

ABSTRACT
Multiple subunits of the hepatitis B virus (HBV) core protein (HBc) assemble into an icosahedral capsid that packages the viral pregenomic RNA (pgRNA). The N-terminal domain (NTD) of HBc is sufficient for capsid assembly, in the absence of pgRNA or any other viral or host factors, under conditions of high HBc and/or salt concentrations. The C-terminal domain (CTD) is deemed dispensable for capsid assembly although it is essential for pgRNA packaging. We report here that HBc expressed in a mammalian cell lysate, rabbit reticulocyte lysate (RRL), was able to assemble into capsids when (low-nanomolar) HBc concentrations mimicked those achieved under conditions of viral replication in vivo and were far below those used previously for capsid assembly in vitro. Furthermore, at physiologically low HBc concentrations in RRL, the NTD was insufficient for capsid assembly and the CTD was also required. The CTD likely facilitated assembly under these conditions via RNA binding and protein-protein interactions. Moreover, the CTD underwent phosphorylation and dephosphorylation events in RRL similar to those seen in vivo which regulated capsid assembly. Importantly, the NTD alone also failed to accumulate in mammalian cells, likely resulting from its failure to assemble efficiently. Coexpression of the full-length HBc rescued NTD assembly in RRL as well as NTD expression and assembly in mammalian cells, resulting in the formation of mosaic capsids containing both full-length HBc and the NTD. These results have important implications for HBV assembly during replication and provide a facile cell-free system to study capsid assembly under physiologically relevant conditions, including its modulation by host factors.
IMPORTANCE Hepatitis B virus (HBV) is an important global human pathogen and the main cause of liver cancer worldwide. An essential component of HBV is the spherical capsid composed of multiple copies of a single protein, the core protein (HBc). We have developed a mammalian cell-free system in which HBc is expressed at physiological (low) concentrations and assembles into capsids under near-physiological conditions. In this cell-free system, as in mammalian cells, capsid assembly depends on the C-terminal domain (CTD) of HBc, in contrast to other assembly systems in which HBc assembles into capsids independently of the CTD under conditions of nonphysiological protein and salt concentrations. Furthermore, the phosphorylation state of the CTD regulates capsid assembly and RNA encapsidation in the cell-free system in a manner similar to that seen in mammalian cells. This system will facilitate detailed studies on capsid assembly and RNA encapsidation under physiological conditions and identification of antiviral agents that target HBc.
INTRODUCTION
Hepatitis B virus (HBV) remains an important global pathogen that chronically infects hundreds of millions of people worldwide and causes hepatitis, cirrhosis, and liver cancer (1, 2). HBV is an enveloped virus with an inner capsid shell which, in turn, encloses a small (3.2-kbp) DNA genome. As a member of the Hepadnaviridae family, which also includes related animal viruses such as the duck hepatitis B virus (DHBV), HBV replicates its DNA genome via reverse transcription of an RNA intermediate called pregenomic RNA (pgRNA) (3–5). HBV assembly begins with the formation of a nucleocapsid (NC) that packages specifically a copy of pgRNA, in complex with the virally encoded reverse transcriptase (RT) protein (6, 7). The pgRNA is then converted to DNA within NCs by RT.
The icosahedral HBV capsid shell enclosing the viral RNA or DNA is composed of multiple copies of a single viral protein, the HBV core (capsid) protein (HBc). This small (ca. 21-kDa) protein has multiple essential functions in the viral life cycle. In addition to capsid assembly and packaging of pgRNA and RT as mentioned above, HBc functions also include regulation of viral reverse transcription, NC envelopment and virion secretion, and nuclear import of the viral DNA (2, 5, 8). The HBc can be divided into an N-terminal domain (NTD) (from position 1 to position 140) responsible for capsid assembly (thus also called the assembly domain) and a C-terminal domain (CTD) (position 149 to position 183 or 185, depending on the strains), which are connected by a linker region (position 140 to position 149) (Fig. 1A). The basic building blocks of the HBV capsid are HBc dimers, with 90 or 120 dimers self-assembling into a T = 3 or T = 4 icosahedral capsid (9–11). In heterologous overexpression systems, including bacterial and insect cells and in vitro assembly reactions using purified protein, the NTD alone, without the CTD, is clearly sufficient for assembly into capsids that are morphologically similar to authentic capsids assembled from full-length HBc (11–14). On the other hand, the highly basic and arginine-rich (protamine-like) CTD shows nonspecific RNA and DNA binding and nucleic acid chaperone activities (15, 16), plays an essential role in viral RNA packaging (17) and DNA synthesis (17, 18), and regulates HBc nuclear localization (19, 20).
FIG 1.

Expression of WT and mutant HBc proteins in RRL. (A) Schematic diagram of HBc domain structure and the CTD sequence. The three major phosphorylation sites in CTD (S155, S162, and S170) are marked above the sequence and the four minor sites (T160, S168, S176, and S178) below. pCI-HBc-3A (3A) and 3E have the three major sites changed to A and E, respectively, whereas pCI-HBc-7A (7A) and 7E have all seven sites substituted. (B) Estimation of HBc concentration as expressed in RRL. Each lane contained 3 μl of the translation reaction mixture. The 35S-labeled HBc proteins were detected by autoradiography (top) and by Western blot analysis using the anti-HBc NTD MAb (bottom). (C) Translation in RRL and capsid assembly schemes. The five different assembly conditions are listed at the bottom. See the text for details. C, WT HBc protein; C149, C-terminally truncated HBc protein (terminated at position 149); CIAP, calf intestine alkaline phosphatase; PPI, phosphatase inhibitors.
The HBc CTD undergoes dynamic phosphorylation and dephosphorylation events that regulate its nucleic acid binding, subcellular localization, and other functions such as pgRNA packaging and DNA synthesis (19–27). The nonspecific RNA binding activity of CTD allows incorporation of nonspecific RNAs into capsids assembled in vitro using purified HBc or in bacteria (11, 13, 28, 29). How capsid assembly discriminates the specific viral pgRNA versus nonspecific RNAs during viral replication remains to be elucidated. It is known, however, that CTD phosphorylation is required for specific viral RNA packaging (25, 26, 30). Also, in insect or mammalian cells where CTD is phosphorylated, HBV capsids assembled from full-length HBc with intact CTD package little to no nonspecific RNA (12, 31, 32), in contrast to those assembled in bacteria where HBc is unphosphorylated, consistent with the inhibition of CTD RNA binding by its phosphorylation (21, 22).
The CTD contains three major S phosphorylation sites (S155, S162, and S170) (19, 33, 34), and three additional S/T phosphorylation sites (T160, S168, and S176) have recently been identified (30) (Fig. 1A). Besides these six known phosphorylation sites, another potential CTD phosphorylation site (S178) (Fig. 1A) is also conserved among most HBV isolates (20). As the virus does not encode any protein kinase, CTD phosphorylation is mediated exclusively by host cell kinases, including cyclin-dependent kinase 2 (CDK2) and protein kinase C (PKC) (22, 34). To date, no information has been available on the cellular phosphatase(s) that mediates CTD dephosphorylation, which accompanies viral reverse transcription and is required during second-strand DNA synthesis, as shown in DHBV (23, 24, 35). HBc dephosphorylation is thought to maintain the interior charge balance as more-negative charges inside the NC build up due to the conversion of the single-stranded pgRNA to the double-stranded DNA (23, 36, 37).
We report here the development of a cell-free system that supports HBV capsid assembly based on a widely used mammalian cell-free translation extract, rabbit reticulocyte lysate (RRL). We found that, in contrast to previous results obtained using overexpression systems and nonphysiological in vitro assembly reactions, the HBc CTD, in addition to the NTD, was required to facilitate capsid assembly under conditions mimicking viral replication in the mammalian cell lysate as well as in mammalian cells. Furthermore, we found that CTD phosphorylation dynamics, as regulated by endogenous cellular kinases and phosphatases, could regulate capsid assembly in the cell lysate.
MATERIALS AND METHODS
Plasmids.
pCI-HBc, pCI-HBc-3A, pCI-HBc-3E, pCI-HBc-7A, and pCI-HBc-7E were constructed by inserting into the pCI vector (Promega) between the NheI and SalI sites the wild-type (WT) or mutant HBc coding sequences, which were amplified by PCR using primers containing the designated serine/threonine-to-alanine or glutamic acid substitutions (Fig. 1A) (20). pCI-HBc-182, -149, and -149cys, which are used to express, respectively, a mutant HBc truncated at position 182 missing the last Cys residue, HBc truncated at position 149, and HBc truncated at position 149 plus a terminal Cys residue, were similarly constructed via PCR mutagenesis. The pCI plasmids were used to express the HBc proteins in mammalian cells as well as during in vitro transcription and translation.
In vitro translation in rabbit reticulocyte lysate.
A TNT-coupled rabbit reticulocyte lysate (RRL) in vitro translation system (Promega) was used to express the HBc proteins as recommended in the manufacturer's protocol. In vitro-translated proteins were labeled by the use of [35S]methionine. The translation reaction mixture contained ca. 125 mM KCl and 2.5 mM Mg++ (Promega).
In vitro capsid assembly in RRL.
Unless specifically indicated otherwise, the general assembly reaction mixtures included 1 to 3 μl of translation products per 10 μl final reaction volume in 1× buffer 3 (100 mM NaCl, 50 mM Tris-HCl, 10 mM MgCl2, 1 mM dithiothreitol, pH 7.9; New England BioLabs, or NEB) supplemented with 1× EDTA-free protease inhibitor cocktail (Roche) and 0.8 U/μl RNasin Plus RNase inhibitor (Promega). The reaction mixtures were incubated overnight (16 h) at 37°C unless indicated otherwise. Where indicated, additional enzymatic manipulations were performed as described below using the same reaction buffer.
(i) Exogenous phosphatase treatment.
One unit of calf intestinal alkaline phosphatase (CIAP) (New England BioLabs) per microliter of the final reaction volume was added at the end of translation, and the reaction mixture was incubated for 16 h at 37°C.
(ii) Phosphatase inhibitor treatment.
A cocktail of nonspecific phosphatase inhibitors (10 mM NaF, 50 mM β-glycerophosphate, 10 mM sodium pyrophosphate, and 2 mM sodium orthovanadate; all final concentrations) was added at the end of the translation, and the reaction mixture was incubated for 16 h at 37°C.
(iii) RNase A digestion.
RNase A (100 μg/ml final concentration) was added at the end of the translation and incubated for 16 h at 37°C or was added at the end of the 16-h assembly reaction, and the reaction mixture was incubated for another 1 h.
Agarose gel electrophoresis and capillary transfer.
35S-labeled translation reaction mixtures were resolved by 1% agarose gel electrophoresis (100 V for ∼3 h) in 1× TAE (40 mM Tris, 20 mM acetic acid, 1 mM EDTA; pH 8.3), using 6× DNA Loading Buffer Blue (New England BioLabs). The gels were then soaked in 10× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) (pH 7.0) for approximately 30 min and transferred overnight to nitrocellulose membrane by capillary transfer. The next day, the membranes were UV cross-linked and dried in a vacuum oven for 2 h. The translation products were analyzed by autoradiography and Western blot analysis with the mouse monoclonal antibody (MAb) against the HBc NTD (32). Cytoplasmic lysates of transfected cells containing HBV capsids were resolved similarly by agarose gel electrophoresis and detected by Western blot analysis using the HBc NTD MAb as previously described (34, 38). Viral RNA associated with the capsids was detected by using an antisense HBV riboprobe as described before (38, 39). To detect total RNA associated with the capsids and the capsid protein signal by nucleic acid and protein staining, using Sybr gold nucleic acid gel stain and Sypro ruby, respectively (32, 34), cytoplasmic lysate was first treated with 0.125 U/μl micrococcal nuclease and 200 μg/μl RNase A for 1 h and 30 min at 37°C to degrade RNAs not protected by the capsids, followed by proteinase K digestion (0.5 mg/ml) for 1 h at 37°C to remove the bulk of cytoplasmic proteins but not the capsids (38, 40). The treated lysate was then resolved similarly by agarose gel electrophoresis and imaged using a Bio-Rad GelDoc MP system after the staining.
Sucrose gradient fractionation of HBc capsids assembled in RRL.
HBc (WT or phosphorylation site mutants) was translated and subjected to 35S labeling in a 100-μl volume in RRL. Where indicated, the translation products were subjected to the indicated enzymatic reactions that were determined to induce capsid assembly most efficiently. The reaction mixtures were layered over a 15% to 30% continuous sucrose gradient (5 ml) and spun in an SW55Ti rotor at 27,000 rpm for 4 h at 4°C (24, 32). Fractions (200 μl) were collected from top to bottom. Individual fractions (10 μl per fraction) along with 0.5 μl of translation reaction input were resolved on a 1% agarose gel. The gel was transferred by capillary transfer to nitrocellulose membrane, and the capsids were detected by autoradiography or Western blot analysis using the NTD MAb as described above.
Cell cultures and transient transfections.
Cells from the HEK293 human embryonic kidney cell line (a gift from Zhijun Luo at Boston University) were cultured in Dulbecco's modified Eagle's medium (DMEM)-F12 medium supplemented with 10% fetal bovine serum (FBS) (HyClone), as previously described (41). HEK293 cells were transfected using a CalPhos mammalian transfection kit (Clontech) (42).
SDS-PAGE and Western blot analysis.
35S-labeled translation reaction mixtures were resolved by SDS-PAGE on 15% or 12.5% polyacrylamide gels. The resolved proteins were transferred to a polyvinylidene difluoride (PVDF) membrane and detected by autoradiography or Western blot analysis as described above. Cytoplasmic lysates of transfected cells containing HBV capsids were resolved similarly and detected by Western blot analysis using the HBc NTD MAb as described above. Where indicated, the phosphorylation state of HBc CTD was analyzed using a rabbit polyclonal antibody (C170) raised against a nonphosphorylated CTD peptide (from position 170 to the C terminus) (32, 43) or using two rabbit MAbs, 25-7 and 6-1, which were custom-made (by Abcam) using a CTD peptide (from residue 152 to the C terminus) as an immunogen.
Large-scale purification of HBV capsids from transfected cells.
HEK293 cells were transfected with pCI-HBc, -3A, or -3E. Cytoplasmic lysate preparation and subsequent sucrose gradient fractionation were performed as described before (24, 32, 34). Briefly, the cell lysate was loaded on top of a linear 15% to 30% sucrose gradient. Ultracentrifugation was then performed in an SW32 rotor at 27,000 rpm for 4 h at 4°C. RNA and protein signals associated with purified capsids, following resolution on an agarose gel, were detected by staining with Sybr gold nucleic acid gel stain and Sypro ruby, respectively, as described above. To concentrate the capsids from the sucrose gradient fraction, approximately 200 μl of the peak capsid fractions was mixed with 0.8 ml TNE (10 mM Tris, 150 mM NaCl, 1 mM EDTA, pH 8.0)–1% NP-40–0.05% β-mercaptoethanol supplemented with protease inhibitors. NaCl and polyethylene glycol (PEG) were added to reach 500 mM and a 10% final concentration, respectively, followed by rotation at room temperature for ca. 30 min to dissolve the PEG. The capsids were then precipitated on ice overnight and recovered by centrifugation at 12,000 rpm for 20 min at 4°C in a microcentrifuge. The capsid pellet was resuspended in 10 to 20 μl TNE–1% NP-40–0.05% β-mercaptoethanol supplemented with protease inhibitors.
IP.
The sucrose capsid peak fraction was diluted in the immunoprecipitation (IP) buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1 mM EDTA, 1% NP-40, 1× protease inhibitor cocktail [Roche]). The anti-CTD rabbit MAb (6-1) or a control IgG antibody prebound to protein A/G beads (Sigma) was used to immunoprecipitate the full-length HBc and any associated C149 in native capsids, as previously described (33, 38). Bound proteins were eluted by boiling the beads in SDS sample buffer, resolved by SDS-PAGE, and detected by Western blot analysis using the HBc NTD MAb (32).
RESULTS
Capsid assembly from HBc expressed in RRL required CTD and could be modulated by manipulating the state of CTD phosphorylation.
We were intrigued by previous reports that HBV capsid could assemble efficiently in Xenopus oocytes when HBc concentrations were less than 1 μM (44), whereas the concentrations required for assembly in vitro with purified HBc NTD at physiological salt concentrations were much higher (ca. 40 to 80 μM) (11, 45). We decided to test the possibility that HBc translated in RRL may also be able to assemble into capsids, even if its concentrations were much lower than those required for assembly using purified HBc. RRL was chosen for in vitro expression due to its ability not only to efficiently translate a variety of proteins but also to facilitate their folding and posttranslational modifications (see, e.g., references 34 and 46). To allow sufficient time for assembly to occur, we incubated HBc in the translation mixture overnight at 37°C, after dilution in a buffer with physiological salt concentrations (see Materials and Methods) (Fig. 1C). As we have recently shown that RRL contains protein kinases, including CDK2, that can phosphorylate DHBc at the same authentic CTD sites as those phosphorylated in vivo (33, 34), we considered it possible that HBc could be phosphorylated by a cellular kinase(s) in RRL as well. Furthermore, since the CTD phosphorylation state might influence capsid assembly (e.g., by affecting CTD interactions with nonspecific RNA), we tested not only the wild type (WT) HBc but also a number of HBc mutants with CTD substitutions that mimic either the nonphosphorylated state (S/T to A) or phosphorylated state (S/T to E) (Fig. 1A) (20, 34). In addition, we included the HBc truncation mutation (C149) containing only the assembly domain with the entire CTD removed, which has been widely used for capsid assembly in bacteria and from purified proteins.
We first estimated the concentrations of the various HBc proteins when they were translated in RRL. A quantitative Western blot analysis was performed using a serial dilution of recombinant HBV capsids purified from bacteria as standards (Fig. 1B, bottom). As HBc translated in RRL could be labeled with [35S]methionine, it could be detected easily also by autoradiography (Fig. 1B, top). We found that ca. 0.5 to 1 ng HBc was made per microliter of translation mixture (Fig. 1B, lanes 1 to 5) independently of the CTD phosphorylation site substitutions, giving an apparent HBc concentration in RRL of ca. 25 to 50 nM (monomer). The CTD-deleted construct, C149, was expressed at a somewhat (ca. 2-fold) higher concentration, i.e., 100 nM (Fig. 1B, lane 6). Thus, the HBc concentrations achieved by translation in RRL were close to but still below the HBc concentrations in Xenopus oocytes (ca. 1 μM) and were far below the threshold required to trigger assembly from purified HBc NTD (40 to 80 μM). Furthermore, as the translation reaction mixtures were diluted ca. 5-fold during the overnight incubation following translation, the HBc concentration during this (“assembly”) incubation period was even lower (ca. 5 to 10 nM). We then attempted to detect any capsids that might have formed by resolving the translation mixture on an agarose gel, in parallel with a capsid standard purified from bacteria as a control (Fig. 2A). A distinct HBc band migrating at the same position on the agarose gel as the capsid standard (lane 11) was detected by both autoradiography (top) and Western blot analysis (bottom) in the translation reaction mixtures of mutants HBc-3A (3A) (lane 3) and 3E (lane 5), after the overnight incubation in the absence of any additional manipulation. The 3A and 3E mutants had the three major CTD phosphorylation sites changed to A or E but retained the four minor phosphorylation sites (Fig. 1A). In contrast, WT HBc migrated much more slowly as a smear near the top of the gel (lane 1). The CTD-deleted C149 ran above the capsid band also but did so much more quickly than WT HBc (lane 8). As described in detail below, the HBc band comigrating with the capsid standard on the agarose gel was indeed verified to represent assembled capsids by sucrose gradient centrifugation; all HBc signals running above this band, including WT HBc and C149, represented unassembled subunits.
FIG 2.

HBV capsid assembly in RRL and effects of exogenous phosphatase and phosphatase inhibitors on assembly. The WT and mutant HBc proteins or the control luciferase (Luc) was translated in RRL. All samples were resolved by agarose gel electrophoresis. (A) The indicated protein translated in RRL was incubated overnight at 37°C in 1× NEB restriction digestion buffer 3 alone (lanes 1, 3, 5, 7, and 9) or with CIAP (lanes 2, 4, 6, 8, and 10) before resolution on the gel. The recombinant HBV capsid (rHBc) purified from E. coli was loaded in lane 11. (B) The indicated translation reaction mixture was loaded directly following translation upon dilution in double-distilled water (dH2O) and without the overnight incubation (i.e., no assembly reaction) (lanes 1, 5, 9, and 13), after dilution in NEB buffer 3 (buffer 3) but without the overnight incubation (lanes 2, 6, 10, and 14), after dilution in buffer 3 and with incubation overnight at 37°C (lanes 3, 7, 11, and 15), or after overnight incubation in buffer 3 and with CIAP (lanes 4, 8, 12, and 16). (C) The indicated translation reaction mixture was loaded directly following translation upon dilution in dH2O and without the overnight incubation (i.e., no assembly reaction) (lanes 1, 5, and 9), after dilution in dH2O and with incubation overnight at 37°C (lanes 2, 6, and 10), after dilution in buffer 3 and with incubation overnight at 37°C (lanes 3, 7, and 11), or after overnight incubation at 37°C in buffer 3 and with a mixture of phosphatase inhibitors (PPI) (lanes 4, 8, and 12). Each lane contained 3 μl translation product except that 3.125 ng rHBc was loaded in lane 11 of panel A. 35S signals were detected by autoradiography (top). The HBc proteins were also detected by the MAb antibody against the NTD (bottom). C/3A/3E, WT, 3A, or 3E HBc subunits (i.e., not present in the capsid); C149, C-terminally truncated HBc protein (terminated at position 149); C-deP, dephosphorylated WT HBc subunits; Ca, capsids.
Given the likelihood of HBc phosphorylation in RRL as mentioned above, the apparent differences in assembly between the WT and the 3A or 3E phosphorylation mutant HBc further suggested that the phosphorylation state of the CTD, at the three major sites, could affect HBc assembly in RRL. We thus decided to treat the translation mixture during the overnight assembly period (i.e., following translation) with an exogenous phosphatase, CIAP, to see if dephosphorylation of the different HBc proteins mediated by CIAP could affect capsid assembly (Fig. 1C). Remarkably, CIAP treatment induced the assembly of WT HBc (Fig. 2A, lane 2) but did not have any significant effect on the assembly of the 3A or 3E mutant (Fig. 2A, lanes 4 and 6), which assembled even without CIAP treatment as described above. CIAP treatment also did not affect the mobility of the C149 truncation mutant or the unrelated luciferase protein (negative control) translated in RRL (Fig. 2A, lanes 7 to 10), indicating that the effect of CIAP treatment was mediated through the HBc CTD.
Because the buffer components were also modified somewhat in both the mock- and CIAP-treated reaction mixtures whose results are shown in Fig. 2A (and repeated in Fig. 2B [lanes 3, 7, 11, and 15 for mock incubation and lanes 4, 8, 12, and 16 for CIAP incubation]), due to the addition of the incubation buffer (“buffer 3”) (see Materials and Methods), which might have, by itself, affected HBc assembly or migration, we added the same buffer to another set of translation reaction mixtures but omitted the overnight incubation (Fig. 2B, lanes 2, 6, 10, and 14). In addition, we ran on the same gel another set of samples that neither had the buffer components nor underwent the overnight incubation (Fig. 2B, lanes 1, 5, 9, and 13). The results seen with the mock and CIAP incubations represented in Fig. 2B were the same as those represented in Fig. 2A for the WT, 3A, 3E, and C149 mutants (lanes 3, 4, 7, 8, 11, 12, 15, and 16); i.e., 3A and 3E could assemble with or without CIAP, WT assembly required CIAP, and C149 did not assemble even with CIAP, following the overnight incubation in the buffer (see also Fig. 4, lanes 2, 3, 8, 9, 14, 15, 32, and 33). Inclusion of the assembly buffer components alone, without the overnight incubation, was insufficient to induce assembly of any of the HBc proteins tested, and the migration of the unassembled HBc proteins with the buffer components was similar to that seen without the buffer components (Fig. 2B, lanes 1, 2, 5, 6, 9, 10, 13, and 14). Interestingly, the mobility of WT HBc was affected by the mock treatment (no CIAP) overnight incubation, which caused it to migrate even more slowly and as a broader smear (Fig. 2B, lane 3 versus lane 2; see also Fig. 4, lane 2 versus lane 1). As in Fig. 2A, the mobility of C149 was not affected by any treatment (Fig. 2B, lanes 13 to 16; see also Fig. 4, lanes 31 to 33), indicating again that the effect of overnight mock incubation or CIAP treatment was mediated through the CTD.
FIG 4.

Effects of exogenous phosphatase and RNase treatment on capsid assembly in RRL. The indicated HBc proteins were translated in RRL, and the translation reaction mixtures were resolved by agarose gel electrophoresis (top panels) or SDS-PAGE (bottom panels) without any further treatment (lanes 1, 7, 13, 19, 25, and 31) or were treated with NEB buffer 3 alone overnight at 37°C (buffer) (lanes 2, 8, 14, 20, 26, and 32), with buffer 3 plus CIAP overnight at 37°C (CIAP) (lanes 3, 9, 15, 21, 27, and 33), with buffer 3 plus CIAP overnight at 37°C followed by RNase treatment for one additional hour (CIAP-RNase) (lanes 4, 10, 16, 22, 28, and 34), with RNase for 1 h followed by buffer 3 plus CIAP overnight at 37°C (lanes 5, 11, 17, 23, 29, and 35), or with the mixture of phosphatase inhibitors overnight at 37°C (lanes 6, 12, 18, 24, 30, and 36). All lanes contained 2 μl translation products. 35S-labeled HBc proteins were detected by autoradiography. C, 3A, and 3E/7A, WT or mutant HBc subunits; C149, C-terminally truncated HBc protein (terminated at position 149); C-deP, dephosphorylated HBc subunits; Ca, capsids.
The effects of the 37°C mock incubation and the exogenous phosphatase on HBc migration or capsid assembly suggested that an endogenous phosphatase(s) present in RRL might have affected these parameters. We thus tested the effects of phosphatase inhibitors on the mobility and assembly by the WT and mutant HBc proteins in RRL, all in the absence of any exogenous phosphatase (Fig. 1C). We used a mixture of nonspecific phosphatase inhibitors (see Materials and Methods) for this purpose. As shown in Fig. 2C, addition of the phosphatase inhibitors during the assembly incubation period completely blocked capsid assembly by 3E (lane 12; see also Fig. 4, lane 18), slightly decreased assembly by 3A (lane 8; see also Fig. 4, lane 12), and prevented the mobility change of WT HBc (lane 4; see also Fig. 4, lane 6). These results thus confirmed our suspicion that RRL indeed contained an endogenous cellular phosphatase(s) that could mediate CTD dephosphorylation and consequently was responsible for inducing capsid assembly by 3E and, to a lesser extent, by 3A. In the case of WT HBc, the presence of the putative endogenous phosphatase, in contrast to that of the exogenous CIAP, was apparently insufficient to trigger capsid assembly but could induce the mobility upshift, presumably as a result of dephosphorylation that was less complete than that caused by the exogenous CIAP treatment.
To verify CTD phosphorylation in RRL and dephosphorylation by CIAP treatment, we employed a polyclonal anti-CTD antibody (C170) raised against a synthetic peptide corresponding to the last 14 residues of nonphosphorylated HBc (43), which is predicted to react preferentially with nonphosphorylated CTD. As anticipated, the C170 antibody detected readily the nonphosphorylated HBc expressed in Escherichia coli, like the anti-NTD MAb (Fig. 3A, lane 1, top panel). However, C170 failed to detect HBc translated in RRL (Fig. 3A, lane 2, top panel) but could do so after the translated HBc was treated with CIAP (Fig. 3A, lane 5, top panel). This was the case despite the finding that more untreated HBc than CIAP-treated HBc was loaded, as detected by the anti-NTD MAb (Fig. 3A, lanes 2 and 5, lower middle panels), which should detect the various HBc proteins independently of the state of phosphorylation of the CTD. Since the C170 antibody used for the experiments whose results are presented in Fig. 3A was raised against the CTD peptide consisting of positions 170 to 183, these results indicated that at least S170, S176, and/or S178 in CTD was indeed phosphorylated in RRL and dephosphorylated by CIAP treatment. Furthermore, we confirmed these results with a rabbit monoclonal antibody raised against CTD, 25-7 (see Materials and Methods for details), which again reacted preferentially with nonphosphorylated CTD and produced essentially the same results as C170 (Fig. 3A, lanes 2 and 5, upper middle panels). Although the exact epitope recognized by 25-7 remains to be defined, these results confirmed that CTD was phosphorylated in RRL and that CIAP caused CTD dephosphorylation as expected. Another monoclonal anti-CTD antibody, 6-1, which reacts with the extreme C-terminal sequence of CTD (see Fig. 7), detected the HBc proteins the same way as the anti-NTD MAb (Fig. 3A, bottom panels), indicating that 6-1, like the anti-NTD MAb, could detect HBc independently of the state of phosphorylation of the CTD. This is consistent with the lack of any potential sites of phosphorylation within the last 5 residues of HBc (Fig. 1A).
FIG 3.
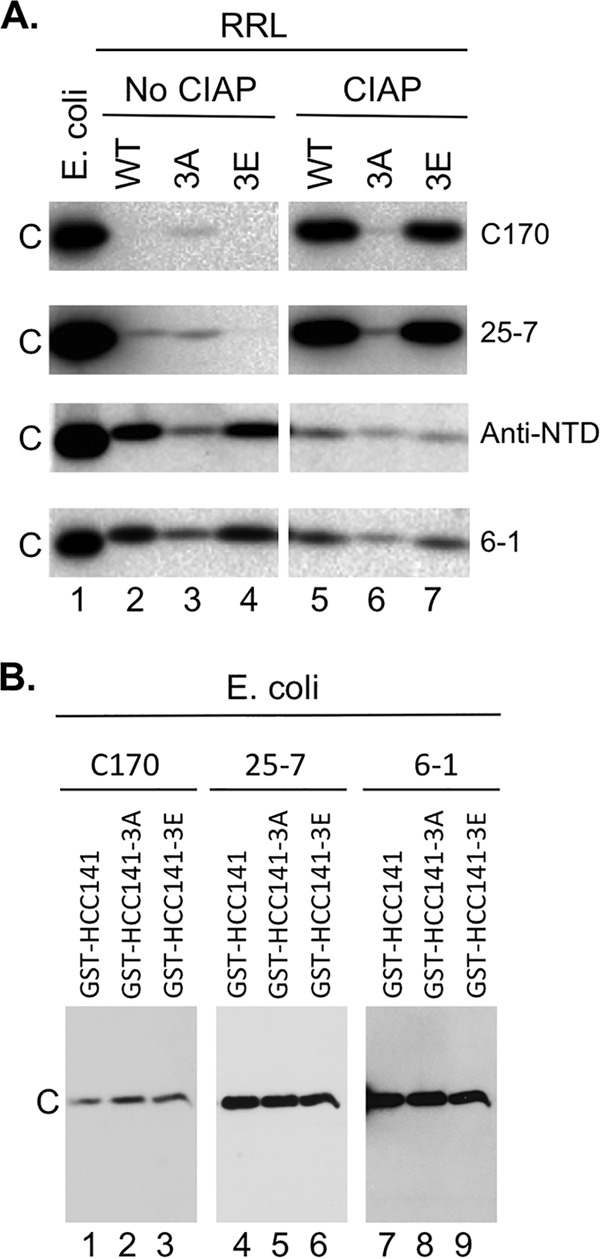
Confirmation of HBc phosphorylation in RRL and dephosphorylation by CIAP using CTD antibodies. (A) WT HBc (lanes 2 and 5), 3A (lanes 3 and 6), or 3E (lanes 4 and 7) was expressed in RRL and resolved by SDS-PAGE, with prior CIAP treatment (lanes 5 to 7) or without prior CIAP treatment (lanes 2 to 4), and was detected by the anti-CTD antibodies (C170, top panels; 25-7, upper middle panels; 6-1, bottom panels) or the anti-NTD MAb (lower middle panels). WT HBc expressed in E. coli (lane 1) was analyzed in parallel. (B) Equal amounts (15 ng) of the indicated GST-CTD fusion proteins purified from E. coli (thus all nonphosphorylated) were resolved by SDS-PAGE and detected by the C170 (lanes 1 to 3), 25-7 (lanes 4 to 6), or 6-1 (lanes 7 to 9) antibody. GST-HCC141 contains HBc CTD sequences from 141-183, and GST-HCC141–3A or –3E contains the same sequences except that the three major S phosphorylation sites (Fig. 1A) are substituted with A or E, respectively. C, WT or mutant HBc.
FIG 7.
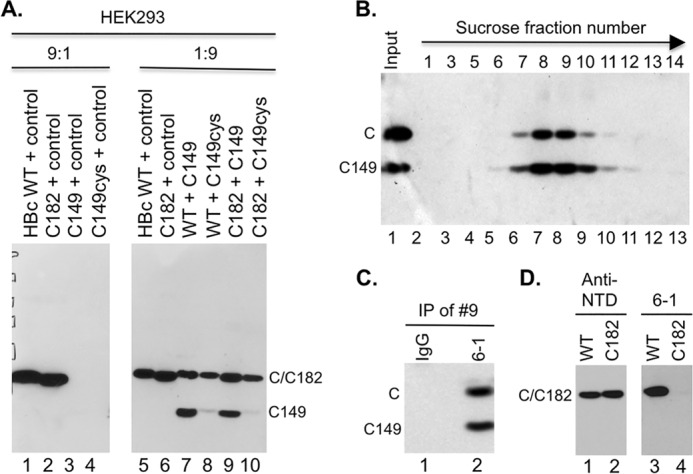
Rescue of expression and assembly of CTD-deleted HBc by WT HBc in HEK293 cells. (A) Cytoplasmic lysate (20 μl) from HEK293 cells transiently transfected with the indicated plasmids was resolved by SDS-PAGE and detected by Western blot analysis using the anti-HBc NTD MAb. The control plasmid expresses the neomycin resistance gene and was used as a filler. The ratios (9:1 and 1:9) refer to the amount of the first plasmid (always expressing an HBc protein) relative to the amount of the second plasmid (expressing either no HBc or the indicated HBc protein). (B) Capsids in cytoplasmic lysate of HEK293 cells cotransfected with both the WT HBc- and C149-expressing plasmids were fractionated on a linear 15% to 30% sucrose gradient in an SW32 rotor at 27,000 rpm for 4 h at 4°C (the direction of centrifugation is indicated by the arrow). The indicated fractions (lanes 2 to 13), along with the input lysate (lane 1), were resolved by SDS-PAGE, and WT HBc and C149 were detected by Western blot analysis using the anti-HBc NTD MAb. (C) Coimmunoprecipitation of C149 and WT HBc in mosaic capsids. The capsid peak fraction (fraction 9) from the sucrose gradient was subjected to immunoprecipitation using the anti-CTD MAb (6-1) (lane 2) or a control IgG (lane 1). The immunoprecipitates were detected by the anti-NTD MAb following SDS-PAGE and Western blot analysis. (D) Cytoplasmic lysate from HEK293 cells transiently transfected with WT (lanes 1 and 3) or C182 (lanes 2 and 4) was resolved by SDS-PAGE and detected by Western blot analysis using the anti-NTD MAb (lanes 1 and 2) or anti-CTD MAb 6-1 (lanes 3 and 4). C, WT HBc monomer; C149 monomer, C-terminally truncated HBc protein (terminated at position 149); IP, immunoprecipitation. C182 comigrated with WT HBc.
Like WT HBc, 3E also was barely detectable without CIAP treatment by the C170 or 25-7 antibodies (Fig. 3A, lane 4, top and upper middle panels). Also similarly to WT HBc, 3E reactivity with C170 or 25-7 dramatically increased upon CIAP treatment (Fig. 3A, lane 7, top and upper middle panels), indicating that one or more non-SP sites (see Fig. 1A) of CTD phosphorylation were utilized in RRL. After normalization to the signal obtained using the anti-NTD MAb, we found that 3A, without any CIAP treatment, was more reactive with C170 or 25-7 than WT HBc or 3E, indicating that 3A was less phosphorylated (at the non-SP site[s]) than 3E or WT HBc. Thus, whereas the anti-NTD MAb showed that the amount of 3A was less than that of WT HBc or 3E (Fig. 3A, lanes 2 to 4, lower middle panel), the amount of 3A detected by C170 or 25-7 was actually higher than that of WT HBc or 3E (Fig. 3A, lanes 2 to 4, top and upper middle panels). Furthermore, CIAP treatment did not significantly affect the reactivity of 3A with either C170 or 25-7 (Fig. 3A, lane 6 versus lane 3), in contrast to WT HBc or 3E. Whereas the anti-NTD MAb showed that the amount of 3A loaded was approximately the same as that of WT HBc or 3E (Fig. 3A, lanes 5 to 7, lower middle panel), 3A was detected by C170 or 25-7 at much lower levels than WT HBc or 3E (Fig. 3A, lane 5 to 7, top and upper middle panels), indicating that 3A, though less phosphorylated in RRL than WT HBc or 3E, remained phosphorylated even after CIAP treatment. Note that the S-to-A/E substitutions in 3A or 3E, on their own, had little effect on the CTD reactivity with either C170 or 25-7, as shown by the equal levels of detection by these two antibodies, as well as the phosphorylation-independent 6-1 MAb, of nonphosphorylated, glutathione S-transferase (GST)–CTD fusion proteins purified from E. coli (34) (Fig. 3B).
Thus, whereas 3A was less phosphorylated in RRL, the phosphorylation sites in 3A were also more resistant to dephosphorylation by CIAP than those in WT HBc or 3E. This was possibly due to RNA binding by 3A, which was much stronger than that by WT HBc or 3E (see below), preventing CIAP from accessing the CTD phosphorylation sites in 3A. Alternatively or additionally, 3A might have assembled faster than WT HBc or 3E such that its sites of phosphorylation became inaccessible to CIAP; the rate of assembly of capsids (and any putative intermediates) remains to be defined. Although the exact status of CTD phosphorylation at each site remains to be more precisely determined by further analyses using additional antibodies that can differentiate phosphorylated CTD from nonphosphorylated CTD at each individual site and mass spectrometry as we did for DHBc (24), our results here clearly demonstrated that WT HBc and 3E and, to a less extent, 3A were phosphorylated by endogenous RRL kinase(s) and that CIAP treatment caused dephosphorylation of WT HBc and 3E. The higher degree of CTD phosphorylation of the CTD phosphomutant 3E versus 3A at the remaining phosphor sites could also account for the need of dephosphorylation (by endogenous RRL phosphatases or CIAP) to trigger assembly by 3E but not by 3A (Fig. 2 and 4).
To further test the role of CTD phosphorylation state in capsid assembly, we included two additional CTD phosphorylation mutants, HBc-7A (7A) and 7E, which have all seven (six confirmed and one putative) phosphorylation sites in CTD changed to A and E, respectively (Fig. 1A). These two mutants would mimic the two extreme states of CTD phosphorylation, i.e., complete dephosphorylation and complete phosphorylation, respectively. Furthermore, as the CTD of these two mutants could not be phosphorylated at all, they served as appropriate controls to dissect direct effects on CTD phosphorylation per se versus indirect effects (e.g., via effects on cellular factors in RRL) that any manipulations (e.g., CIAP treatment) had on capsid assembly. Thus, if CIAP treatment affected capsid assembly via direct effects on CTD phosphorylation, it was not expected to affect assembly by either 7A or 7E. On the other hand, if CIAP treatment exerted its effect on capsid assembly indirectly through effects on some RRL factors, it might still affect 7A or 7E assembly. Similarly to 3A, 7A was able to assemble into capsids and this assembly was independent of exogenous phosphatase but dependent on the 37°C incubation (Fig. 4, top, lanes 19 to 21). Inhibition of the endogenous phosphatase had little effect on 7A assembly (Fig. 4, top, lane 24), consistent with its loss of all CTD phosphorylation sites. Interestingly, the 7E mutant, uniquely among all the HBc proteins tested, appeared to have already undergone substantial assembly by the end of the translation reaction such that most 7E migrated at the authentic capsid position without the assembly period at 37°C (Fig. 4, top, lane 25). Neither the addition of the exogenous phosphatase nor inhibition of the endogenous phosphatase after the translation reaction had any significant effect on 7E assembly (Fig. 4, top, lanes 26, 27, and 30). These results were thus most consistent with a direct effect of CIAP or endogenous RRL phosphatases (and RRL kinases) on the phosphorylation state of the CTD affecting capsid assembly.
Whereas the assembled capsids, from either the WT or mutant HBc proteins, all migrated to the same position as the bacterially derived capsid standard on the native agarose gel, the mobility of the unassembled HBc subunits was apparently affected by their phosphorylation state. In addition to the mobility upshift of the WT HBc protein induced by the endogenous phosphatase as described above, unassembled 3A and 7A, mimicking CTD dephosphorylation, migrated mostly just above the capsid band and comigrated with C149, whereas the phosphomimetic 3E mutant protein migrated mostly on top of the gel above the WT HBc protein but could also run as a broad smear from above the assembled capsids to above the unassembled WT HBc subunits (Fig. 2 and Fig. 4, top). Assembly of 7E was so fast that little unassembled protein was detected on the agarose gel (Fig. 4, top, lane 25). The exact physical state of the unassembled HBc proteins (subunits) remains to be fully characterized, but they were presumably dimers, as determined on the basis of the rapid HBc dimerization observed in the Xenopus oocyte (9) and in vitro from purified proteins (11). Furthermore, as described below, the unassembled HBc proteins were probably associated with RNA nonspecifically in RRL, which would, at least to some extent, account for their mobility on the agarose gel. The fact that they stayed in the top fractions during sucrose gradient centrifugation also suggests that these complexes were much smaller than the capsids which sedimented to the middle of the gradient (Fig. 5).
FIG 5.

Analysis of capsid assembly in RRL by sucrose gradient centrifugation. The indicated RRL translation reaction mixtures (100 μl), either left untreated or treated as indicated, were separated over a linear 15% to 30% sucrose gradient spun in an SW55 rotor at 27,000 rpm for 4 h at 4°C. The indicated sucrose fractions (10 μl each) or input RRL translation mixtures (either left untreated or treated as indicated) (0.5 μl) were resolved by agarose gel electrophoresis. The direction of centrifugation is indicated by the arrows. 35S-labeled HBc proteins were detected by autoradiography. Capsids purified from E. coli (unlabeled) in panel C were detected by Western blot analysis using the anti-HBc NTD MAb. Fract #, sucrose fraction number; mock, overnight incubation in the assembly buffer alone; C and 3E, full-length HBc proteins; C149, C-terminally truncated HBc protein (terminated at position 149); Ca, capsids.
The role of RNA in capsid assembly in RRL depended on the CTD phosphorylation state.
Since the CTD is known to have nonspecific RNA binding activity that is modulated by its phosphorylation state, we were interested in any potential effects of RNA on core protein mobility and, more importantly, on capsid assembly in RRL. We thus treated the translation reaction mixtures with RNase immediately following translation and before conducting the assembly reaction or, alternatively, treated the reaction mixtures with RNase following assembly (Fig. 1C). With respect to WT HBc, RNase treatment before CIAP treatment, which was needed to induce WT HBc assembly (Fig. 2 and 4), prevented capsid assembly (Fig. 4, top, lane 5), thus indicating that RNA indeed played a critical role in the assembly of WT HBc. RNase pretreatment also prevented assembly of the 3E mutant (Fig. 4, top, lane 17). On the other hand, RNase treatment had no effect on assembly of the 3A mutant (Fig. 4, top, lane 11). In contrast to the 3A results, 7A assembly was abolished by RNase treatment (Fig. 4, top, lane 23), indicating an essential role for RNA in 7A assembly. On the other hand, RNase treatment had no effect on 7E assembly (Fig. 4, top, lane 29). In contrast to RNase treatment before assembly, RNase treatment following the completion of capsid assembly showed little effect on any of the assembled capsids (Fig. 4, top, lanes 4, 10, 16, 22, and 28), suggesting that the assembled capsids were no longer sensitive to RNase and were able to protect the RNA if it was packaged during assembly (see Discussion).
RNase digestion also affected the mobility of the unassembled HBc subunits, especially the WT, 3E, and 7A. Indeed, these unassembled proteins became mostly undetectable on the agarose gel (Fig. 4, top, lanes 5, 17, and 23), indicating that they either ran off the agarose gel or failed to enter the gel. Even C149, with the CTD entirely removed, was affected by the RNase treatment, albeit to a smaller degree (Fig. 4, top, lanes 34 and 35). This is consistent with the previous finding that C149 retains some RNA binding activity (13). The proteins were not simply degraded during the RNase treatment, since the amounts of total core proteins detected by SDS-PAGE were not affected by RNase digestion or any of the manipulations tested (Fig. 4, bottom). As the core proteins are highly basic, they presumably failed to migrate toward the anode side (i.e., failed to enter the agarose gel) without bound RNA.
Analysis of capsid assembly in RRL by sucrose gradient centrifugation.
To further verify capsid assembly in RRL, the capsids assembled from the WT and mutant HBc proteins that were translated in RRL were analyzed by sucrose gradient centrifugation, in parallel with a capsid standard purified from bacteria, which was reconstituted into mock-translated RRL to mimic the conditions of capsids assembled in RRL. Based on the results described above, the WT and various HBc mutants were allowed to assemble into capsids under the optimal conditions appropriate for each (Fig. 5). Aliquots of unfractionated RRL translation reaction mixtures, which either did not undergo any further treatment or were treated with CIAP, were loaded in parallel. As expected, the capsid standard sedimented into the gradient and peaked at around fraction 11 (Fig. 5C, lane 10). C149, which did not show any sign of assembly under any conditions in the preceding figures, also showed no evidence of assembly as judged by the gradient centrifugation (Fig. 5H). C149 stayed on the top of the gradient (Fig. 5H, lanes 2 to 3), and the protein from the top of the gradient migrated quickly (above assembled capsids) on the agarose gel, just as it did in unfractionated RRL (Fig. 5H, lane 1, and Fig. 2 and 3). C149 was incubated with CIAP before the sucrose gradient analysis, as the other HBc proteins were mostly assembled into capsids under the CIAP treatment condition before gradient analysis.
WT HBc did not assemble into any capsids in the absence of exogenous CIAP and stayed on the top of the gradient (Fig. 5A). The unassembled WT HBc proteins from the top fractions of the gradient also migrated much more slowly on the agarose gel than assembled capsids (Fig. 5A, lanes 2 and 3), as shown for the unfractionated translation reaction mixtures (Fig. 5A, lane 1; see also Fig. 2 and 4). In contrast, capsids assembled from CIAP-treated WT HBc sedimented into the gradient with a peak at fractions 10 to 11 (Fig. 5B, lanes 9 to 10), similarly to the capsid standard purified from bacteria. Capsids assembled from 3A and 3E similarly peaked in fraction 11 (Fig. 5D, lane 11, and Fig. 5E, lane 10). Interestingly, the capsids assembled from 7A appeared to sediment significantly faster into the gradient, peaking at fraction 12 (Fig. 5F, lane 12), whereas the 7E capsids sedimented the slowest, peaking at fraction 9 (Fig. 5G, lane 9). We have shown previously that the sedimentation of capsids on the sucrose gradient is affected by their interior nucleic acid content, e.g., double-stranded DNA-containing capsids sediment faster than single-stranded DNA- or pgRNA-containing capsids (24). Thus, the differences in sedimentation characteristics on the gradient may suggest that the WT and mutant capsids assembled had different interior RNA contents; in particular, the fairly large difference between 7A and 7E might have reflected the difference in nonspecific RNA packaging in these capsids (see below and Discussion).
Rescue of NTD assembly in RRL by coexpression of WT HBc.
Since CTD-deleted C149 is clearly able to assemble into capsids at a sufficiently high concentration and CTD was able to facilitate capsid assembly at physiologically low HBc concentrations, we reasoned that C149 assembly under low-concentration conditions might be rescued when WT HBc was present. To test this possibility, we cotranslated WT and C149 proteins together in RRL and assayed for capsid assembly by sucrose gradient centrifugation and agarose gel electrophoresis. Interestingly, upon resolution of the cotranslation reaction on the agarose gel, a novel HBc species appeared that migrated between WT HBc and C149 subunits (indicated by an asterisk [*] in Fig. 6A, bottom panel), suggesting that WT and C149 HBc proteins could indeed interact, presumably through the NTD assembly domain in both proteins. Upon CIAP treatment to induce assembly, a fast-migrating species on the agarose gel that sedimented into the capsid position in the sucrose gradient, corresponding to the assembled capsids, appeared in the HBc-alone reaction (Fig. 6A, top panel) but not in the C149-alone reaction (Fig. 6A, middle panel), as shown above. In the mixed-translation reaction, CIAP also induced capsid assembly (Fig. 6A, bottom). To ascertain if the capsids formed in the mixed translation might also contain C149, in addition to WT HBc, we analyzed the capsid fractions purified from the mixed-translation reaction mixture by SDS-PAGE. Indeed, C149 was detected in the same capsid peak fractions as WT HBc, in contrast to the C149-alone translation where no C149 protein sedimented into the capsid position (Fig. 6B). These results thus indicated that the assembly defect of C149 could indeed be rescued by WT HBc, likely by coassembly of mosaic capsids containing both WT HBc and C149 mediated by NTD-NTD interactions (see below also). The amount of capsid assembled in the presence of C149 appeared to be somewhat less than that in the absence of C149 (Fig. 6), suggesting that C149 might have also exerted some interference (i.e., a dominant-negative effect) on WT HBc assembly besides being rescued in the form of assembled capsids by WT HBc.
FIG 6.
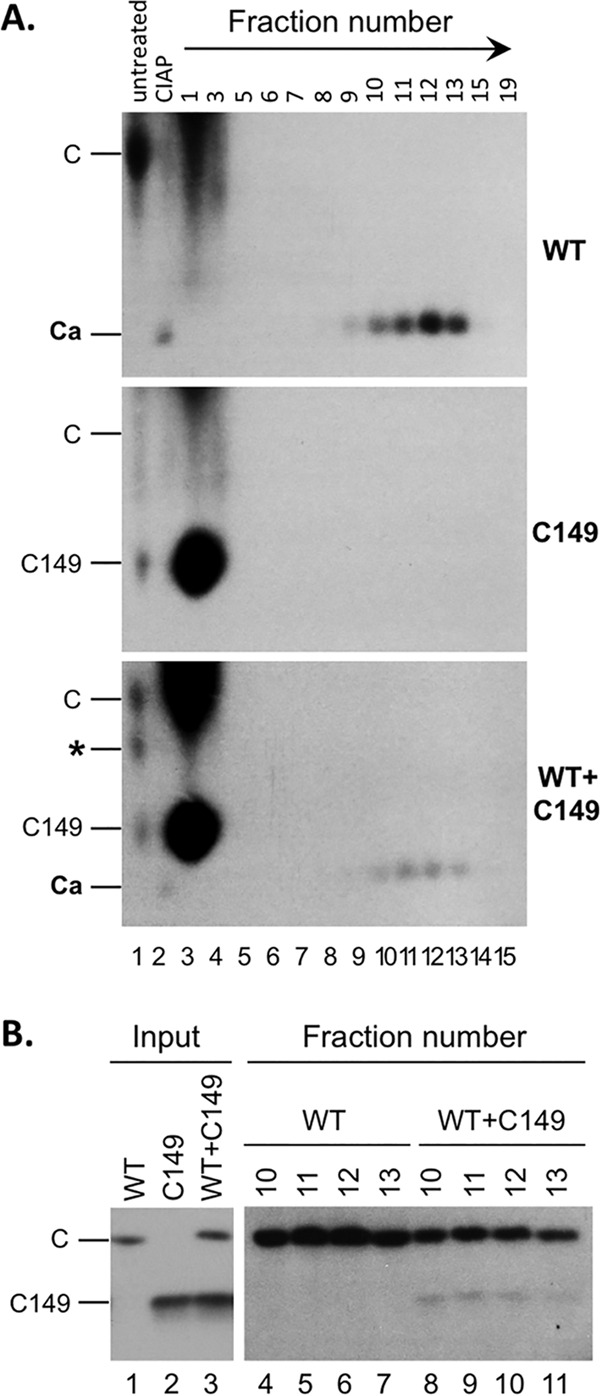
Rescue of capsid assembly of CTD-deleted HBc by WT HBc in RRL. WT or C149 HBc was translated alone (A, top or middle, respectively; B, lane 1 or lane 2, respectively), or WT HBc and C149 HBc were translated together (A, bottom; B, lanes 3) in RRL. The capsids were induced to assemble using CIAP treatment, and the assembled capsids were resolved by sucrose gradient centrifugation as described for Fig. 4. The translation reaction mixtures (A, lane 1, 0.5 μl), the assembly reaction mixtures (i.e., the input to the gradient; CIAP) (A, lane 2, 0.5 μl; B, lanes 1 to 3, 5 μl), and the indicated sucrose fractions (A, lanes 3 to 15, 10 μl; B, lanes 4 to 11, 50 μl) were resolved by agarose gel electrophoresis (A) or SDS-PAGE (B) and detected by autoradiography. C, WT HBc subunits; C149, C-terminally truncated HBc protein (terminated at position 149); Ca, capsids; *, novel band appearing only in the mixed translation and migrating between WT and C149 HBc subunits. The direction of centrifugation is indicated by the arrow in panel A.
Failure of NTD to accumulate in mammalian cells and rescue of NTD expression and assembly in mammalian cells by WT HBc.
Given the surprising result described above that showed that C149 was unable to assemble in RRL (Fig. 2 and 4 to 6), we were interested in determining if C149 was able to assemble in mammalian cells. Surprisingly, C149 expression was barely detectable in HEK293 cells (Fig. 7A, lane 3) or in HepG2 or Huh7 cells (data not shown). Given that unassembled core subunits are known to be less stable than assembled capsids (47) and that C149 was unable to assemble in RRL, it was likely that the low expression level of C149 was a consequence of its failure to assemble into capsids and thus of its rapid degradation as subunits.
To test if coexpression of WT HBc might rescue the expression and assembly of C149 in cells, we cotransfected both the WT HBc-expressing and C149-expressing constructs into HEK293 cells. Indeed, coexpression with WT HBc dramatically increased the levels of C149 in cells (Fig. 7A, lane 7 versus lane 3), suggesting that WT HBc could indeed rescue the expression of C149 in cells, presumably through interactions mediated by the NTD. Sucrose gradient ultracentrifugation demonstrated that the C149 protein detected in the cotransfected cells sedimented to the same fractions as WT HBc (Fig. 7B), indicating that C149 was assembled into capsids under cotransfection conditions. Given that C149 was expressed at the same levels as or at even higher levels than WT HBc in RRL and yet failed to assemble into capsids (Fig. 2 and 4 to 6), the rescue of C149 expression levels in HEK293 cells by WT HBc was likely a result of C149 coassembly into mosaic capsids (and the consequent stabilization) with the coexpressed WT HBc.
To detect the putative mosaic capsids containing both the WT HBc and C149 proteins, the anti-CTD MAb, 6-1, was used to pull down WT HBc in native capsids, and the presence of any C149 in the immunoprecipitated capsids was then detected by SDS-PAGE and Western blot analysis using the ant-NTD MAb (Fig. 7C). Indeed, both WT HBc and C149 were readily detected in the immunoprecipitates, confirming that C149 was able to coassemble with WT HBc into mosaic capsids. This result was thus consistent with the notion that all the information required for capsid assembly is contained in C149, which was also folded appropriately in mammalian cells in a manner similar to that seen in bacteria, but the concentrations reached in mammalian cells or in RRL were insufficient for C149 to assemble, in contrast to bacterial and insect cell overexpression systems. The presence of CTD donated by WT HBc, in a fraction of assembling subunits, was thus sufficient to lower the threshold concentration for assembly, leading to the generation of mosaic capsids incorporating both WT and CTD-truncated HBc subunits. Successful precipitation of the capsid particles by the anti-CTD antibody further indicated that at least a fraction of the CTD was exposed on the surface of these empty capsids assembled in mammalian cells, consistent with previous results of experiments performed on empty capsids assembled using purified subunits in vitro (48). As the 6-1 MAb recognizes an epitope at the very C-terminal end of CTD—the removal of the last Cys residue of HBc (in C182) led to a complete loss of 6-1 reactivity (Fig. 7D)—this result further indicated that the very C-terminal end of HBc is exposed on the capsid surface.
Since the C-terminal Cys (i.e., Cys183) is known to be involved in cross-linking HBc dimers in the capsids and, though nonessential for assembly, can stabilize the capsids (11, 14, 49–51) and since this residue is missing from C149, we were interested in testing the role of C183 in capsid formation in the mammalian cell system. First, we added a terminal Cys residue to C149 to make C149cys such that it was still missing the entire CTD except for the last Cys. However, C149cys was no better than C149 in terms of expression or assembly (Fig. 7A, lanes 3 and 4), suggesting that the terminal Cys alone was insufficient to substitute for the CTD in capsid assembly. We then tested the reciprocal of C149cys, C182, which contains the entire CTD except the terminal Cys residue. C182 expression (and assembly; not shown) behaved just like that seen with WT HBc (Fig. 7A, lanes 1, 2, 5, and 6). Furthermore, C182 was as effective as WT HBc in rescuing C149 expression (Fig. 7A, lanes 7 and 9). Thus, there was no need for the C-terminal Cys to be present in order for HBc to assemble or to rescue C149 in our expression system. In fact, C149cys expression could not be rescued as effectively as C149 expression by either WT HBc or C182 (Fig. 7A, lanes 8 and 10 versus 7 and 9). This result suggested that the effect of the last Cys on capsid assembly was context dependent and that it might interfere with assembly under certain conditions, perhaps via inappropriate disulfide formation.
Analysis of nonspecific RNA packaging by WT and mutant HBV capsids.
The results shown in Fig. 4 and 5 and described above suggested that the capsids assembled in RRL might package RNA in a manner dependent on the state of CTD phosphorylation. Due to the small amounts of capsids assembled in RRL, it was difficult to measure accurately RNA packaging by capsids in RRL (see Discussion below). To look into nonspecific RNA packaging during the assembly of the WT and mutant capsids in detail, we turned to the mammalian cell system (HEK293 cells) for producing large amounts of capsids from either the WT HBc or CTD phosphorylation mutants. As shown in Fig. 8A (lane 1), capsids assembled from WT HBc, expressed without the viral RT or pgRNA, in HEK293 cells did not package detectable levels of viral RNA (i.e., the HBc mRNA expressed from the HBc expression plasmid) as determined by the Northern blot assay following resolution of capsids in the cell lysate by agarose gel electrophoresis. This was expected since specific viral RNA packaging is known to depend on the viral RT and the RNA package signal, ε, neither of which was present in these systems and since WT HBV capsids package little to no RNA nonspecifically when expressed alone in mammalian cells or insect cells (i.e., they are empty) (12, 31, 32). Similarly, the 7E capsids, and especially the 3E capsids, assembled in the mammalian cells packaged only low levels of RNA nonspecifically (Fig. 8A, lanes 3 and 5). In contrast, the 3A and 7A capsids from HEK293 cells packaged high levels of HBc mRNA nonspecifically (Fig. 8A, lanes 2 and 4). Interestingly, we detected consistently lower levels of 7A capsid accumulation (Fig. 8A, lane 4) in HEK293 cells. To detect all the RNAs (not just the HBc mRNA) packaged in the capsids, we stained the capsid-associated RNAs following resolution by agarose gel electrophoresis with a nonspecific RNA detecting dye, Sybr gold, and the capsid protein signal was subsequently detected by the use of Sypro ruby protein dye on the same gel (Fig. 8B), as we reported previously (32). The levels of total packaged RNA, as detected by the staining method, were consistent with the levels of HBc mRNA packaged in the various capsids shown in Fig. 8A. Upon normalization to the capsid protein levels, the amounts of RNA packaging by the 3A and 7A capsids in HEK293 cells were similar to that in the (unphosphorylated) capsids assembled in E. coli and that by 7E was ca. 2-fold lower and that by 3E even lower (by 8-fold) (Fig. 8B, graph). In agreement with the lack of detection of HBV-specific RNA in WT capsids assembled in HEK293 cells, the total RNA staining approach also failed to detect nonspecific RNA packaging by WT capsids in HEK293 cells (Fig. 8B, lane 3).
FIG 8.

Analysis of nonspecific RNA packaging by capsids assembled in mammalian and bacterial cells. The indicated WT and mutant HBc expression constructs were transfected into HEK293 cells. (A) Cytoplasmic lysates (5 μl) from transfected cells were resolved on an agarose gel and transferred to a nitrocellulose membrane. Packaged HBc mRNA was detected by 32P-labeled antisense HBV riboprobe (top) and the capsid (Ca) by the anti-HBc NTD MAb (bottom). (B) Total RNA packaged in capsids in the HEK293 lysates (15 μl), along with the capsid standard purified from E. coli, was also detected by Sypro gold staining and the capsid protein signal detected by Sypro ruby staining following treatment of the lysate with micrococcal nuclease and proteinase K (see Materials and Methods for details) and agarose gel electrophoresis (gel images). Green fluorescent protein (GFP) (lane 2) represented a negative control for the staining that contained the lysate from cells transfected with a GFP-expressing plasmid. Note that duplicate 3E (lanes 7 and 8), 3A (lanes 9 and 10), and 7A (lanes 5 and 6) samples from two separate transfections were loaded. The RNA staining signals normalized to the protein signals are presented in the graph. (C) Capsids purified by sucrose gradient centrifugation from transfected HEK293 cells (lanes 1 to 5) or E. coli (lanes 6 to 12) were resolved on an agarose gel and detected by Sypro ruby staining (top) and their associated nucleic acids by Sybr gold staining (bottom). (D) Nucleic acid isolated from the purified capsids from HEK293 cells (3A, lane 2; 3E, lane 3) or E. coli (lane 4) was isolated and resolved on an agarose gel and detected by Sybr gold staining. The RNA marker and tRNA were also loaded as size standards (lanes 1 and 5, respectively). Ca, capsids; kb, sizes of RNA markers.
To look further into the nature of nonspecific RNA packaged by the mutant capsids, we purified large amounts of WT, 3A, and 3E capsids assembled in HEK293 cells and compared the quantity and nature of RNA packaged in these capsids to the quantity and nature of that assembled in bacteria. Capsids purified by sucrose gradient centrifugation from the different sources were resolved on an agarose gel, and the amounts of protein and RNA associated with the capsids were detected by Sypro ruby and Sybr gold staining, respectively. As shown in Fig. 8C, the levels of RNA packaging in the 3A capsids from HEK293 cells (lane 3) were similar to those in the bacterially derived capsids (lane 7), in agreement with the results obtained using crude cell lysate as described above (Fig. 8B). In contrast, no RNA was detectable in WT capsids from HEK293 cells even when they were loaded in larger amounts than the 3A capsids (Fig. 8C, lanes 1 and 2). The levels of RNA in WT HEK293 capsids were thus at least 30-fold lower than those in the 3A HEK293 capsids or WT capsids from bacteria. Low levels of RNA were detectable in the 3E capsids purified from HEK293 cells (Fig. 8C, lane 4), but they were at least 5-fold lower than those in the capsids from bacteria. To look into the nature of the RNA being packaged, the capsid-associated RNA was extracted by phenol-chloroform and resolved on an agarose gel (Fig. 8D). As reported previously (11), the RNA packaged into the 3A (and, to lesser extent, 3E) capsids from HEK293 cells and WT capsids from bacteria was mostly small (comigrating with tRNA and mostly below 200 nucleotides [nt]), although larger (4 kb and above) RNAs could also be detected.
DISCUSSION
We have demonstrated here that the HBc NTD assembly domain, alone, was insufficient, or at least inefficient, for capsid assembly in a mammalian cell extract (RRL) or in mammalian cells (both hepatoma and HEK293 cells) when it was expressed at physiologically relevant concentrations. This is in contrast to previous results obtained using heterologous overexpression systems and nonphysiological in vitro assembly studies. Under our conditions, which were near physiological, the HBc CTD was required to stimulate capsid assembly, which was further modulated by the CTD state of phosphorylation as controlled by the host protein kinase(s) and phosphatase(s). Our results further indicate that nonspecific RNA binding by CTD likely played an important role in facilitating capsid assembly but that CTD may also facilitate assembly independently of its RNA binding activity.
HBV capsid assembly is well known to be concentration dependent. The HBc concentration for assembly in vitro using purified proteins is rather high (ca. 40 to 80 μM) at physiological salt concentrations (11, 45). Under these conditions, the NTD is clearly sufficient for capsid assembly, with no need for the CTD. In the RRL expression system used here, the HBc concentrations were lower by 3 orders of magnitude (ca. 50 nM in the translation mixture and ca. 10 nM during the assembly reaction). Under conditions incorporating these low HBc concentrations, the full-length HBc, but not NTD alone, was able to assemble into capsids. Importantly, the HBc concentrations required to support CTD-independent capsid assembly are unlikely to be reached in infected hepatocytes or transfected hepatoma host cells where HBV assembly and replication readily occur. An estimate of the steady-state HBc concentration by quantitative Western blot analysis in stably transfected hepatoma cells suggested that it was ca. 300 nM (ca. 600 ng of HBc per 107 HepG2 cells, assuming a volume of 10−5 μl per cell) (L. Ludgate and J. Hu, unpublished results), far below the concentration needed for in vitro capsid assembly mediated by the NTD alone. Nevertheless, with these low concentrations, HBc is able to assemble efficiently into empty capsids with no need for, or incorporation of, the viral pgRNA (32, 38) (also see below), as well as the replication-competent nucleocapsids packaging the specific viral RT-pgRNA complex. It was also reported that virtually all HBc produced in hepatocytes in the livers of HBV-infected patients are in the assembled capsids, with few free subunits (52). Although the exact HBc concentration in an infected hepatocyte is difficult to measure directly, it is presumably similar to those reached in transfected hepatoma cells in culture. HBc was also reported to assemble efficiently into capsids at concentrations of ca. 1 μM or lower in Xenopus oocytes (44, 53). Importantly, we found that the HBc NTD alone, without the CTD, also failed to assemble in HEK293 or human hepatoma cells, indicating that the conclusions derived from studies using RRL are likely to be applicable to living cells under conditions that support efficient capsid assembly and viral replication.
The CTD may facilitate HBc assembly under physiological concentrations in a number of ways. First, it likely functions through its well-known interactions with nonspecific RNA, which would effectively lower the threshold concentration required for capsid assembly (29). Even in systems where high HBc concentrations make the CTD dispensable for assembly, CTD-RNA interactions are thought to stabilize the capsids (13, 28). In addition to providing the protein-RNA interactions to drive assembly, RNA also helps to neutralize the positive charges of CTDs, minimizing the electrostatic repulsion that would impede assembly (29). The 3A and 7A HBc mutants, mimicking nonphosphorylated HBc, indeed packaged nonspecific RNAs when assembled in mammalian cells at levels as high as those shown by WT (unphosphorylated) capsids assembled in E. coli, consistent with a role for RNA-CTD interactions in stimulating capsid assembly in the mammalian system at physiological HBc concentrations. Previously, it was reported that removal of packaged RNA from unphosphorylated capsids (assembled in bacteria) leads to capsid disruption and that they can reassemble following the addition of polyanions in vitro (54), also supporting the idea of a role of RNA, and specifically electrostatic (ionic) interactions, in facilitating capsid assembly and stabilization.
There was apparently no strict correlation between the requirement of RNA for assembly, as assayed by the effect of RNase pretreatment on assembly in RRL, and the amount of RNA packaged in the assembled capsids. Although the small amounts of capsids assembled in RRL made it difficult to accurately quantify the levels of RNA packaged into those capsids, the levels of HBc RNA (used for in vitro translation) packaged as detected by Northern blot analysis were similar to those measured in the cell-derived capsids (K. Liu and J. Hu, unpublished results). 3A and 7A capsids packaged high levels of RNA, as anticipated from their high levels of positive CTD charges. However, 3A assembly in RRL was not inhibited by RNase treatment following translation and before assembly. The fact that RNase pretreatment before 3A assembly did not completely remove all the RNA from the 3A capsids suggests that that some RNA was already bound to 3A at the end of translation and was protected from RNase digestion in some yet-to-be-defined precapsid structure (a putative assembly intermediate). 3E capsids packaged only very low levels of RNA, and yet RNase treatment abolished its assembly, indicating that the RNA binding by 3E, though reduced relative to that by 3A or 7A, was nevertheless critical for its assembly. Similarly, assembly of WT HBc in RRL, following dephosphorylation treatment, was also inhibited by RNase treatment before dephosphorylation, and the amount of RNA packaged by WT capsids was undetectable (i.e., was even lower than that seen with 3E). These results suggest that WT HBc was likely dephosphorylated by CIAP only partially before it started to assemble, mimicking the 3E mutant, and was unlikely to be fully dephosphorylated, which would have led to WT HBc behaving like 7A in packaging high levels of nonspecific RNA. Interestingly, 7E had amounts of RNA that were clearly larger than those seen with 3E or WT. Thus, CTD phosphorylation in the case of WT and 3E may inhibit nonspecific RNA binding and packaging beyond the negative charges that were mimicked by the 7E mutation; i.e., the effects of S phosphorylation at the CTD sites could not be completely mimicked by the E substitutions. Furthermore, as WT and 3E required dephosphorylation to assemble in RRL, too-extensive CTD phosphorylation could apparently inhibit assembly, by decreasing CTD-RNA interactions and perhaps also protein-protein interactions (see below).
Second, beyond the protein-RNA interaction mediated by the CTD, protein-protein interactions mediated by the CTD, in addition to the well-defined interactions mediated by the NTD, may also facilitate capsid assembly. This idea is supported by the assembly of the CTD phosphomimetic 7E mutant, which has all seven potential sites of phosphorylation substituted with the acidic E residue. 7E assembled rapidly upon translation in RRL and in mammalian cells and may assemble independently of RNA. Indeed, 7E appeared to assemble more quickly and efficiently in RRL than the WT HBc and all other mutant HBc proteins tested here, highlighting the important contributions to capsid assembly that the CTD-mediated protein-protein interactions can make. Also, WT and 3E capsids packaged very little RNA, i.e., at least 30-fold less than the amount packaged in the nonphosphorylated capsid assembled in E. coli or its mimetic 3A and 7A in mammalian cells. Although the assembly of WT and 3E still required some RNA, the very small amounts of RNA packaged are unlikely to be sufficient to neutralize all the CTD-positive charges. Instead, the phosphorylated CTD likely plays an important role in assembly via protein-protein interactions. The putative CTD-mediated protein-protein interactions may involve CTD-CTD and perhaps even CTD-NTD interactions. In support of this notion, recent cryoelectron microscopy (cryo-EM) studies have suggested that CTD-CTD interactions may occur that may be further modulated by the CTD state of phosphorylation (55). Another recent study also suggested that the negatively charged phosphorylated S/T residues may interact with the positively charged R residues in the CTD to stabilize the capsids (56).
A potential protein-protein interaction involving CTD in capsid assembly and stability may be mediated through the C-terminal Cys residue. Although highly conserved, the terminal Cys residue is nonessential for assembly or viral replication but can stabilize core/capsids (49, 50), by forming interdimer disulfide cross-links (11, 51, 57, 58). However, we showed here that this C-terminal disulfide cross-linking did not play an essential role in facilitating capsid assembly, at least under our experimental conditions, since the C182 mutant, missing the terminal Cys residue, assembled just like the WT and it also rescued C149 (also missing a terminal Cys residue) as well as the WT.
Finally, C149 retains the linker sequence (position 141 to position 149) connecting the NTD (position 1 to position 140) to the CTD (position 150 to position 183) in WT HBc. The linker is routinely included together with the NTD in bacterial expression and in vitro assembly from purified proteins with high protein and/or salt concentrations and clearly does not interfere with NTD assembly under these conditions. However, it remains possible that the linker sequence, which is highly conserved, retained in C149 somehow interfered with assembly by the NTD under physiological conditions in RRL and mammalian cells and that this inhibitory effect of the linker is overcome by the CTD. Interestingly, HBc mutants with only part of the linker attached to the NTD (truncated at position 147, 145, or 144) accumulated and assembled at differing but detectable levels (17, 59, 60), suggesting that the exact truncation point within the linker region may affect the capacity of the NTD to assemble in the absence of the CTD. Also, the linker is known to affect the dimorphism of capsid assembly (into either T = 3 or T = 4 capsids) and deletion of the linker (and thus fusion of the NTD directly to the CTD) abolished capsid assembly (61). Given these results, further systematic studies into the role of the linker in capsid assembly are warranted.
CTD phosphorylation is well known to play a critical role in HBV pgRNA packaging (25, 26, 30), but how it facilitates pgRNA packaging has remained unclear. Our results presented here suggest that CTD phosphorylation may, at least in part, facilitate viral RNA packaging indirectly, by decreasing competition from nonspecific RNA binding (and packaging). It was shown that when the three S residues in the SP sites were substituted with A, almost no pgRNA packaging was detectable, which was similar to the results seen with 6A (retaining only one potential CTD phosphorylation site [S178 in the strain used here]; Fig. 1A); however, when 3 of the 4 conserved S/T sites in the non-SP motifs (Fig. 1A) were substituted with A, moderate levels of pgRNA packaging were still detectable (25, 26, 30). This is consistent with the results showing that the non-SP sites are less phosphorylated than the SP sites in vivo and that their phosphorylation may play a less important role in pgRNA packaging (19, 30). On the other hand, E substitutions at either group alone (the SP or non-SP phosphorylation sites) showed only small effects on pgRNA packaging whereas more-extensive E substitutions at both classes of sites (6E, analogous to the 7E used here) showed a severe defect in pgRNA packaging (25, 26, 30). Thus, too much phosphorylation of the CTD may not be productive for viral pgRNA packaging either, e.g., by reducing the RNA binding affinity too much, so as to interfere with specific packaging of the pgRNA-RT complex. To facilitate the specific packaging of the pgRNA-RT RNP complex, CTD needs to be partially phosphorylated, more on the SP sites than the non-SP sites, such that the positive charges on CTD are reduced by this partial phosphorylation to allow the assembling HBc to discriminate the viral RNP complex from nonspecific RNA by demonstration of a higher affinity for the former than the latter. The degree and dynamics of CTD phosphorylation, as determined by the balance of the host kinase and phosphatase activities acting on the CTD, can thus determine the proportion of empty (i.e., no viral or cellular RNA) versus replication-competent (packaging the pgRNA-RT complex) capsids (Fig. 9), both of which are assembled during viral replication in cell cultures and in the infected liver (32, 62, 63). Recently, it was shown that the kinetics of capsid assembly could be modulated by NTD mutations, which in turn affect pgRNA packaging (45, 64). It is possible that capsid assembly kinetics can also be modulated by the CTD and its state of phosphorylation, as suggested by the rapid assembly of the 7E mutant in RRL. Therefore, the CTD phosphorylation state may also regulate pgRNA packaging by modulating the capsid assembly kinetics.
FIG 9.

Regulation of specific versus nonspecific RNA packaging of HBV capsids by the degree of CTD phosphorylation and dephosphorylation. The HBc CTD state of phosphorylation is proposed to be regulated dynamically by the host protein kinase(s) and phosphatase(s). The letter P on the capsids denotes phosphorylation. The specific viral pgRNA is depicted as a thick and long wavy line inside the capsid (diamond), and the nonspecific RNAs are depicted as thin and short wavy lines. The dashed arrow indicates that under natural replication conditions in human cells, HBc is unlikely to be in a nonphosphorylated state upon expression and thus that few capsids with nonspecific RNA packaging would form, in contrast to capsid assembly in E. coli. See the text for details. For clarity, RT packaging is not shown.
The RRL system described here represents a more physiologically relevant mammalian system, which allows the study of HBV capsid assembly under cell-free conditions that more closely mimic the in vivo host cellular environment. In particular, the role of CTD and its state of phosphorylation on assembly can be dissected. The role of specific host factors in modulating capsid assembly, by mediating HBc modifications or via other mechanisms, can also be studied in detail. In particular, the RRL HBc assembly system provides an experimentally tractable system to study further the role of the cellular kinases such as CDK2 and phosphatases in modulation of the CTD state of phosphorylation and, consequently, in capsid assembly and RNA packaging, a role which remains completely unknown at present. Efforts in this direction have been hampered so far by the multiple important roles of these host factors in cell biology and the resultant pleiotropic and often toxic effects associated with manipulating these factors in living cells (33, 34). In addition, this facile assembly system should facilitate antiviral development targeted at capsid assembly. To date, only the NTD has been targeted to modulate capsid assembly (8, 47). Our results here suggest that the CTD and host factors that modulate CTD modifications and functions may also serve as viable targets for antiviral development to inhibit capsid assembly as well as the other multiple essential functions of CTD in HBV replication (8, 20).
ACKNOWLEDGMENTS
We thank Ju-Tao Guo and Dongliang Yang for antibodies.
REFERENCES
- 1.Trépo C, Chan HL, Lok A. 2014. Hepatitis B virus infection. Lancet 384:2053–2063. doi: 10.1016/S0140-6736(14)60220-8. [DOI] [PubMed] [Google Scholar]
- 2.Seeger C, Zoulim F, Mason WS. 2013. Hepadnaviruses, p 2185–2221. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott, Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 3.Hu J. 2016. Hepatitis B virus virology and replication, p 1–34. In Liaw Y-F, Zoulim F (ed), Hepatitis B virus in human diseases. Humana Press, New York, NY. [Google Scholar]
- 4.Summers J, Mason WS. 1982. Replication of the genome of a hepatitis B–like virus by reverse transcription of an RNA intermediate. Cell 29:403–415. doi: 10.1016/0092-8674(82)90157-X. [DOI] [PubMed] [Google Scholar]
- 5.Hu J, Seeger C. 2015. Hepadnavirus genome replication and persistence. Cold Spring Harb Perspect Med 5:a021386. doi: 10.1101/cshperspect.a021386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bartenschlager R, Schaller H. 1992. Hepadnaviral assembly is initiated by polymerase binding to the encapsidation signal in the viral RNA genome. EMBO J 11:3413–3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu J, Lin L. 2009. RNA-protein interactions in hepadnavirus reverse transcription. Front Biosci 14(Landmark Ed):1606–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zlotnick A, Venkatakrishnan B, Tan Z, Lewellyn E, Turner W, Francis S. 2015. Core protein: a pleiotropic keystone in the HBV lifecycle. Antiviral Res 121:82–93. doi: 10.1016/j.antiviral.2015.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou S, Standring D. 1992. Hepatits B virus capsids are assembled from core protein dimer precursors. Proc Natl Acad Sci U S A 89:10046–10050. doi: 10.1073/pnas.89.21.10046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wynne SA, Crowther RA, Leslie AG. 1999. The crystal structure of the human hepatitis B virus capsid. Mol Cell 3:771–780. doi: 10.1016/S1097-2765(01)80009-5. [DOI] [PubMed] [Google Scholar]
- 11.Wingfield PT, Stahl SJ, Williams RW, Steven AC. 1995. Hepatitis core antigen produced in Escherichia coli: subunit composition, conformational analysis, and in vitro capsid assembly. Biochemistry 34:4919–4932. doi: 10.1021/bi00015a003. [DOI] [PubMed] [Google Scholar]
- 12.Lanford RE, Notvall L. 1990. Expression of hepatitis B virus core and precore antigens in insect cells and characterization of a core-associated kinase activity. Virology 176:222–233. doi: 10.1016/0042-6822(90)90247-O. [DOI] [PubMed] [Google Scholar]
- 13.Birnbaum F, Nassal M. 1990. Hepatitis B virus nucleocapsid assembly: Primary structure requirements in the core protein. J Virol 64:3319–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gallina A, Bonelli F, Zentilin L, Rindi G, Muttini M, Milanesi G. 1989. A recombinant hepatitis B core antigen polypeptide with the protamine-like domain deleted self-assembles into capsid particles but fails to bind nucleic acids. J Virol 63:4645–4652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hatton T, Zhou S, Standring D. 1992. RNA- and DNA-binding activities in hepatitis B virus capsid protein: a model for their role in viral replication. J Virol 66:5232–5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chu TH, Liou AT, Su PY, Wu HN, Shih C. 2014. Nucleic acid chaperone activity associated with the arginine-rich domain of human hepatitis B virus core protein. J Virol 88:2530–2543. doi: 10.1128/JVI.03235-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nassal M. 1992. The arginine-rich domain of the hepatitis B virus core protein is required for pregenome encapsidation and productive viral positive-strand DNA synthesis but not for virus assembly. J Virol 66:4107–4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu M, Summers J. 1994. Multiple functions of capsid protein phosphorylation in duck hepatitis B virus replication. J Virol 68:4341–4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liao W, Ou JH. 1995. Phosphorylation and nuclear localization of the hepatitis B virus core protein: significance of serine in the three repeated SPRRR motifs. J Virol 69:1025–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu K, Ludgate L, Yuan Z, Hu J. 2015. Regulation of multiple stages of hepadnavirus replication by the carboxyl-terminal domain of viral core protein in trans. J Virol 89:2918–2930. doi: 10.1128/JVI.03116-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Machida A, Tsuda O, Yoshikawa A, Hoshi Y, Tanaka T, Kishimoto S, Akahane Y, Miyakawa Y, Mayumi M. 1991. Phosphorylation in the carboxyl-terminal domain of the capsid protein of hepatitis B virus: evaluation with a monoclonal antibody. J Virol 65:6024–6030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kann M, Gerlich WH. 1994. Effect of core protein phosphorylation by protein kinase C on encapsidation of RNA within core particles of hepatitis B virus. J Virol 68:7993–8000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Basagoudanavar SH, Perlman DH, Hu J. 2007. Regulation of hepadnavirus reverse transcription by dynamic nucleocapsid phosphorylation. J Virol 81:1641–1649. doi: 10.1128/JVI.01671-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perlman DH, Berg EA, O'Connor PB, Costello CE, Hu J. 2005. Reverse transcription-associated dephosphorylation of hepadnavirus nucleocapsids. Proc Natl Acad Sci U S A 102:9020–9025. doi: 10.1073/pnas.0502138102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gazina EV, Fielding JE, Lin B, Anderson DA. 2000. Core protein phosphorylation modulates pregenomic RNA encapsidation to different extents in human and duck hepatitis B viruses. J Virol 74:4721–4728. doi: 10.1128/JVI.74.10.4721-4728.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lan YT, Li J, Liao W, Ou J. 1999. Roles of the three major phosphorylation sites of hepatitis B virus core protein in viral replication. Virology 259:342–348. doi: 10.1006/viro.1999.9798. [DOI] [PubMed] [Google Scholar]
- 27.Lewellyn EB, Loeb DD. 2011. Serine phosphoacceptor sites within the core protein of hepatitis B virus contribute to genome replication pleiotropically. PLoS One 6:e17202. doi: 10.1371/journal.pone.0017202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Porterfield JZ, Dhason MS, Loeb DD, Nassal M, Stray SJ, Zlotnick A. 2010. Full-length hepatitis B virus core protein packages viral and heterologous RNA with similarly high levels of cooperativity. J Virol 84:7174–7184. doi: 10.1128/JVI.00586-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zlotnick A, Porterfield JZ, Wang JC. 2013. To build a virus on a nucleic acid substrate. Biophys J 104:1595–1604. doi: 10.1016/j.bpj.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jung J, Hwang SG, Chwae YJ, Park S, Shin HJ, Kim K. 2014. Phosphoacceptors threonine 162 and serines 170 and 178 within the carboxyl-terminal RRRS/T motif of the hepatitis B virus core protein make multiple contributions to hepatitis B virus replication. J Virol 88:8754–8767. doi: 10.1128/JVI.01343-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hilditch CM, Rogers LJ, Bishop DH. 1990. Physicochemical analysis of the hepatitis B virus core antigen produced by a baculovirus expression vector. J Gen Virol 71(Pt 11):2755–2759. doi: 10.1099/0022-1317-71-11-2755. [DOI] [PubMed] [Google Scholar]
- 32.Ning X, Nguyen D, Mentzer L, Adams C, Lee H, Ashley R, Hafenstein S, Hu J. 2011. Secretion of genome-free hepatitis B virus–single strand blocking model for virion morphogenesis of para-retrovirus. PLoS Pathog 7:e1002255. doi: 10.1371/journal.ppat.1002255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ludgate L, Adams C, Hu J. 2011. Phosphorylation state-dependent interactions of hepadnavirus core protein with host factors. PLoS One 6:e29566. doi: 10.1371/journal.pone.0029566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ludgate L, Ning X, Nguyen DH, Adams C, Mentzer L, Hu J. 2012. Cyclin-dependent kinase 2 phosphorylates s/t-p sites in the hepadnavirus core protein C-terminal domain and is incorporated into viral capsids. J Virol 86:12237–12250. doi: 10.1128/JVI.01218-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pugh J, Zweidler A, Summers J. 1989. Characterization of the major duck hepatitis B virus core particle protein. J Virol 63:1371–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Le Pogam S, Chua PK, Newman M, Shih C. 2005. Exposure of RNA templates and encapsidation of spliced viral RNA are influenced by the arginine-rich domain of human hepatitis B virus core antigen (HBcAg 165–173). J Virol 79:1871–1887. doi: 10.1128/JVI.79.3.1871-1887.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chua PK, Tang FM, Huang JY, Suen CS, Shih C. 2010. Testing the balanced electrostatic interaction hypothesis of hepatitis B virus DNA synthesis by using an in vivo charge rebalance approach. J Virol 84:2340–2351. doi: 10.1128/JVI.01666-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nguyen DH, Hu J. 2008. Reverse transcriptase- and RNA packaging signal-dependent incorporation of APOBEC3G into hepatitis B virus nucleocapsids. J Virol 82:6852–6861. doi: 10.1128/JVI.00465-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu J, Flores D, Toft D, Wang X, Nguyen D. 2004. Requirement of heat shock protein 90 for human hepatitis B virus reverse transcriptase function. J Virol 78:13122–13131. doi: 10.1128/JVI.78.23.13122-13131.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cui X, Ludgate L, Ning X, Hu J. 2013. Maturation-associated destabilization of hepatitis B virus nucleocapsid. J Virol 87:11494–11503. doi: 10.1128/JVI.01912-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gao W, Hu J. 2007. Formation of hepatitis B virus covalently closed circular DNA: removal of genome-linked protein. J Virol 81:6164–6174. doi: 10.1128/JVI.02721-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nguyen DH, Gummuluru S, Hu J. 2007. Deamination-independent inhibition of hepatitis B virus reverse transcription by APOBEC3G. J Virol 81:4465–4472. doi: 10.1128/JVI.02510-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guo H, Mao R, Block TM, Guo JT. 2010. Production and function of the cytoplasmic deproteinized relaxed circular DNA of hepadnaviruses. J Virol 84:387–396. doi: 10.1128/JVI.01921-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seifer M, Zhou S, Standring D. 1993. A micromolar pool of antigenically distinct precursors is required to initiate cooperative assembly of hepatitis B virus capsids in Xenopus oocytes. J Virol 67:249–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tan Z, Maguire ML, Loeb DD, Zlotnick A. 2013. Genetically altering the thermodynamics and kinetics of hepatitis B virus capsid assembly has profound effects on virus replication in cell culture. J Virol 87:3208–3216. doi: 10.1128/JVI.03014-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hu J, Seeger C. 1996. Hsp90 is required for the activity of a hepatitis B virus reverse transcriptase. Proc Natl Acad Sci U S A 93:1060–1064. doi: 10.1073/pnas.93.3.1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deres K, Schroder CH, Paessens A, Goldmann S, Hacker HJ, Weber O, Kramer T, Niewohner U, Pleiss U, Stoltefuss J, Graef E, Koletzki D, Masantschek RN, Reimann A, Jaeger R, Gross R, Beckermann B, Schlemmer KH, Haebich D, Rubsamen-Waigmann H. 2003. Inhibition of hepatitis B virus replication by drug-induced depletion of nucleocapsids. Science 299:893–896. doi: 10.1126/science.1077215. [DOI] [PubMed] [Google Scholar]
- 48.Chen C, Wang JC, Zlotnick A. 2011. A kinase chaperones hepatitis B virus capsid assembly and captures capsid dynamics in vitro. PLoS Pathog 7:e1002388. doi: 10.1371/journal.ppat.1002388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou S, Standring DN. 1992. Cys residues of the hepatitis B virus capsid protein are not essential for the assembly of viral core particles but can influence their stability. J Virol 66:5393–5398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nassal M. 1992. Conserved cysteines of the hepatitis B virus core protein are not required for assembly of replication-competent core particles nor for their envelopment. Virology 190:499–505. doi: 10.1016/0042-6822(92)91242-M. [DOI] [PubMed] [Google Scholar]
- 51.Nassal M, Rieger A, Steinau O. 1992. Topological analysis of the hepatitis B virus core particle by cysteine-cysteine cross-linking. J Mol Biol 225:1013–1025. doi: 10.1016/0022-2836(92)90101-O. [DOI] [PubMed] [Google Scholar]
- 52.Naoumov NV, Antonov KA, Miska S, Bichko V, Williams R, Will H. 1997. Differentiation of core gene products of the hepatitis B virus in infected liver tissue using monoclonal antibodies. J Med Virol 53:127–138. doi: 10.1002/(SICI)1096-9071(199710)53:2<127::AID-JMV4>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 53.Zhou S, Yang S, Standring D. 1992. Characterization of hepatitis B virus capsid particle assembly in Xenopus oocytes. J Virol 66:3086–3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Newman M, Chua PK, Tang FM, Su PY, Shih C. 2009. Testing an electrostatic interaction hypothesis of hepatitis B virus capsid stability by using an in vitro capsid disassembly/reassembly system. J Virol 83:10616–10626. doi: 10.1128/JVI.00749-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang JC, Dhason MS, Zlotnick A. 2012. Structural organization of pregenomic RNA and the carboxy-terminal domain of the capsid protein of hepatitis B virus. PLoS Pathog 8:e1002919. doi: 10.1371/journal.ppat.1002919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Selzer L, Kant R, Wang JC, Bothner B, Zlotnick A. 2015. Hepatitis B virus core protein phosphorylation sites affect capsid stability and transient exposure of the C-terminal domain. J Biol Chem 290:28584–28593. doi: 10.1074/jbc.M115.678441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zheng J, Schodel F, Peterson DL. 1992. The structure of hepadnaviral core antigens. Identification of free thiols and determination of the disulfide bonding pattern. J Biol Chem 267:9422–9429. [PubMed] [Google Scholar]
- 58.Newman M, Suk FM, Cajimat M, Chua PK, Shih C. 2003. Stability and morphology comparisons of self-assembled virus-like particles from wild-type and mutant human hepatitis B virus capsid proteins. J Virol 77:12950–12960. doi: 10.1128/JVI.77.24.12950-12960.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chou SF, Tsai ML, Huang JY, Chang YS, Shih C. 2015. The dual role of an ESCRT-0 component HGS in HBV transcription and naked capsid secretion. PLoS Pathog 11:e1005123. doi: 10.1371/journal.ppat.1005123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jung J, Kim HY, Kim T, Shin BH, Park GS, Park S, Chwae YJ, Shin HJ, Kim K. 2012. C-terminal substitution of HBV core proteins with those from DHBV reveals that arginine-rich 167RRRSQSPRR175 domain is critical for HBV replication. PLoS One 7:e41087. doi: 10.1371/journal.pone.0041087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Watts NR, Conway JF, Cheng N, Stahl SJ, Belnap DM, Steven AC, Wingfield PT. 2002. The morphogenic linker peptide of HBV capsid protein forms a mobile array on the interior surface. EMBO J 21:876–884. doi: 10.1093/emboj/21.5.876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sakamoto Y, Yamada G, Mizuno M, Nishihara T, Kinoyama S, Kobayashi T, Takahashi T, Nagashima H. 1983. Full and empty particles of hepatitis B virus in hepatocytes from patients with HBsAg-positive chronic active hepatitis. Lab Invest 48:678–682. [PubMed] [Google Scholar]
- 63.Luckenbaugh L, Kitrinos KM, Delaney WE IV, Hu J. 2015. Genome-free hepatitis B virion levels in patient sera as a potential marker to monitor response to antiviral therapy. J Viral Hepat 22:561–570. doi: 10.1111/jvh.12361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tan Z, Pionek K, Unchwaniwala N, Maguire ML, Loeb DD, Zlotnick A. 2015. The interface between hepatitis B virus capsid proteins affects self-assembly, pregenomic RNA packaging, and reverse transcription. J Virol 89:3275–3284. doi: 10.1128/JVI.03545-14. [DOI] [PMC free article] [PubMed] [Google Scholar]