the samples at 100°C for 10 min. The samples were size fractionated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) with 4 to 12% bis-Tris-acrylamide (NuPAGE; Invitrogen, Carlsbad, CA) and transferred to a polyvinylidene difluoride (PVDF) membrane. The membrane was blocked with 5% nonfat dry milk (Bio-Rad Laboratories, Hercules, CA) for 30 min. Hamster prion protein was detected using the mouse monoclonal anti-PrP antibody 3F4 (1:10,000; Chemicon, Temecula, CA). The Western blot was developed with Pierce SuperSignal West Femto maximum-sensitivity substrate, according to the manufacturer's instructions (Pierce, Rockford, IL), and imaged on a Kodak 4000R Imaging Station (Kodak, Rochester, NY) as previously described (53).
PrPSc conformational stability assay.
The conformational stability of PrPSc was determined as described previously (28). Brain homogenates were combined with 0.04 g/ml SDS or DPBS to a final concentration of 7.5% (wt/vol) homogenate and 1% (wt/vol) SDS or 0% (wt/vol) SDS, respectively. The resulting mixtures were incubated at 70°C for 10 min. Proteinase K (Roche Diagnostics, Indianapolis) was added to a final concentration of 2.0 U/ml, and the samples were incubated at 37°C for 30 min with shaking. Samples were brought to a final volume of 200 μl in DPBS, and 125-μg brain equivalents were analyzed for PrPSc content by 96-well immunoassay as previously described (54) using the monoclonal anti-PrP antibody 3F4 (1:10,000; Chemicon, Temecula, CA). To standardize between samples, the amount of PK-resistant PrPSc in the 1% (wt/vol) SDS group was divided by the amount of PK-resistant PrPSc in the 0% (wt/vol) SDS group (i.e., untreated), resulting in the relative percentage of 1% (wt/vol) SDS PK-resistant PrPSc compared to the total (0% [wt/vol] SDS) PK-resistant PrPSc. The value of the standardized 1% (wt/vol) SDS PK-resistant HY TME PrPSc sample was adjusted to 100%, and the remaining sample values were similarly adjusted to the relative percentage of this control. The background was set to the signal intensity of an uninfected brain homogenate treated with 1% (wt/vol) SDS followed by digestion with PK.
RESULTS
Retrograde transsynaptic transport of 139H PrPSc.
Sciatic nerve inoculation of the HY TME agent results in retrograde axonal transport along the same four descending motor pathways (51). To investigate whether the 139H strain of hamster-adapted scrapie uses the same pathways, the temporal and spatial spread of PrPSc in the lumbar spinal cord, brain stem, and brain was determined following sciatic nerve inoculation. At 25 days postinfection (p.i.), IHC failed to detect PrPSc in the CNS (Table 1). At 50 days p.i., PrPSc immunoreactivity was detected in lamina IX of the lumbar spinal cord ipsilateral to the side of sciatic nerve inoculation in 2 of the 3 inoculated animals (Table 1; Fig. 1O) and in the magnocellular aspect of the red nucleus contralateral to the side of sciatic nerve inoculation in one of the three 139H-inoculated hamsters (Table 1; Fig. 1F). At 75 days p.i., PrPSc immunoreactivity in the lumbar spinal cord and red nucleus was detected in all three animals with a bilateral or contralateral pattern of immunoreactivity, respectively (Table 2). At 75 days p.i., PrPSc was first detected in the reticular formation (Fig. 1L), lateral vestibular nucleus (Fig. 1I), hind limb motor cortex (Fig. 1C), and interposed nucleus and reticular thalamic nucleus (Table 1). At 100 days p.i., increased PrPSc immunoreactivity was observed in the previously listed structures (Table 1) and was also detected in the ventroposterior thalamic nucleus (Table 1). At clinical disease, PrPSc immunoreactivity had become widespread throughout the CNS. In the mock-infected hamsters we failed to detect PrPSc at any time point p.i. (Fig. 1B, E, H, K, and N). Overall, the temporal and spatial spread of PrPSc is consistent with retrograde spread along the reticulospinal, vestibulospinal, rubrospinal, and corticospinal tracks.
TABLE 1.
Temporal and spatial spread of PrPSc in the CNS of hamsters infected in the sciatic nerve with the 139H strain of hamster-adapted scrapie
| Central nervous system region | PrPSc immunoreactivity at day postinfectiona: |
||||
|---|---|---|---|---|---|
| 25 | 50 | 75 | 100 | Clinical | |
| Spinal cord, L2-L4 | − | +b | ++c | +++ | +++ |
| Medulla-pons | |||||
| Reticular formation | − | − | + | ++ | ++++ |
| Lateral vestibular nucleus | − | − | + | ++ | +++ |
| Cerebellum, interposed nucleus | − | − | + | ++ | +++ |
| Mesencephalon, red nucleus | − | +d,e | +d | +++ | ++++ |
| Diencephalon | |||||
| Reticular thalamic nucleus | − | − | +b | ++ | +++ |
| Ventroposterior thalamic nucleus | − | − | − | ++ | +++ |
| Telencephalon, hind limb cortex | − | − | + | ++ | +++ |
Relative intensities of PrPSc immunoreactivity: −, none; +, rare; ++, weak; +++, moderate; ++++, heavy.
PrPSc immunoreactivity is ipsilateral to side of inoculation in 2 of 3 animals.
PrPSc immunoreactivity is bilateral.
PrPSc immunoreactivity is contralateral to the side of inoculation.
PrPSc immunoreactivity in 1 of 3 animals.
FIG 1.

Distribution of PrPSc in spinal cord, brain stem, and brain of hamsters infected in the sciatic nerve with the 139H agent. (A to C) Nissl staining (A) and PrPSc immunostaining (B and C) of the contralateral hind limb motor cortex from mock-infected (B) or 139H-infected (C) hamsters at 75 days postinfection. (D) Nissl staining of the mesencephalon containing the red nuclei (dashed outline). (E and F) PrPSc immunoreactivity was detected in the ventrolateral portion of the contralateral red nucleus at 75 days postinfection (F) but was not detected in mock-infected animals (E). (G) Hematoxylin and eosin (H&E) staining of the pons ipsilateral to the side of inoculation that contains the lateral vestibular nucleus. (H and I) At 100 days postinfection, PrPSc immunoreactivity is detected in the 139H-infected tissue (I) but is not detected in mock-infected animals (H). (J to L) H&E-stained section of the pons (J) containing the reticular formation contains PrPSc immunoreactivity in 139H-infected (L) but not in mock-infected (K) hamsters. (M) Nissl-stained section of lumbar spinal cord that contains ventral motor neurons (VMNs) whose axons are contained in the sciatic nerve. (N and O) PrPSc immunoreactivity of lumbar spinal cord from uninfected animals failed to detect PrPSc (N), while in 139H-infected hamsters at 50 days postinfection, PrPSc deposits were associated with VMNs ipsilateral to the side of inoculation (O). Scale bars, 200 μm in main panels and 100 μm in insets.
TABLE 2.
Incubation periods and clinical signs in hamsters inoculated with the 139H strain prior to superinfection with the HY TME agent
| First inoculation | Interval (days) between inoculations | Second inoculation | Clinical signs | PrPSc | No. affected/no. inoculated | Onset of clinical symptoms (avg days ± SD) after: |
|
|---|---|---|---|---|---|---|---|
| First inoculation | Second inoculation | ||||||
| Mock | 50 | HY TME | HY TME | HY | 5/5 | NAa | 82 ± 3 |
| 139H | 25 | HY TME | HY TME | HY/139H | 5/5 | 112 ± 3 | 78 ± 3b |
| 139H | 50 | HY TME | HY TME | HY/139H | 5/5 | 137 ± 6 | 87 ± 6b |
| 139H | 75 | HY TME | HY TME | HY | 1/5 | 162 | 87 |
| 139H | 75 | HY TME | 139H | 139H | 4/5 | 188 ± 3c | 113 ± 3 |
| 139H | 50 | Mock | 139H | 139H | 5/5 | 188 ± 3 | NA |
NA, not applicable.
Incubation period similar to that for control animals inoculated with the HY TME agent alone (P > 0.05).
Incubation period similar to that for control animals inoculated with the 139H agent alone (P > 0.05).
The rates of 139H PrPSc spread from the site of inoculation to ventral motor neurons, lateral vestibular nucleus, red nucleus, and hind limb motor cortex were 1.29, 1.60, 2.59, and 1.75 mm/day, respectively. The overall average rate of 139H PrPSc spread was 1.80 ± 0.27 mm per day and was significantly (P < 0.05) different than our previously reported rates of 4.14 ± 0.35 and 1.10 ± 0.11 for HY or drowsy (DY) PrPSc, respectively (51).
139H and HY TME strain interference.
To investigate whether 139H could interfere with HY TME, hamsters were infected in the sciatic nerve with 139H agent at 25, 50, or 75 days prior to superinfection with HY TME agent (Table 2). The sciatic nerve route was chosen since both HY TME and 139H are transported by the same descending motor tracks (Table 1; Fig. 1); therefore, both of these strains will be targeted to the same population of neurons in each brain area. The interval time points were based on the temporal and spatial spread of PrPSc in hamsters infected in the sciatic nerve with 139H (Table 1).
Five hamsters were inoculated in the sciatic nerve with uninfected brain homogenate (mock) 50 days prior to infection with HY TME to serve as HY TME positive controls. All 5 positive-control animals developed clinical signs of HY TME (hyperexcitability and ataxia) at 82 ± 3 days p.i. (Table 2). The 139H positive-control group consisted of 5 hamsters inoculated in the sciatic nerve with the 139H agent and then inoculated 50 days later with uninfected brain homogenate (mock). In this group, all 5 hamsters developed clinical signs consistent with 139H infection (ataxia and weight gain) at 188 ± 3 days p.i. (Table 2).
Three experimental groups were generated by first inoculating the animals in the sciatic nerve with the 139H agent and then superinfecting the same sciatic nerve with HY TME at 25, 50, and 75 days after 139H infection. In the 25- and 50-day interval groups, all 5 animals developed clinical signs of hyperexcitability and ataxia at 78 ± 3 and 87 ± 6 days postinfection, respectively, which does not significantly differ (P > 0.05) from the results for the HY TME positive-control-infected group (Table 2). In the 75-day interval group, 1 hamster developed clinical signs of hyperexcitability and ataxia at 87 days postinfection, consistent with HY TME infection. The remaining 4 hamsters developed clinical signs of ataxia and weight gain at 188 ± 3 days p.i., which does not significantly differ (P > 0.05) from the results for the positive-control 139H-infected group (Table 2). Western blot analysis of 250-μg brain equivalents of PK-digested brain homogenate from all clinically affected animals contained PrPSc, confirming the clinical diagnosis of prion disease (Fig. 2). PK-digested PrPSc from both HY TME- and 139H-infected hamsters had similar glycoform ratios and an unglycosylated PrPSc polypeptide that migrated at 21 kDa; therefore, it was not possible to confirm which strain caused disease by using migration or glycoform ratio (Fig. 2). Overall, we conclude that 139H can interfere with HY TME.
FIG 2.
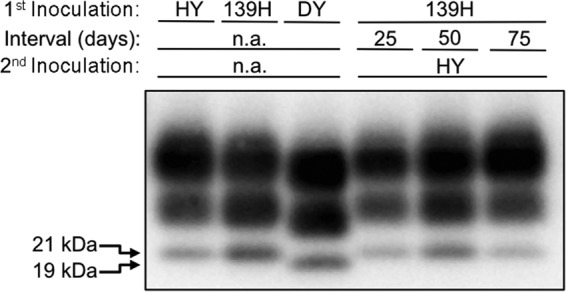
Western blot detection of PrPSc from HY TME- and 139H-coinfected hamsters. Brain homogenates from hamsters infected with either the hyper (HY) 139H or drowsy (DY) TME strain at terminal disease were digested with proteinase K, and the unglycosylated PrPSc polypeptide migrates at 21 kDa for the HY and 139H strains and at 19 kDa for the DY strain. In animals first infected with 139H and then superinfected with HY at 25, 50, or 75 days after 139H infection, PrPSc was detected in the brains of all animals. Due to the similar PrPSc Western blot properties for HY and 139H, it is not possible to determine which strain caused disease. The 19- and 21-kDa unglycosylated PrPSc polypeptides are indicated at the left. n.a., not applicable.
Prion coinfection can alter levels of PrPSc in both the blocking and superinfecting prion strains.
To determine whether 139H or HY TME caused disease in the coinfected hamsters, we utilized a modified PrPSc conformational stability assay. HY PrPSc has a higher conformational stability in SDS than 139H PrPSc, with [SDS]1/2 values of 1.14% ± 0.03% (wt/vol) versus 0.50% ± 0.01%, respectively (28). Brain homogenates are treated with or without 1% (wt/vol) SDS, followed by treatment with PK. The remaining PrPSc in the 1% (wt/vol) SDS-treated sample was represented as percentage of that for the 0% (wt/vol) SDS sample. Since each sample was normalized to its respective 0% (wt/vol) SDS control, reductions in PrPSc concentration in the 1% (wt/vol) SDS group were due to increased sensitivity to PK digestion under these conditions and not to differences in the starting amount of PrPSc. Setting the relative PrPSc concentration in the 1% (wt/vol) SDS-treated HY TME control as 100% allows us to determine if HY or 139H PrPSc is present in the CNS of the coinfected hamsters (Fig. 3A). The conformational stability assay can identify mixtures of 139H and HY PrPSc. We compared the relative 1% (wt/vol) SDS-resistant PrPSc of a mixture of 139H- and HY TME-infected brain homogenates (50% 139H and 50% HY) to that of homogenates with 139H (100% 139H) or HY TME (100% HY) alone (Fig. 3A). Due to the presence of PrPSc from the two different strains, the relative 1% (wt/vol) SDS-resistant PrPSc of the 139H/HY homogenate mixture was not similar to that of either the homogenate with HY TME alone (P < 0.05) or the homogenate with 139H alone (P < 0.05).
FIG 3.
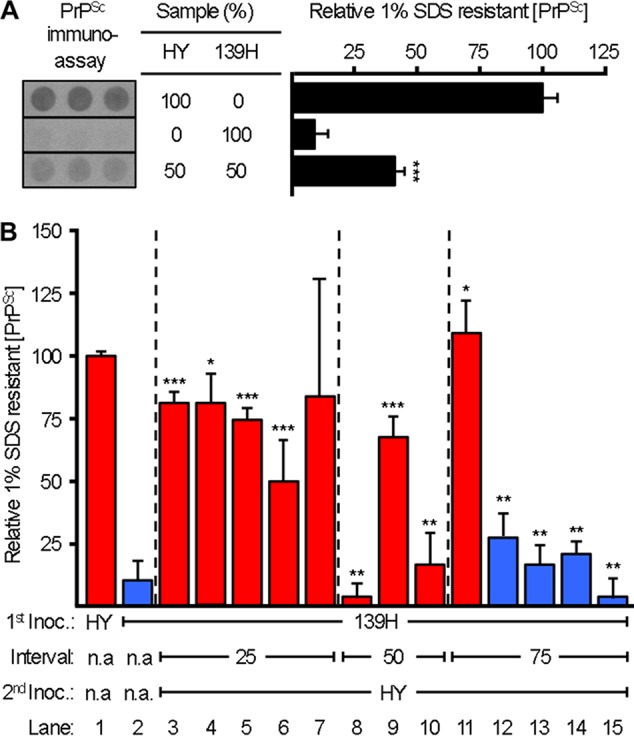
Alteration of blocking and superinfecting strain PrPSc in coinfected animals. (A) The relative amount of PrPSc (average ± standard deviation) that remained after treatment with 1% (wt/vol) SDS followed by PK digestion was determined for the brain homogenates of HY TME- or 139H-infected hamsters using a 96-well immunoassay. Under the treatment conditions, samples with a mixture of 139H and HY PrPSc are distinguishable from samples containing HY or 139H PrPSc alone. The immunoassay panel was assembled from the same exposure of a single plate. (B) Relative amounts of 1% (wt/vol) SDS-resistant PrPSc remaining in brain homogenates from animals infected with HY TME alone, from animals infected with 139H alone, or from animals first inoculated with 139H and then superinfected with HY TME at 25, 50, or 75 days postinfection. Red and blue bars indicate animals that developed clinical signs and incubation periods consistent with a HY TME or 139H infection, respectively. This experiment was repeated three times with similar results. *, similar (P > 0.05) to HY TME control; **, similar (P > 0.05) to 139H TME control; ***, not similar (P < 0.05) to HY TME or 139H control. n.a., not applicable.
In the 75-day strain interference interval group, 1 animal had clinical signs and an incubation period consistent with HY TME infection (Table 2) that contained high-stability PrPSc similar (P > 0.05) to that in the HY TME control (Fig. 3B, lane 11). The remaining 4 hamsters in the 75-day group had clinical signs and incubation periods that were consistent with 139H infection (Table 2), and they contained low-stability PrPSc which was similar (P > 0.05) to that in the 139H control (Fig. 3B, lanes 12 to 15). In the 50-day interval group, all three animals tested had clinical signs and incubation periods that were consistent with HY TME infection (Table 2). In the 50-day interval group, two animals contained low-stability PrPSc similar (P > 0.05) to that in the 139H control (Fig. 3B, lanes 8 and 10), and the third tested animal had relative 1% (wt/vol) SDS-resistant PrPSc that was not similar to that in either the HY TME (P < 0.05) or 139H (P < 0.05) control (Fig. 3B, lane 9). Frozen tissue samples were unavailable for the remaining 2 animals in the 50-day group. In the 25-day interval group, all 5 animals had incubation periods and clinical signs consistent with HY TME infection (Table 2). In this group one animal contained high-stability PrPSc similar (P > 0.05) to that in the HY TME control (Fig. 3B, lane 4), three animals contained relative 1% (wt/vol) SDS-resistant PrPSc that was not similar to that in either the HY TME (<0.05) or 139H (P < 0.05) control (Fig. 3B, lanes 3, 5, and 6), and one animal (Fig. 3B, lane 7) had a high variance that precluded meaningful statistical analysis.
Overall, in the 75-day interval group, the strain-specific PrPSc stability properties corresponded with the clinical signs and incubation period of disease (Table 2; Fig. 3). In the 25- and 50-day interval groups, all of the hamsters exhibited incubation periods and clinical signs indistinguishable from those in HY TME-infected control animals (Table 2), but a subset of these animals contained 1% (wt/vol) SDS-resistant PrPSc that was intermediate between those in HY TME- and 139H-infected controls (Fig. 3), suggesting that they contained a mixture of both 139H and HY PrPSc.
DISCUSSION
Transsynaptic neuronal transport of PrPSc along known neuroanatomical pathways has been reported following peripheral inoculation into several different targets and appears to be a characteristic aspect of prion neuroinvasion and transport. Ocular inoculation of prions results in a sequential spread of spongiform degeneration that is consistent with anterograde transport along well-defined neuroanatomical tracts (55, 56). Intradental inoculation of prions resulted in transport of the agent to the ipsilateral trigeminal ganglia, consistent with transport along the mandibular branch of the trigeminal nerve (57). Moreover, prion inoculation into the tongue results in retrograde axonal transport via the hypoglossal nerve ipsilateral to the hypoglossal nucleus in the medulla, followed by subsequent transport to brain areas that project axons to this nucleus, providing further evidence for transsynaptic axonal transport along both peripheral nerves and CNS tracts (58). Our group and others have shown that sciatic nerve inoculation results in direct neuronal spread of prions in rats, mice, and hamsters (51, 59–61). Detailed analysis of the temporal and spatial spread of PrPSc in the peripheral nervous system and CNS of hamsters inoculated with either the HY or DY TME strain indicated that both of these strains were retrogradely transported along the same 4 descending neuroanatomical pathways. In the current study, the initial deposition of 139H PrPSc in the lumbar cord was associated with ventral motor neurons in lamina IX, similar to the case for HY and DY TME-infected hamsters (Fig. 1; Table 1) (51). The temporal and spatial spread of 139H PrPSc followed the same 4 descending motor pathways: the reticulospinal, vestibulospinal, rubrospinal, and corticospinal tracks (Fig. 1; Table 1). These data are consistent with the hypothesis that PrPSc is able to cross the synaptic cleft between synaptically connected neurons within a pathway.
There are strain specific differences in the rate of prion spread in the nervous system. The rate of HY PrPSc spread in the nervous system appears faster compared to DY PrPSc (51). The rate of HY PrPSc formation is greater than DY PrPSc, therefore, it is unclear if the observed higher rate of HY PrPSc spread along neuroanatomical pathways is due to faster axonal transport, or due to the faster formation of HY PrPSc. Interestingly, the rate of PrPSc formation is similar between 139H and DY TME, yet 139H has a higher rate of spread (28). This observation suggests that factors in addition to the rate of PrPSc formation are involved in strain specific differences in prion transport.
Our results showed that the 139H strain can interfere with, or completely block, the emergence of HY TME. A previous study determined that intracerebral inoculation of 139H up to 63 days prior to superinfection with Sc237 failed to extend the incubation period of Sc237 (62). This result indicated that 139H was unable to interfere with the superinfecting strain. The authors hypothesized that the failure of 139H to interfere with Sc237 was due to conversion of 139H and Sc237 in different populations of cells (62). In the current study, the sciatic nerve route of inoculation was found to direct both 139H and HY TME to the same population of neurons (Fig. 1; Table 1) (51) and to result in 139H interfering with HY TME. These data indicate that the failure of 139H to interfere with Sc237 was not due to an intrinsic inability of 139H to act as a blocking strain but is consistent with the hypothesis that prion strains must infect common populations of neurons for interference to occur (48). We cannot exclude the possibility, however, that differences between HY TME and Sc237 could account for the contradictory interference results, although this is unlikely since Sc237 and HY TME are similar, if not identical, strains.
The blocking strain can inhibit conversion of the superinfecting strain PrPSc. Lesion profile, clinical signs, incubation period of disease, and strain-specific differences in PrPSc Western blot profiles can be used to determine the predominant strain in the coinfected animals at terminal disease. Under conditions where the blocking strain extends the incubation period of the superinfecting strain, there is evidence that the blocking strain inhibits the rate of accumulation of superinfecting PrPSc (48). In situations where the blocking strain completely inhibited the superinfecting strain from causing disease, the blocking strain suppressed conversion of the superinfecting strain (48, 63). The results for the 75-day interval group in the current study are consistent with these previous findings. In the 75-day interval group, 139H was able to completely block HY TME from causing disease and suppressed HY PrPSc accumulation in 4 of the 5 animals (Table 2; Fig. 3). The fact that one animal in this group had the HY PrPSc and incubation period may be due to an incomplete inhibition by 139H infection. In previous studies in hamsters coinfected with the DY and HY TME strains under conditions where DY TME was able to completely block HY TME emergence, protein misfolding cyclic amplification (PMCA) of brain homogenate from these animals indicated that a small amount of HY TME persisted in these animals (63). While we did not determine whether low levels of HY TME persist in this study, taken together, these results suggest that the blocking strain can inhibit, but not extinguish, superinfecting strain conversion. In this model, altering the conditions of prion formation can lead to the emergence of different strains (64–68).
Strain interference can alter the accumulation of the blocking prion strain. Using a modification of the PrPSc conformational stability assay, we were able to determine whether the brain contained HY TME, 139H, or a mixture of PrPSc from each strain. It is important to emphasize that under the assay conditions, we can only conclude whether a mixture of HY and 139H PrPSc is present and cannot quantify the amount of PrPSc from each strain. With this limitation in mind, we found evidence of a mixed population of HY and 139H PrPSc in the 25- and 50-day interval groups (Fig. 3). In both of these interval groups, the clinical signs and incubation period of disease did not differ from those for animals inoculated with HY TME alone, indicating that 139H was not able to extend the incubation period of HY TME under these conditions. Previous strain interference studies have indicated that the presence of PrPSc in the lumbar spinal cord corresponds with the ability of the blocking strain DY TME to interfere with or completely block superinfection with HY TME (48). At 25 and 50 days 139H infection, PrPSc is not detected or is weakly positive in the lumbar spinal cord (Table 1). In coinfected animals, in the 25- and 50-day interval groups, 139H does not extend the incubation period of HY TME, consistent with our previous work (Table 2) (48). Interestingly, in the 25- and 50-day interval groups, where 139H was not able to extend the incubation period of HY TME, a mixture of both 139H and HY PrPSc was present in these animals at terminal disease (Fig. 3). These data indicate that in coinfected animals where the blocking strain does not affect the incubation period or clinical signs of disease, interference between the blocking and superinfecting strains can still occur. In this scenario, 139H is able to interfere with HY TME conversion but is unable to delay HY TME from reaching and affecting the HY TME clinical target areas.
We interpret the intermediate PrPSc stability profile as a mixture of PrPSc from both the HY TME and 139H strains; however, we cannot exclude the possibility that the intermediate PrPSc stability profile is the result of a strain other than 139H or HY TME. In this scenario, it is possible that the presence of both 139H and HY PrPSc favors the de novo formation of a new strain-specific conformation of PrPSc or that interactions between 139H and HY TME allow for the emergence of a preexisting substrain (64, 69). Future serial passage experiments will differentiate between these possibilities. Finally, the mixed-strain PrPSc profile in hamsters with a single-strain phenotype may provide a basis for the observation in human prion disease where more than one PrPSc strain profile is observed with the clinical presentation of disease (40, 70).
Overall, these studies demonstrate that in mixed-strain infections, the dynamics of PrPSc formation of each strain are more complex than previously appreciated. This work adds to the growing body of literature that suggests that prions are a dynamic mixture of substrains (64). The ratio of strains can be altered depending on the environment and, as demonstrated here, the initial ratio of strains present. Importantly, in coinfected animals, each strain can persist regardless of the outcome of infection.
ACKNOWLEDGMENTS
We thank Maria Christensen for excellent technical support.
Funding Statement
K.A.L. was supported by the Institute of General Medical Sciences, National Institutes of Health, Institutional Development Award (IDeA) Networks of Biomedical Research Excellence (INBRE) Program and by the Clare Boothe Luce Program.
REFERENCES
- 1.Marsh R, Bessen R, Lehmann S, Hartsough G. 1991. Epidemiological and experimental studies on a new incident of transmissible mink encephalopathy. J Gen Virol 72:589–594. doi: 10.1099/0022-1317-72-3-589. [DOI] [PubMed] [Google Scholar]
- 2.Williams E, Young S. 1980. Chronic wasting disease of captive mule deer: a spongiform encephalopathy. J Wildl Dis 16:89–98. doi: 10.7589/0090-3558-16.1.89. [DOI] [PubMed] [Google Scholar]
- 3.Zlotnik I, Stamp JT. 1961. Scrapie disease of sheep. World Neurol 2:895–907. [PubMed] [Google Scholar]
- 4.Cuillé J, Chelle PL. 1936. Cuillé: la maladie dite tremblante du mouton est-elle Inoculable? C R Hebd Seances Acad Sci 203:1552. [Google Scholar]
- 5.Hope J, Reekie L, Hunter N, Multhaup G, Beyreuther K, White H, Scott A, Stack M, Dawson M, Wells G. 1988. Fibrils from brains of cows with new cattle disease contain scrapie-associated protein. Nature 336:390–392. doi: 10.1038/336390a0. [DOI] [PubMed] [Google Scholar]
- 6.Wells G. 1987. A novel progressive spongiform enchephalopathy in cattle. Vet Rec 121:419–420. doi: 10.1136/vr.121.18.419. [DOI] [PubMed] [Google Scholar]
- 7.Alpers M, Gajdusek DC. 1965. Changing patterns of kuru: epidemiological changes in the period of increasing contact of the Fore people with western civilization. Am J Trop Med Hyg 14:852–879. [DOI] [PubMed] [Google Scholar]
- 8.Tateishi J, Brown P, Kitamoto T, Hoque Z, Roos R, Wollman R, Cervenakova L, Gajdusek D. 1995. First experimental transmission of fatal familial insomnia. Nature 376:434–435. doi: 10.1038/376434a0. [DOI] [PubMed] [Google Scholar]
- 9.Basler K, Oesch B, Scott M, Westaway D, Walchli M, Groth DF, McKinley MP, Prusiner SB, Weissmann C. 1986. Scrapie and cellular PrP isoforms are encoded by the same chromosomal gene. Cell 46:417–428. doi: 10.1016/0092-8674(86)90662-8. [DOI] [PubMed] [Google Scholar]
- 10.Bolton D, McKinley M, Prusiner S. 1982. Identification of a protein that purifies with the scrapie prion. Science 218:1309–1311. doi: 10.1126/science.6815801. [DOI] [PubMed] [Google Scholar]
- 11.Prusiner S. 1982. Novel proteinaceous infectious particles cause scrapie. Science 216:136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 12.Deleault N, Harris B, Rees J, Supattapone S. 2007. Formation of native prions from minimal components in vitro. Proc Natl Acad Sci U S A 104:9741–9746. doi: 10.1073/pnas.0702662104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang F, Wang X, Yuan C-G, Ma J. 2010. Generating a prion with bacterially expressed recombinant prion protein. Science 327:1132–1135. doi: 10.1126/science.1183748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Caughey B, Raymond G. 1991. The scrapie-associated form of PrP is made from a cell surface precursor that is both protease- and phospholipase-sensitive. J Biol Chem 266:18217–18223. [PubMed] [Google Scholar]
- 15.Pan K, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, Mehlhorn I, Huang Z, Fletterick R, Cohen F, al E. 1993. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci U S A 90:10962–10966. doi: 10.1073/pnas.90.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moore RA, Timmes AG, Wilmarth PA, Safronetz D, Priola SA. 2011. Identification and removal of proteins that co-purify with infectious prion protein improves the analysis of its secondary structure. Proteomics 11:3853–3865. doi: 10.1002/pmic.201100253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prusiner S. 1991. Molecular biology of prion diseases. Science 252:1515–1522. doi: 10.1126/science.1675487. [DOI] [PubMed] [Google Scholar]
- 18.Kimberlin R, Walker C. 1986. Pathogenesis of scrapie (strain 263K) in hamsters infected intracerebrally, intraperitoneally or intraocularly. J Gen Virol 67:255–263. doi: 10.1099/0022-1317-67-2-255. [DOI] [PubMed] [Google Scholar]
- 19.Kimberlin R, Walker C. 1982. Pathogenesis of mouse scrapie: patterns of agent replication in different parts of the CNS following intraperitoneal infection. J R Soc Med 75:618–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kimberlin R, Walker C. 1979. Pathogenesis of mouse scrapie: dynamics of agent replication in spleen, spinal cord and brain after infection by different routes. J Comp Pathol 89:551–562. doi: 10.1016/0021-9975(79)90046-X. [DOI] [PubMed] [Google Scholar]
- 21.Bruce ME. 1985. Agent replication dynamics in a long incubation period model of mouse scrapie. J Gen Virol 66:2517–2522. doi: 10.1099/0022-1317-66-12-2517. [DOI] [PubMed] [Google Scholar]
- 22.Dickinson A, Meikle V. 1971. Host-genotype and agent effects in scrapie incubation: change in allelic interaction with different strains of agent. Mol Gen Genet 112:73–79. doi: 10.1007/BF00266934. [DOI] [PubMed] [Google Scholar]
- 23.Dickinson A, Fraser H. 1969. Genetical control of the concentration of ME7 scrapie agent in mouse spleen. J Comp Pathol 79:363–366. doi: 10.1016/0021-9975(69)90051-6. [DOI] [PubMed] [Google Scholar]
- 24.Dickinson A, Meikle V. 1969. A comparison of some biological characteristics of the mouse-passaged scrapie agents, 22A and ME7. Genet Res 13:213–225. doi: 10.1017/S0016672300002895. [DOI] [PubMed] [Google Scholar]
- 25.Dickinson A, Meikle V, Fraser H. 1968. Identification of a gene which controls the incubation period of some strains of scrapie agent in mice. J Comp Pathol 78:293–299. doi: 10.1016/0021-9975(68)90005-4. [DOI] [PubMed] [Google Scholar]
- 26.Kascsak R, Rubenstein R, Merz P, Tonna-DeMasi M, Fersko R, Carp R, Wisniewski H, Diringer H. 1987. Mouse polyclonal and monoclonal antibody to scrapie-associated fibril proteins. J Virol 61:3688–3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caughey B, Raymond GJ, Bessen RA. 1998. Strain-dependent differences in beta-sheet conformations of abnormal prion protein. J Biol Chem 273:32230–32235. doi: 10.1074/jbc.273.48.32230. [DOI] [PubMed] [Google Scholar]
- 28.Ayers JI, Schutt CR, Shikiya RA, Aguzzi A, Kincaid AE, Bartz JC. 2011. The strain-encoded relationship between PrP replication, stability and processing in neurons is predictive of the incubation period of disease. PLoS Pathog 7:e1001317. doi: 10.1371/journal.ppat.1001317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Colby DW, Giles K, Legname G, Wille H, Baskakov IV, Dearmond SJ, Prusiner SB. 2009. Design and construction of diverse mammalian prion strains. Proc Natl Acad Sci U S A 106:20417–20422. doi: 10.1073/pnas.0910350106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Supattapone S. 2014. Synthesis of high titer infectious prions with cofactor molecules. J Biol Chem 289:19850–19854. doi: 10.1074/jbc.R113.511329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miller MB, Wang DW, Wang F, Noble GP, Ma J, Woods VL, Li S, Supattapone S. 2013. Cofactor molecules induce structural transformation during infectious prion formation. Structure 21:2061–2068. doi: 10.1016/j.str.2013.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deleault NR, Walsh DJ, Piro JR, Wang F, Wang X, Ma J, Rees JR, Supattapone S. 2012. Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proc Natl Acad Sci U S A 109:E1938–46. doi: 10.1073/pnas.1206999109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pattison IH, Millson GC. 1961. Scrapie produced experimentally in goats with special reference to the clinical syndrome. J Comp Pathol 71:101–109. doi: 10.1016/S0368-1742(61)80013-1. [DOI] [PubMed] [Google Scholar]
- 34.Thackray AM, Hopkins L, Lockey R, Spiropoulos J, Bujdoso R. 2011. Emergence of multiple prion strains from single isolates of ovine scrapie. J Gen Virol 92:1482–1491. doi: 10.1099/vir.0.028886-0. [DOI] [PubMed] [Google Scholar]
- 35.Thackray AM, Lockey R, Beck KE, Spiropoulos J, Bujdoso R. 2012. Evidence for co-infection of ovine prion strains in classical scrapie isolates. J Comp Pathol 147:316–329. doi: 10.1016/j.jcpa.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 36.Bartz JC, Bessen RA, McKenzie D, Marsh RF, Aiken JM. 2000. Adaptation and selection of prion protein strain conformations following interspecies transmission of transmissible mink encephalopathy. J Virol 74:5542–5547. doi: 10.1128/JVI.74.12.5542-5547.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parchi P, Castellani R, Capellari S, Ghetti B, Young K, Chen S, Farlow M, Dickson D, Sima A, Trojanowski J, Petersen R, Gambetti P. 1996. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol 39:767–778. doi: 10.1002/ana.410390613. [DOI] [PubMed] [Google Scholar]
- 38.Parchi P, Zou W, Wang W, Brown P, Capellari S, Ghetti B, Kopp N, Schulz-Schaeffer WJ, Kretzschmar HA, Head MW, Ironside JW, Gambetti P, Chen SG. 2000. Genetic influence on the structural variations of the abnormal prion protein. Proc Natl Acad Sci U S A 97:10168–10172. doi: 10.1073/pnas.97.18.10168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Telling GC, Parchi P, DeArmond S, Cortelli P, Montagna P, Gabizon R, Mastrianni J, Lugaresi E, Gambetti P, Prusiner S. 1996. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science 274:2079–2082. doi: 10.1126/science.274.5295.2079. [DOI] [PubMed] [Google Scholar]
- 40.Polymenidou M, Stoeck K, Glatzel M, Vey M, Bellon A, Aguzzi A. 2005. Coexistence of multiple PrPSc types in individuals with Creutzfeldt-Jakob disease. Lancet Neurol 4:805–814. doi: 10.1016/S1474-4422(05)70225-8. [DOI] [PubMed] [Google Scholar]
- 41.Notari S, Capellari S, Langeveld J, Giese A, Strammiello R, Gambetti P, Kretzschmar HA, Parchi P. 2007. A refined method for molecular typing reveals that co-occurrence of PrP(Sc) types in Creutzfeldt-Jakob disease is not the rule. Lab Invest 87:1103–1112. doi: 10.1038/labinvest.3700676. [DOI] [PubMed] [Google Scholar]
- 42.Schoch G, Seeger H, Bogousslavsky J, Tolnay M, Janzer RC, Aguzzi A, Glatzel M. 2006. Analysis of prion strains by PrPSc profiling in sporadic Creutzfeldt-Jakob disease. PLoS Med 3:e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Puoti G, Giaccone G, Rossi G, Canciani B, Bugiani O, Tagliavini F. 1999. Sporadic Creutzfeldt-Jakob disease: co-occurrence of different types of PrP(Sc) in the same brain. Neurology 53:2173–2176. doi: 10.1212/WNL.53.9.2173. [DOI] [PubMed] [Google Scholar]
- 44.Notari S, Capellari S, Giese A, Westner I, Baruzzi A, Ghetti B, Gambetti P, Kretzschmar HA, Parchi P. 2004. Effects of different experimental conditions on the PrPSc core generated by protease digestion: implications for strain typing and molecular classification of CJD. J Biol Chem 279:16797–16804. doi: 10.1074/jbc.M313220200. [DOI] [PubMed] [Google Scholar]
- 45.Dickinson A, Fraser H, Meikle V, Outram G. 1972. Competition between different scrapie agents in mice. Nat New Biol 237:244. [DOI] [PubMed] [Google Scholar]
- 46.Dickinson AG, Fraser H, McConnell I, Outram GW, Sales DI, Taylor DM. 1975. Extraneural competition between different scrapie agents leading to loss of infectivity. Nature 253:556. [DOI] [PubMed] [Google Scholar]
- 47.Manuelidis L. 1998. Vaccination with an attenuated Creutzfeldt-Jakob disease strain prevents expression of a virulent agent. Proc Natl Acad Sci U S A 95:2520–2525. doi: 10.1073/pnas.95.5.2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bartz JC, Kramer ML, Sheehan MH, Hutter JAL, Ayers JI, Bessen RA, Kincaid AE. 2007. Prion interference is due to a reduction in strain-specific PrPSc levels. J Virol 81:689–697. doi: 10.1128/JVI.01751-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
- 50.Wilson M, McBride P. 2000. Technical aspects of tracking scrapie infection in orally dosed rodents. J Cell Pathol 5:17–22. [Google Scholar]
- 51.Ayers J, Kincaid AE, Bartz JC. 2009. Prion strain targeting independent of strain-specific neuronal tropism. J Virol 83:81–87. doi: 10.1128/JVI.01745-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kincaid AE, Bartz JC. 2007. The nasal cavity is a route for prion infection in hamsters. J Virol 81:4482–4491. doi: 10.1128/JVI.02649-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shikiya RA, Eckland TE, Young AJ, Bartz JC. 2014. Prion formation, but not clearance, is supported by protein misfolding cyclic amplification. Prion 8:415–420. doi: 10.4161/19336896.2014.983759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kramer ML, Bartz JC. 2009. Rapid, high-throughput detection of PrPSc by 96-well immunoassay. Prion 3:44–48. doi: 10.4161/pri.3.1.8442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Scott J, Fraser H. 1989. Transport and targeting of scrapie infectivity and pathology in the optic nerve projections following intraocular infection. Prog Clin Biol Res 317:645–652. [PubMed] [Google Scholar]
- 56.Fraser H, Dickinson A. 1985. Targeting of scrapie lesions and spread of agent via the retino-tectal projection. Brain Res 346:32–41. doi: 10.1016/0006-8993(85)91091-1. [DOI] [PubMed] [Google Scholar]
- 57.Ingrosso L, Pisani F, Pocchiari M. 1999. Transmission of the 263K scrapie strain by the dental route. J Gen Virol 80:3043–3047. doi: 10.1099/0022-1317-80-11-3043. [DOI] [PubMed] [Google Scholar]
- 58.Bartz JC, Kincaid AE, Bessen RA. 2003. Rapid prion neuroinvasion following tongue infection. J Virol 77:583–591. doi: 10.1128/JVI.77.1.583-591.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bartz JC, Kincaid AE, Bessen RA. 2002. Retrograde transport of transmissible mink encephalopathy within descending motor tracts. J Virol 76:5759–5768. doi: 10.1128/JVI.76.11.5759-5768.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bassant M, Baron H, Gumpel M, Cathala F, Court L. 1986. Spread of scrapie agent to the central nervous system: study of a rat model. Brain Res 383:397–401. doi: 10.1016/0006-8993(86)90048-X. [DOI] [PubMed] [Google Scholar]
- 61.Kimberlin R, Hall S, Walker C. 1983. Pathogenesis of mouse scrapie. Evidence for direct neural spread of infection to the CNS after injection of sciatic nerve. J Neurol Sci 61:315–325. [DOI] [PubMed] [Google Scholar]
- 62.Hecker R, Taraboulos A, Scott M, Pan KM, Yang SL, Torchia M, Jendroska K, Dearmond SJ, Prusiner SB. 1992. Replication of distinct scrapie prion isolates is region specific in brains of transgenic mice and hamsters. Genes Dev 6:1213–1228. doi: 10.1101/gad.6.7.1213. [DOI] [PubMed] [Google Scholar]
- 63.Shikiya RA, Ayers JI, Schutt CR, Kincaid AE, Bartz JC. 2010. Coinfecting prion strains compete for a limiting cellular resource. J Virol 84:5706–5714. doi: 10.1128/JVI.00243-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Collinge J, Clarke A. 2007. A general model of prion strains and their pathogenicity. Science 318:930–936. doi: 10.1126/science.1138718. [DOI] [PubMed] [Google Scholar]
- 65.Mahal SP, Browning S, Li J, Suponitsky-Kroyter I, Weissmann C. 2010. Transfer of a prion strain to different hosts leads to emergence of strain variants. Proc Natl Acad Sci U S A 107:22653–22658. doi: 10.1073/pnas.1013014108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li J, Browning S, Mahal SP, Oelschlegel AM, Weissmann C. 2010. Darwinian evolution of prions in cell culture. Science 327:869–872. doi: 10.1126/science.1183218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gonzalez-Montalban N, Lee YJ, Makarava N, Savtchenko R, Baskakov IV. 2013. Changes in prion replication environment cause prion strain mutation. FASEB J 27:3702–3710. doi: 10.1096/fj.13-230466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Makarava N, Savtchenko R, Baskakov IV. 2013. Selective amplification of classical and atypical prions using modified protein misfolding cyclic amplification. J Biol Chem 288:33–41. doi: 10.1074/jbc.M112.419531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nilsson KPR, Joshi-Barr S, Winson O, Sigurdson CJ. 2010. Prion strain interactions are highly selective. J Neurosci 30:12094–12102. doi: 10.1523/JNEUROSCI.2417-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yull HM, Ritchie DL, Langeveld JPM, van Zijderveld FG, Bruce ME, Ironside JW, Head MW. 2006. Detection of type 1 prion protein in variant Creutzfeldt-Jakob disease. Am J Pathol 168:151–157. doi: 10.2353/ajpath.2006.050766. [DOI] [PMC free article] [PubMed] [Google Scholar]