

ABSTRACT
Extensive studies of orthoretroviral capsids have shown that many regions of the CA protein play unique roles at different points in the virus life cycle. The N-terminal domain (NTD) flexible-loop (FL) region is one such example: exposed on the outer capsid surface, it has been implicated in Gag-mediated particle assembly, capsid maturation, and early replication events. We have now defined the contributions of charged residues in the FL region of the Rous sarcoma virus (RSV) CA to particle assembly. Effects of mutations on assembly were assessed in vivo and in vitro and analyzed in light of new RSV Gag lattice models. Virus replication was strongly dependent on the preservation of charge at a few critical positions in Gag-Gag interfaces. In particular, a cluster of charges at the beginning of FL contributes to an extensive electrostatic network that is important for robust Gag assembly and subsequent capsid maturation. Second-site suppressor analysis suggests that one of these charged residues, D87, has distal influence on interhexamer interactions involving helix α7. Overall, the tolerance of FL to most mutations is consistent with current models of Gag lattice structures. However, the results support the interpretation that virus evolution has achieved a charge distribution across the capsid surface that (i) permits the packing of NTD domains in the outer layer of the Gag shell, (ii) directs the maturational rearrangements of the NTDs that yield a functional core structure, and (iii) supports capsid function during the early stages of virus infection.
IMPORTANCE The production of infectious retrovirus particles is a complex process, a choreography of protein and nucleic acid that occurs in two distinct stages: formation and release from the cell of an immature particle followed by an extracellular maturation phase during which the virion proteins and nucleic acids undergo major rearrangements that activate the infectious potential of the virion. This study examines the contributions of charged amino acids on the surface of the Rous sarcoma virus capsid protein in the assembly of appropriately formed immature particles and the maturational transitions that create a functional virion. The results provide important biological evidence in support of recent structural models of the RSV immature virions and further suggest that immature particle assembly and virion maturation are controlled by an extensive network of electrostatic interactions and long-range communication across the capsid surface.
INTRODUCTION
Infectious retrovirus particles are the end products of a complex multistage assembly process. In the early phase, the Gag polyprotein binds virion genomic RNA and is targeted to the plasma membrane, where it assembles a near-spherical immature protein lattice. As the virion leaves the cell by a budding process, the virally encoded protease PR is activated, initiating maturation. Cleavage of the Gag polyprotein into the structural proteins MA (matrix), CA (capsid), and NC (nucleocapsid) results in a large-scale reorganization that leaves MA associated with the viral envelope while NC, the genome, and replication enzymes condense to form the reverse transcription complex (RTC). Newly liberated CA protein rearranges around the nascent RTC, forming a closed polyhedral capsid. Since the orderly completion of the capsid shell is essential for the early stages of the replication cycle (1–3), the Gag lattice and the maturation process pose attractive targets for pharmacological intervention (4–10).
CA provides the protein-protein interactions that organize both the immature Gag lattice and the capsid structure of the mature core, thereby determining the size, shape, and functionality of both structures. The protein structure is highly conserved despite considerable CA sequence variation across the orthoretrovirus families, including human immunodeficiency virus (HIV) (a lentivirus), Rous sarcoma virus (RSV) (an alpharetrovirus), murine leukemia virus (MLV) (a gammaretrovirus), and Mason-Pfizer monkey virus (M-PMV) (a betaretrovirus) (www.ncbi.nlm.nih.gov/genome/viruses/retroviruses). In each virus, the CA monomer comprises two predominantly alpha-helical domains, a 6- to 7-helix N-terminal domain (NTD), and a 4-helix C-terminal domain (CTD). The helical folds of the two domains are largely identical in both the mature capsid form and the Gag precursor, with the major distinguishing feature being the β-hairpin structure that precedes the first α-helix of the mature NTD. This β-hairpin forms only after CA is cleaved from the upstream Gag domain. In the NTD, helices α1 to α4 and the last helix form a tight bundle, while the loops and short helices between α4 and the last helix comprise a surface-exposed flexible-loop (FL) region that is the least conserved region in the CA sequence and exhibits considerable structural variation across retroviral families (11–19, 87).
The sequence diversity in the FL region across virus families has been driven in part by species-specific interactions between the viruses and their hosts, since the region is displayed on the outer capsid surface and exposed to the cytosol after entry into a new cell (20, 21). At least three cellular proteins are known to bind the HIV-1 FL region: cyclophilin A, tripartite motif 5α (TRIM5α), and cleavage and polyadenylation specific factor 6 (CPSF6) (22–29). The MLV CA interacts with the cellular Friend virus susceptibility factor 1 (Fv1) (30). Binding of these proteins to capsids in the cytosol affects several postentry events (22, 23, 30–40), suggesting that the FL serves both structural and regulatory roles in the establishment of a new infection. As yet, no CA-binding host proteins have been identified in the alpharetroviruses.
Diversity in the FL sequences is also likely related to the unique structural roles that the region plays in the Gag lattices of evolutionarily distant viruses. The immature structures are comprised of Gag protein hexamers, with the CA NTD and CTD of each molecule vertically stacked and both interhexameric and intrahexameric contacts mediated horizontally (i.e., NTD-NTD and CTD-CTD) (41–43). Recent high-resolution studies of immature virions of M-PMV and HIV allowed unequivocal positioning of the alpha helices of the two domains. The packing of CTDs in the inner CA layer is highly conserved across virus families (42, 44, 45, 47, 82) and provides for the critical role of the CTD and immediately downstream spacer peptide in creating the interhexameric contacts that form the Gag lattice. In contrast, NTD packing is strikingly different between the two viruses. In M-PMV, the FL region and helix α7 form an extensive dimeric interface that provides prominent interhexamer interactions (44). NTD packing in the HIV lattice features more extensive intrahexamer interactions that include a modest contribution of the FL, while interhexamer interactions are formed by helices α1 and α2 (45). Two new models of the RSV Gag lattice developed by all-atom molecular dynamics (MD) modeling and by cryo-electron tomography of in vitro-assembled protein now indicate that the RSV lattice bears more similarity to the M-PMV Gag model than to that of HIV (46, 47).
In all the orthoretroviruses, the extensive lattice interfaces formed by both domains must separate during maturation to allow repositioning of the domains and formation of the mature capsid (44–47). These mature structures are closed polyhedral shells formed of CA hexamers and punctuated by 12 points of pentameric symmetry (20, 48, 49). Unlike in the immature lattice, the interhexameric interactions in mature capsids are contributed entirely by adjacent CTDs. Additionally, a unique NTD-CTD interface (15, 20, 21, 49, 50) and a CTD trimeric interface (51, 52) distinguish the mature from the immature lattice.
Genetic studies in RSV and other retroviruses support the dominant role of the CTD and adjacent SP in driving Gag assembly (53–58). However, biological evidence from several labs has also suggested a contribution of the FL in the assembly of functional virions, consistent with the recent structural studies. Deletions within the FL region of a protease-deficient RSV Gag yielded particles of highly heterogeneous size (59). Similarly, in MLV and HIV, various point mutations, deletions, or insertions involving the FL region resulted in inefficient particle release or products of heterogeneous size (3, 60–62). In another RSV study, FL point mutations emerged spontaneously as second-site suppressors of β-hairpin lesions, suggesting a role of the FL residues during either Gag assembly or maturational rearrangements (63).
In the current study, we have examined the contributions of specific FL residues in the assembly of infectious RSV particles in order to provide important functional data needed for the assessment of the evolving models for alpharetrovirus assembly. We designed mutations in the FL region, targeting predominantly charged residues, and assessed their effects on virus replication and particle assembly. The results are consistent with a role for the FL region in the formation of a well-ordered Gag lattice and suggest that this depends upon a network of electrostatic interactions at the NTD-NTD interface and across the lattice surface. Because of the need for such contacts to separate upon proteolytic processing, the orderly rearrangements that create the functional core structure are also expected to require proper management of surface charges.
MATERIALS AND METHODS
Plasmids.
Mutations were created in the replication-competent proviral vector pRS.V8.eGFP, which carries a gag gene derived from the Prague C strain of RSV and the enhanced green fluorescent protein (EGFP) in place of the src gene (64). Wild-type (WT) and mutant proteins were expressed in Escherichia coli using a pET-24(+) plasmid bearing the RSV Prague C CA sequence (residues 1 to 239) and pET3xc containing the ΔMBDΔPR Gag coding sequence (65, 66).
Infectivity assays.
Viral infectivity assays were performed as previously described (67). Viral stocks were produced by transfection of QT6 quail cells with the wild-type or mutant proviral plasmids and harvested after 1 day. Virus suspensions were standardized according to the percentage of green fluorescent protein (GFP)-expressing cells in the producer cultures as measured by flow cytometry and were used to infect DF1 chicken cells. Virus spread was monitored by flow cytometry up to 14 days postinfection. The growth rate for each virus was expressed as the change in ln(percent GFP-positive cells)/time (67, 68).
Particle release assays.
QT6 cells were transfected with proviral plasmids and radiolabeled with [35S]Met-Cys (54, 67). CA-related proteins in cell lysates and medium samples were immunoprecipitated with anti-CA serum (αFCRC12) and analyzed by SDS-PAGE, autoradiography, and densitometry. Relative levels of particle release were calculated as a ratio of CA in medium to Gag in lysate.
Viral core detergent resistance.
Core stability assays were also performed in QT6 cells (63, 69) that were transfected with proviral plasmids and radiolabeled with [35S]Met-Cys. Released virions were pelleted through sucrose solutions containing either 1% Triton X-100 or no detergent. Soluble and pelletable CA-related proteins were immunoprecipitated and quantified as described above and then expressed as a percentage of total CA.
Rate velocity sedimentation.
Virion size distribution was assessed by velocity sedimentation assays (59, 70, 71). Briefly, QT6 cells were transfected with proviral vectors or with pCMV Gag3h (a protease-deficient Gag construct that serves as an internal control) and labeled with [35S]Met-Cys for 7 h. The cell-free medium samples containing harvested virus were spiked with Gag virus-like particles (VLPs), and the mixtures were subjected to rate zonal centrifugation in a 10 to 30% (wt/wt) at sucrose gradient 26,000 rpm for 30 min. The CA proteins present in one-milliliter fractions were analyzed by immunoprecipitation, as described above.
Protein expression and purification.
CA protein was expressed in Escherichia coli BL21(DE3) and purified as described previously (72). Purification of ΔMBDΔPR Gag protein expressed in E. coli was adapted from the method described by Ma and Vogt (65). Cells were lysed and nucleic acid precipitated with 0.15% polyethyleneimine. The Gag protein was ammonium sulfate precipitated, isolated by cation exchange, further purified by size exclusion chromatography (SEC), and stored at ∼ 5 mg/ml in buffer [25 mM HEPES, 0.5 M NaCl, 0.1 mM EDTA, 0.1 mM tris(2-carboxyethyl)phosphine (TCEP), and 0.01 mM ZnSO4, pH 7.5].
In vitro assembly assays.
Assembly was initiated by combining purified CA protein with sodium phosphate (pH 8) to final concentrations of 2 mg/ml and 500 μM, respectively (72). The resulting turbidity (optical density at 450 nm [OD450]) was recorded. Experiments were performed in triplicate. To assess Gag assembly, purified ΔMBDΔPR protein was first diluted to 1 mg/ml with MES [2-(N-morpholino)ethanesulfonic acid] buffer (50 mM MES and 100 mM NaCl, pH 6.0). Assembly was initiated by addition of a 50-mer GT-repeat DNA oligonucleotide at a protein-to-oligonucleotide ratio of 10:1 by weight.
Electron microscopy.
Thin sections of virus-expressing cells were prepared for transmission electron microscopy (TEM), stained with alcoholic uranyl acetate (UA) and lead citrate as described previously (73), and imaged with a 120-kV JOEL 1400 transmission electron microscope at 60 kV. Particle-producing cells were identified by the appearance of 10 or more particles of approximately viral size (between 100 and 150 nm) and/or actively budding virus-like structures. A total of 40 particle-producing cells were examined in each specimen. The perimeter of each cell was scanned carefully for typical virus particles, which were identified as spherical shapes with central, hyperdense condensations (viral RNA) and wispy protrusions (Env) along their circumference.
In vitro-assembled CA and Gag proteins were analyzed by negative-stain EM as previously described (72). Assembled material was collected at the end of the assembly period (at the point of maximum turbidity for CA or after 30 min for Gag), adhered onto Formvar- and carbon-coated grids, and stained with 2% uranyl acetate prior to TEM imaging.
Suppressor mutant isolation.
Serial passaging of virus-infected DF1 cells was used to select for evolved viruses that carry a second-site suppressor of a crippling FL mutation. DF1 cells were infected as described above and then serially passaged. When the proportion of GFP-positive cells reached 90%, the medium was passed onto uninfected DF1 cells. This was repeated until a prominent increase in virus growth rate was observed. Cellular genomic DNA was isolated from the infected cells and sequenced. Suspected suppressors were recreated in proviral plasmid with and without the corresponding FL mutation for confirmation of the suppressor phenotype.
MD simulations.
A wild-type (WT) model and eight models incorporating the mutations (D87A/E/R, E99A/Q, K107D, A134V, and D87E/A134V) were prepared using Visual Molecular Dynamics (VMD) (74) based on a previously published structure (46). Each model was solvated in a sufficiently large water box with salinity set to 150 mM NaCl. All-atom MD simulations, typically involving 1.8 million atoms (60 CA proteins), were performed using NAMD 2.10 (75) for 100 ns with positional restraints applied to the lattice boundary and CTDs. The number and frequency of all intermolecular electrostatic and hydrogen bonds involving the NTD were calculated and are listed in Table S1 in the supplemental material for the WT and all mutant lattices. For each lattice, the occupancy of bonding between each pair of residues was averaged over 6 monomers in the central hexamer. For ease of reading, the prominent interactions (those that occur with ≥30% frequency in either the WT or one of the mutant models) were highlighted. Flexibility of the protein was measured by means of the root mean square fluctuations (RMSF). Additionally, VMD was used to estimate interaction strengths between subunits using the number of hydrogen bonds between monomers as a surrogate measure (74). Electrostatic potentials for the average coordinates of the heavy atoms over the last 50 ns of simulation were determined at 300 K and 150 mM NaCl, using the PDB2PQR/APBS v2.0.0 suite of programs (76). The partition-around-medoids (PAM) algorithm was employed to cluster the structural ensembles from the simulations, and the root-mean-square difference (RMSD) between structures was employed as the criterion for similarity (46). PAM identified representative conformations and quantified the relative population between multiple conformers present during simulation.
Network analysis was performed to quantify the dynamic coupling between residue D87 and the residues at the NTD-NTD dimer interface (77). The value of the dynamic coupling is inversely proportional to the correlation between residues and is employed in network analysis as a measure of allosteric communication between distal residues. Correlation between residues for the central hexamer in the Gag lattice was calculated from the last 50 ns of simulation using CARMA (78). Subsequently, highly correlated paths of residues, with their respective dynamic couplings, between protein sites of interest were calculated employing the subopt feature available in NetworkTools (79). Additionally, a total of 1,000 suboptimal paths between D87 and R145 of each NTD monomer in the central hexamer were identified using the weighted implementation of suboptimal paths (WISP) algorithm (80). Therefore, a total of 6,000 shortest paths were collected for each RSV mutant.
RESULTS
The present study was initiated to provide functional data about the contributions of NTD residues in the formation of immature virions to complement the emerging structural models of immature retrovirions. Our attention was directed to the FL region by a prior study in which compromising mutations in the β-hairpin region of RSV CA were partially suppressed by compensating mutations at R86 and N104 in the FL region (63). We therefore undertook a genetic analysis of FL assembly function in RSV, creating mutations in the proviral plasmid at the codons specifying R86 and N104 (the two positions at which β-hairpin suppressors had arisen), at D87 and R89 at the end of helix α4 and the beginning of the first loop (conserved residues previously implicated in MLV Gag assembly [16]), at K107, which lies in the trimeric NTD interface in RSV Gag lattice (46, 47), and at two intervening polar residues, Q95 and E99 (Fig. 1). The virus growth rate and Gag/CA release efficiency for all mutants are presented in Table 1.
FIG 1.
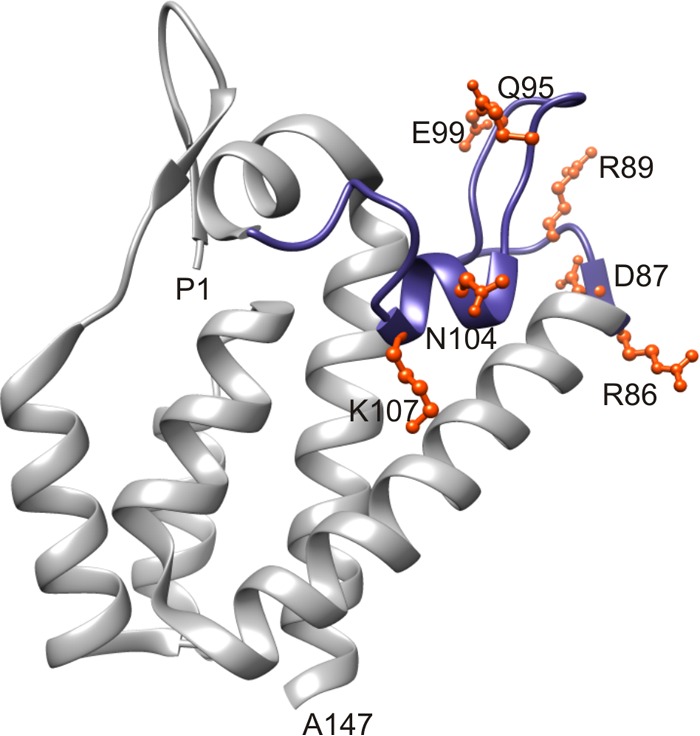
The flexible-loop region in the RSV CA N-terminal domain. The crystal structure of the RSV CA N-terminal domain (PDB 1EM9) is shown with the flexible loop highlighted in blue. Residues mutated in this study are depicted in ball-and-stick form and colored orange.
TABLE 1.
Gag release and virus infectivity phenotypes of CA flexible-loop mutant viruses
| WT residue | Mutant residue | Gag/CA release (mean ± SD)a,b | Rate of spread (mean ± SD)b,c |
|---|---|---|---|
| All | 1.44 ± 0.46 | 0.80 ± 0.17 | |
| R86 | A | 1.38 ± 0.42 | 0.62 ± 0.07 |
| D | 1.28 ± 0.17 | ≤0.05*** | |
| K | 1.84 ± 0.32 | 0.73 ± 0.14 | |
| D87 | A | 0.83 ± 0.73 | 0.08 ± 0.09*** |
| R | 0.00 ± 0.01** | ≤0.05*** | |
| E | 1.92 ± 0.69 | 0.28 ± 0.02*** | |
| N | NDd | 0.14 ± 0.08*** | |
| R89 | A | 1.19 ± 0.75 | 0.57 ± 0.07* |
| D | 1.14 ± 0.60 | ≤0.05*** | |
| K | 1.01 ± 0.02 | 0.58 ± 0.25* | |
| Q95 | A | 1.07 ± 0.31 | 0.89 ± 0.06 |
| E | 0.46 ± 0.44 | 0.78 ± 0.09 | |
| E99 | A | 1.25 ± 0.48 | 0.41 ± 0.04** |
| K | 0.73 ± 0.93 | ≤0.05*** | |
| D | 0.89 ± 0.81 | 0.70 ± 0.02 | |
| Q | ND | 0.19 ± 0.03*** | |
| N104 | A | 0.58 ± 0.43 | 0.59 ± 0.07* |
| R | ND | ≤0.05*** | |
| D | ND | 0.63 ± 0.001* | |
| K107 | A | 1.02 ± 0.85 | ≤0.05*** |
| D | 0.33 ± 0.46 | ≤0.05*** | |
| R | 1.75 ± 0.45 | 0.30 ± 0.04*** |
Particle release was quantified from measurements of 35S-labeled CA in supernatants of transfected cells, normalized for Gag expression, as described in Materials and Methods.
Statistical analyses are described in the legend to Fig. 2C. ***, P < 0.001; **, P = 0.001 to 0.01; *, P = 0.01 to 0.05.
Rates of virus spread were calculated from values for the percentage of GFP-expressing DF1 cells during the exponential phase of virus spread at 37°C and are expressed as Δln(percent green cells) per day (60).
ND, not done.
An alanine mutagenesis screen was conducted first, and the growth of mutant viruses in DF1 cells was monitored over 14 days by detection of the virus-encoded GFP (Fig. 2A and B). For a quantitative assessment, the rate of virus spread through the cell population was calculated from the change in the fraction of green cells over time (Fig. 2C) (67, 68). Of the seven alanine mutants tested at 37°C, only two were severely crippled, the K107A and D87A mutants. Spread of the K107A mutant was undetectable over 2 weeks, and that of the D87A mutant was ∼10% of the WT rate (Fig. 2B and C). In contrast, the R86A, N104A, R89A, and Q95A viruses spread at rates of between 60% and 90% of that of the WT virus (Fig. 2A and C). The E99A virus was less robust, spreading at approximately half the WT rate. When the temperature was raised to 42°C, all mutants showed further reduction in infectivity. The E99A mutant was particularly temperature sensitive, with its activity reduced at 42°C to the level of the severely crippled D87A and K107A viruses (Fig. 2C). These data indicate that the side chains at all seven positions influence virus infectivity but that the charged residues D87, E99, and K107 are particularly important.
FIG 2.
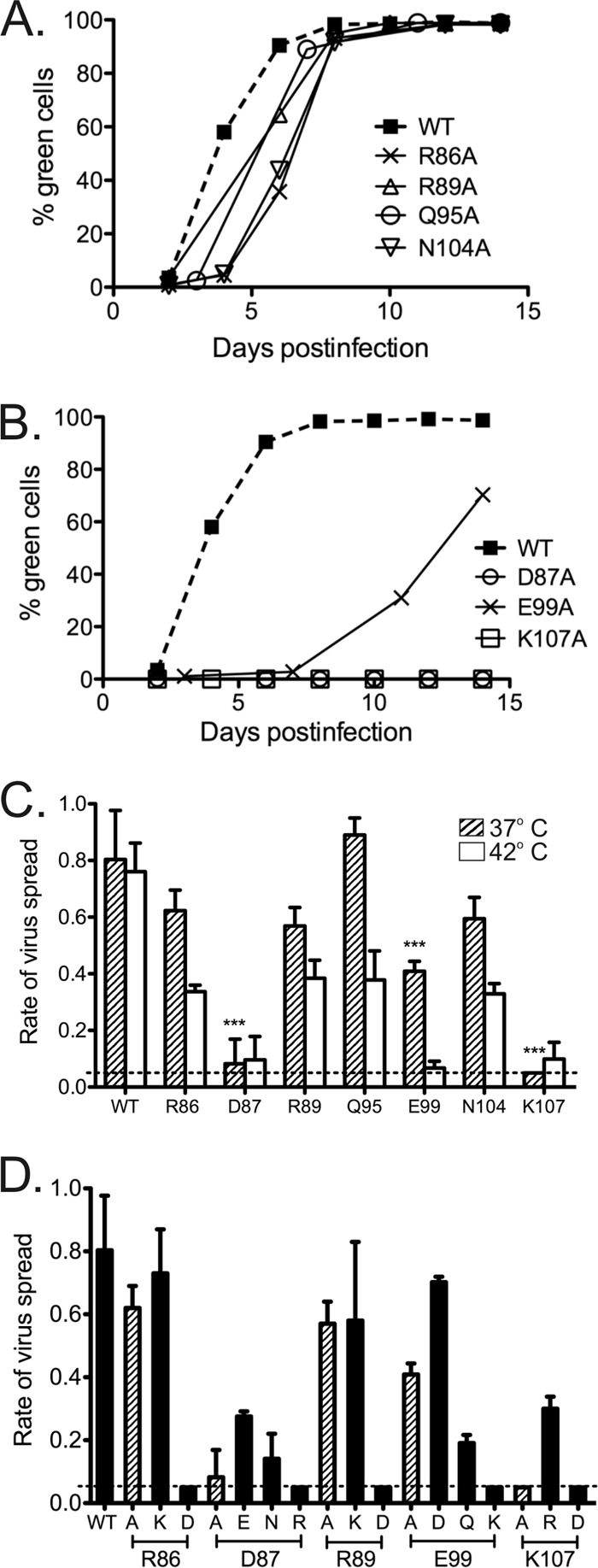
Infectivity of wild-type virus and FL mutant viruses. (A and B) Spread of WT and FL alanine substitution viruses in DF1 cells over 14 days at 37°C detected by GFP expression. (C) The rate of virus spread for WT and Ala mutants at two temperatures was expressed as the Δln(percent green cells) per day over the period of exponential growth. The dotted line marks the practical limit of accurate rate determination. Spreading rates for mutant viruses at 37°C were compared to those for the WT by one-way analysis of variance (ANOVA), with Bartlett's test used to verify equal variances (***, P < 0.001). For each bar, n = 4. (D) Rate of spread for WT and charge-manipulated viruses in DF1 cells at 37°C. Results for the Ala mutants (hatched bars) are reproduced from panel C for comparison.
Preservation of charge is necessary for virus infectivity.
Additional substitutions were created to test the importance of charges in the FL region. Strikingly, placement of the opposite charge at all five charged residues (R86, D87, R89, E99, and K107) resulted in completely noninfectious viruses (Fig. 2D). Similarly, the replacement of N104 with aspartate (which is the natural allele in most strains of RSV) allowed strong replication, while its charged-reversed N104K allele prevented virus spread (Table 1).
Conservative substitutions that preserved the amino acid charge led to the production of viruses that retained considerable infectivity. The D87E, E99D, and K107R mutants were each significantly more infectious than their corresponding alanine variants by a factor of 2- to 6-fold (P < 0.05) (Fig. 2D). Next, the acidic residues (D87 and E99) were replaced with their amide derivatives to remove charge while retaining overall size and polar character. Neither the D87N nor the E99Q virus showed greater replication capability than its alanine counterpart; in fact, the rate of spread of the E99Q virus was only half that of the E99A virus. Thus, the presence of a side chain capable of hydrogen bonding was insufficient to support function, arguing for the importance of the charge at these positions.
Particle release and morphology.
To assess the effects of the mutations on particle production, transfected QT6 cells were metabolically labeled and CA-related proteins in cell lysates and medium samples were analyzed by immunoprecipitation (Fig. 3 and Table 1). Gag expression levels measured from lysates of all mutants were similar to those of the WT. Gag protein appeared to be processed normally into CA in all mutants, and no abnormal cleavage products were seen in lysates or medium fractions (Fig. 3A and data not shown). Only two mutations, both charge reversals, caused potent effects on particle production: CA release was abolished by the D87R mutation and severely impacted in the K107D virus (Fig. 3B). The E99K charge swap reduced the average CA release, but the experiment-to-experiment variability was high and the difference was not statistically significant (P = 0.05). Most of the other mutants were similarly variable. In total, FL function in particle release is quite tolerant of mutation, with the notable exception of the charge swaps at D87 and K107.
FIG 3.
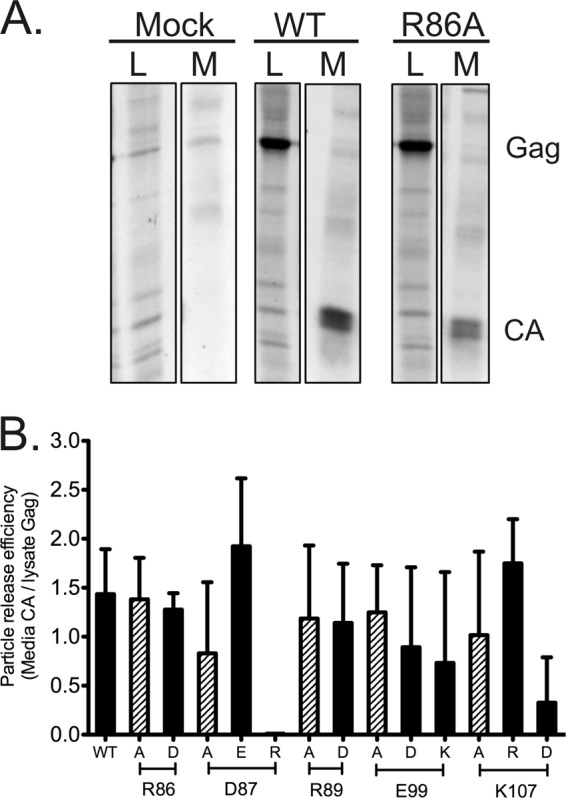
Detection of virus release for wild-type virus and FL mutant viruses. (A) Representative data for Gag/CA release and processing in mock-, WT-, and R86A-transfected QT6 cells. CA-related proteins were immunoprecipitated from cell lysate (L) and medium (M) samples, as described in Materials and Methods. (B) Particle release as quantified from measurements of 35S-labeled CA in supernatants of transfected cells, normalized for Gag expression, as described in Materials and Methods. All particle release experiments were performed in triplicate.
Since alterations at the D87, E99, and K107 positions produced the most dramatic effects on infectivity, we next examined the morphology of D87A, E99A, and K107A particles produced by transfected QT6 cells at 37°C by TEM of embedded and thin-sectioned specimens. Typical WT virions of 110 to 140 nm in diameter and roughly spherical morphology with electron-dense central cores were observed in abundance and often in clusters near the cell surface of the expressing cells (Fig. 4A). The capsid shells are not typically preserved in such analyses of RSV; the dense core represents the nucleocapsid component of the virion.
FIG 4.

Representative electron micrographs of thin sections of QT6 cells transfected at 37°C with WT or mutant proviral plasmids. (A to I) Examples of particles with typical morphology are marked with open triangles. Black arrows mark areas of density that may represent Gag accumulation at membranes. Black boxes surround viral particles with heterogeneous features (e.g., large particles or ones with fragmented, multiple, or eccentric nuclei). Bars 500 nm. (J) Size distribution of viral particle diameters measured from electron micrographs using ImageJ software.
Normal-appearing mature virus particles were present in samples of cells infected with the replication-crippled K107A and D87A viruses but were very rare in the latter case (Fig. 4B, C, E, and F). In addition, these cells produced heterogeneous particles of random size, electron-dense membrane features reminiscent of Gag accumulation, and large-scale cell membrane disturbances. In contrast, the E99A virus produced large amounts of normal-appearing particles (Fig. 4D). The diameters of particles with WT-like features were measured for each mutant except for the D87A virus, in which the numbers were vanishingly small. The size distributions of E99A and K107A particles overlapped but were broader than that of the WT, as evidenced by the standard deviations, although the mean diameters were not statistically significantly different (Fig. 4J). The three conservative substitution mutants D87E, E99D, and K107R produced abundant WT-like virions (Fig. 4G to I) without evidence of the membrane disturbances seen with the alanine mutations.
Particle size distribution.
Sucrose gradient rate-velocity sedimentation allows an unbiased comparison of the entire population of CA-containing particles released from cells. We examined the sedimentation of radiolabeled mutant particles (Fig. 5) in 10 to 30% sucrose gradients. Protease-deficient (Gag3h) virus-like particles, which resemble WT RSV in size distribution pattern (59, 81), were mixed with each mutant virus sample as an internal control for the comparison of size distributions and an indicator of reproducibility of the sucrose column in the different tubes. The gradient profile for the D87A mutant was consistent with impressions from the TEM experiment. While a subset of released particles sedimented with the control peak, the total D87A population was markedly more heterogeneous than the internal control, with a considerable proportion of CA protein near the top of the gradient. Only 42% of D87A CA was found in the three central fractions that contained 71% of the internal control (Fig. 5A). In a duplicate gradient, only 23% of the D87A mutant (versus 54% for the control) was present in the central 3 fractions (data not shown). In contrast, the E99A and K107A particles were less heterogeneous than the D87A particles (Fig. 5B and C), while the conservative substitution viruses E99D and K107R were virtually identical to the internal control (Fig. 5E and F). The D87E particles formed a more symmetric peak than did the D87A particles, although the particles were slightly larger than the control (Fig. 5D). In total, the gradient profiles confirm the presence of normal-sized particles in all mutants, consistent with the TEM findings, but also support marked irregularity in the particle size distribution in the case of the D87A mutant.
FIG 5.

Analysis of particle size by rate zonal centrifugation in 10 to 30% sucrose gradients. (A) D87A mutant; (B) D87E mutant; (C) E99A mutant; (D) E99D mutant; (E) K107A mutant; (F) K107R mutant. Dotted vertical lines mark the peak fractions of the Gag 3-h internal control. Arrows indicate the direction of sedimentation. All experiments were performed in triplicate.
In vitro Gag assembly.
To assess whether the disorder in particle regularity described above for the D87A virus could be reproduced in vitro with purified Gag protein, we attempted to test the effects of mutations on the assembly capability of a truncated Gag protein, ΔMBDΔPR, that has been used extensively for RSV Gag studies. The WT ΔMBDΔPR protein (Fig. 6) assembled into spheres of 60 to 65 nm diameter showing the characteristic double-layered organization that is determined by the NTD and CTD CA domains (42, 82). Unfortunately, the D87A mutant protein was expressed in E. coli but precipitated uncontrollably during purification, and its assembly behavior could not be analyzed. The same was true of proteins bearing the D87R and K107D charge swap mutations. In contrast, the D87E and E99A mutations, which allowed robust particle formation in vivo, also permitted the in vitro assembly of Gag structures indistinguishable from the WT, as did the K107A substitution.
FIG 6.
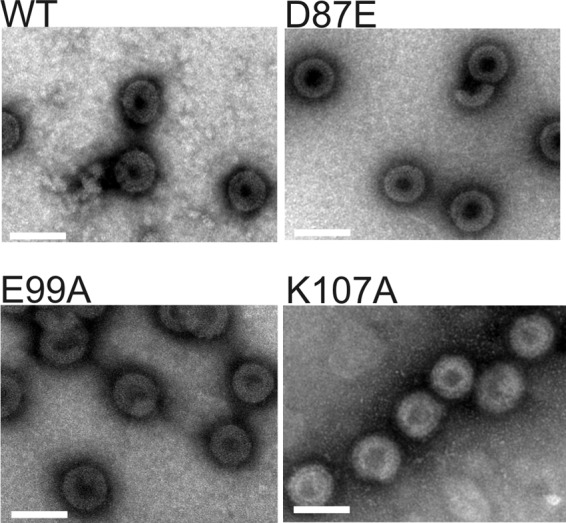
Electron micrographs of negatively stained, in vitro-assembled WT (ΔMBDΔPR) Gag protein and three FL mutant variants. Bars, 100 nm.
Structural consequences of D87 mutations.
MD simulations employing the recent computational model for the WT Gag lattice revealed a number of intermolecular electrostatic and H-bond interactions contributed by the FL residues (see Table S1 in the supplemental material), many of which had been predicted by the p10-CA dimer crystal structure (83) and/or the recent cryo-tomography model (47). One such interaction observed by MD was the transient intrahexameric bonding between a cluster of charged FL residues (R86, D87, and R89) and several residues in the CA N terminus that form the β-hairpin upon proteolytic processing (Fig. 7A; see Table S1 in the supplemental material) previously noted by Schur et al. (47). Besides intermolecular interactions, D87 also forms a prominent intramolecular salt bridge with R89 in the same monomer at 84% likelihood as determined by structural clustering. In addition, residue E99 in the large loop forms an intramolecular interaction with R125 at a 50% likelihood, which may help to stabilize the region by anchoring the loop to helix α7.
FIG 7.

Mutations at residues 87 and 99 alter properties of the immature lattice. The effects of mutations on the Gag lattice structure were predicted based upon the WT MD model of Goh et al. (46). (A) D87A alters the conformations adopted by R89 and its network of interactions. Hydrogen bonds are shown as black lines. (B) The flexibility of residues 90 to 95 in the FL large loop is markedly increased upon D87A mutation as reflected by RMSF. (C) Surface electrostatic potential maps show that the removal of negative charges by D87A or E99Q leads to accumulation of positive charge at the interhexameric interface (black arrows). Only the NTD is shown for clarity. (D) The D87E change alters the interactions of residue E41 at the NTD dimer interface. E41 interacts with R141 and R145 (black lines) across the interface in WT but favors contact with Q138 and R145 in the D87E lattice. Helices 2 and 7 are shown as solid ribbons and the remainder of the NTD as translucent ribbons.
The D87E, D87A, D87R, E99A, and E99Q changes were introduced into the Gag lattice model in silico, and effects on the surface charge distribution and the network of interactions were examined (Fig. 7C; see Table S1 in the supplemental material and Fig. S1 at http://hdl.handle.net/2142/89695). Removal of a negative charge at position 87 or 99 resulted in a noticeable accumulation of positive charges at the interhexameric interfaces of the lattice. In addition, the D87A substitution caused an approximately 2-fold increase in flexibility of the CA monomer in the large loop (residues 90 to 95) compared to the WT protein (Fig. 7B). No similar alterations in flexibility were observed with the other mutants.
The three mutations introduced at position 87 (D87E, D87A, and D87R) each caused unique effects on the intramolecular interaction between D87 and R89 observed in silico. The D87A and D87R mutations effectively reduced the frequency of D87-R89 bonding from 84% to 22% and to 0%, respectively. Furthermore, when R89 was freed from its interaction with D87 by the D87A substitution, it gained the ability to adopt alternate conformations, forming intramolecular and intermolecular interactions with nearby polar residues that were not seen in the WT lattice (see Table S1 in the supplemental material). Additionally, introduction of D87R enables residue 87 to interact with neighboring N-terminal CA residues such as E7, G8, and P9. Thus, D87A and D87R both disturb the network of interaction observed in WT and result in an accumulation of positive charges at the interhexameric interface (Fig. 7A and C; see Fig. S1 at http://hdl.handle.net/2142/89695).
The conservative D87E change, which in vivo allowed the production of abundant amounts of normal appearing but poorly infectious particles, slightly increased the likelihood of interaction with R89 from 84% (WT) to 93%. More striking, however, were long-distance effects of D87E on CA-CA interactions. In the WT lattice, E41 on helix 2 interacts prominently with residues Q138, R141, and R145 in a polar cluster on the lower half of α7, which make the primary point of NTD-NTD contact across the dimer interface (Fig. 7D, left; see Table S1 in the supplemental material). The D87E substitution shifted the frequency of E41 interactions to favor contact with Q138 and R145 rather than R141 (Fig. 7D, right).
A second-site suppressor of D87E.
Spontaneously arising second-site suppressors have proven to be a powerful and unbiased means to reveal functional relationships between residues in RSV CA (67, 69, 84, 85). For this study, three mutant viruses (D87E, E99A, and E99C) were put through a serial passaging protocol to encourage outgrowth of new genetic variants. These experiments were repeated seven times for D87E and three times for E99A. Reversion to the WT residue was observed at position 99 in one E99A trial. The E99C mutant, not otherwise characterized in this study, behaved like E99A and showed a tendency to revert to WT, with no second-site suppressors found. In the case of D87E, seven viruses with improved infectivity were isolated and sequenced. Of these, five had reverted to the WT aspartate, while two independently acquired a second mutation, an alanine-to-valine change at position 134 on helix α7 (A134V).
To test the impact of the suspected suppressor on infectivity, a new A134V proviral vector was created. The resulting A134V virus was moderately compromised compared to the WT (Fig. 8A), consistent with previous observations of suppressors of crippling CTD mutations (67, 69, 85). However, the addition of A134V to a D87E provirus improved virus infectivity convincingly compared to D87E alone. The double mutant virus infected susceptible DF1 cells at a rate 54% of that of the WT, compared to 16% for the D87E virus, confirming A134V as a partial suppressor of the D87E lesion.
FIG 8.

Characterization of an A134V suppressor of the D87E mutation in virus and in vitro CA assembly. (A) Growth curves for the WT and D87E, A134V, and D87E A134V viruses at 37°C. Each line shows an average of at least four repeats with two different viral clones. (B) Detergent stability of WT, D87E, and D87E A134V virus capsids. The percentage of stable CA was calculated from the amount of pelletable CA after detergent exposure relative to the total CA, as described in Materials and Methods. (C) Kinetics of in vitro phosphate-driven assembly of CA proteins. (D) In vitro assembly products imaged by negative staining and EM. Bars, 100 nm.
Relevance of D87 for mature CA function.
Since the near-normal morphology of D87E virions and in vitro assembly products indicates normal Gag assembly functions, we next used detergent extraction of mature virions to determine whether compromised core integrity might contribute to the poor infectivity of D87E (54, 69). The D87E virions were indeed strongly detergent sensitive; only 5.6% (±7.3%) of CA remained pelletable after exposure to Triton X-100, versus 46.7% (±13.2%) for WT virions (Fig. 8B). In the double mutant D87E A134V virus, however, CA detergent resistance was markedly improved to 33.2% (±12.4%), suggesting that the A134V substitution confers near-normal capsid stability to viruses bearing the D87E mutation.
To test whether the capsid stability defect of the D87E virus could be observed during in vitro CA assembly, WT and mutant CA proteins were purified and triggered to self-assemble capsid-like structures by the addition of phosphate (72). The sigmoidal kinetics of assembly for the D87E protein were indistinguishable from those of WT CA (Fig. 8C), and TEM imaging revealed populations of small spherical particles and larger structures similar to the WT CA (Fig. 8D). The D87E A134V double mutant and D87E proteins behaved identically. Thus, consistent with current models for the mature capsid, in which neither D87 nor A134 makes intermolecular contacts, the D87 residue is not critical for formation of mature CA-CA interactions as assessed in vitro. Instead, the compromised core stability in D87E virions (Fig. 8B) may result from disturbances at an earlier stage of maturation when CA proteins, after cleavage from Gag, must separate to allow the rearrangements that lead to capsid formation.
The top and bottom of the NTD are dynamically coupled.
If infectivity and core defects in the D87E virus are related to the altered intermolecular interactions seen with MD modeling of the mutant lattice, we predicted that the addition of the second-site suppressor A134V to the lattice simulation may correct the changes in these interactions. This effect is indeed evident in a D87E-A134V model (see Table S1 in the supplemental material). MD simulations that include the second-site suppressor A134V showed shifts in the frequencies of intermolecular interactions involving residues E41, Q138, R141, and R145 at the point of NTD-NTD contact in the immature lattice, restoring the contacts that were disturbed by D87E (as noted above) to a more WT-like pattern. In the double mutant, A134V reduces the frequency of the E41-R145 interaction while increasing the E41-R141 contact (see Table S1 in the supplemental material). The presence of the A134V mutation also increases the buried surface area in this region of the dimer interface by an average of 15 Å2 per monomer compared to the WT lattice.
The reversal of the core stability and infectivity deficits in D87E virions by the A134V compensatory change suggests that long-range communication occurs between the FL and helix α7. Therefore, we applied dynamical network analysis to our MD models to identify dimer interface residues whose motions are highly correlated (77). This approach identified a route of communication between D87 and R145 along an optimal pathway that in the WT lattice includes residues D87, A83, and V80 near the top of helix 4 and residues S135, Q138, R141, and R145 in the lower part of helix 7 in the same NTD (Fig. 9A and B).
FIG 9.

Network analysis identifies a communication pathway across the NTD. Residues 87 (at the junction of α4 and the FL) and 145 (α47) are dynamically coupled via a communication pathway in the monomeric CA (gray). (A) Residues that constitute the optimal pathway for the WT are labeled. Helix 7 is colored in yellow, while other helices are in light gray. The communication pathway is shown in green. (B) Histogram of the WT residues involved in the pathways connecting D87 and R145, showing the frequency at which a particular residue was identified in one of the 6,000 calculated paths. (C) The average dynamic coupling along all calculated paths connecting residue 87 with residues E41, Q138, R141, and R145 is shown for the WT and mutant lattices. Dynamic coupling is a measure of the length of a highly correlated path of residues between two nodes (77) that is inversely correlated to the strength of communication. The asterisk indicates coupling across the dimer interface, i.e., between D87 of monomer 1 and R145 of monomer 2.
We employed “dynamic coupling” as a relative measure of the strength of communication, inversely related to the degree to which the motion of a pair of residues is correlated. Figure 9C presents the dynamic coupling between D87 and helix 7 residues in the WT lattice (blue bars). The low values for Q138, R141, and R145, as well as their interacting partner E41, indicate a strong correlation between these residues and D87 that is not seen for two residues (V2 and I4 near the CA N terminus) that are not part of the observed pathway.
Analyses of the D87E and D87E-A134V lattices predict effects of the mutations that correlate with the phenotypes in vivo. The primary route of communication was not altered by D87E (see Fig. S2 at http://hdl.handle.net/2142/89695). However, the D87E crippling mutation reduced the strength of the dynamic coupling between D87 and each of four residues in the pathway (E41, Q138, R141, and R145) as indicated by the higher average values compared to those for the WT (Fig. 9, red bars). Also, the high standard deviations observed at points all along the pathway argue that there is increased heterogeneity in the available pathways in this mutant. The latter is evidenced as well by the appearance of residues V40 to L48 from the neighboring monomer in the ensemble of pathways for D87E (see Fig. S2B at http://hdl.handle.net/2142/89695). Importantly, the A134V compensatory mutation, which is located at the point of crossover along the α4-α7 communication path, restored the strength of dynamic coupling that was compromised by D87E (Fig. 9C, cyan bars). In contrast, three residues that are not part of the communication pathway (V2 and I4 in the same monomer and R145 of the adjacent monomer) failed to show similar responses to the D87E and A134V mutations. In total, these findings suggest that A134V suppresses the D87E maturation defect by correcting defects in long-distance communication caused by mutation in the FL.
DISCUSSION
The flexible-loop region, like many parts of CA, plays distinctly different structural and functional roles in immature particles versus the mature infectious virions. In the immature particle, the FL is contained within a larger portion of the NTD region that forms closely packed NTD-NTD contacts (46, 47). In contrast, the FL is displayed on the outer capsid surface in the mature virus and does not appear to form direct protein-protein contacts (Fig. 10). With this study, we sought to assess the contributions of individual FL residues to the assembly pathway and thereby provide important functional data in the context of evolving models for RSV structure. Residue D87 at the beginning of the FL (Fig. 1) was found to play a dual role, making important contributions to the functional Gag lattice and controlling core maturation via long-distance communication with the distal half of helix α7.
FIG 10.

Positions of the key residues in the immature and mature hexamers. Key residues D87 (blue), E99 (red), K107 (yellow), A134 (green), and R145 (black) are marked with spheres. The CA dimer in the immature lattice is enlarged in the boxes in panel A. Residues 87 and 134 reside at the NTD dimer interface in the immature lattice (A), while none of the key residues forms any intermolecular interaction in the mature CA lattice of Cardone et al. (20) (B). Only the NTD and CTD are shown. The intermolecular contacts that are featured in Fig. 7D do not exist in mature virus and are not shown here.
Overall, RSV Gag assembly function proved to be quite tolerant of most single point mutations, with the exception of certain charge reversals. Attention was focused on the three of seven residues that proved most important for virus infectivity. The presence of a subset of particles of normal size and shape seen by TEM and sucrose gradient analyses of the D87A, E99A, and K107A mutants showed that none of these residues is individually essential for Gag to form the higher-order interactions that build the immature lattice. The latter two Gag proteins were also able to assemble particles of normal size in vitro. This retention of Gag assembly capacity despite mutations in prominent CA-CA interfaces suggests that Gag lattice assembly can accommodate certain variations in protein packing and/or that redundancy of interactions in the complex NTD network allows many single point mutations to be introduced with little effect on the overall immature lattice structure. Both are consistent with the large body of structural and genetic data for HIV, M-PMV, and RSV, which has documented that the CA CTD, rather than the NTD, is the primary determinant of Gag organization in immature retrovirus particles (3, 44, 45, 47, 57, 81, 86).
In spite of the relative tolerance of this region to mutation, the severe release deficits with charge reversals at D87 and K107 and the partial dysregulation of particle size and shape seen by TEM and gradient analysis of D87A and K107A viruses are all consistent with the location of these residues at points of NTD-NTD packing in the immature lattice models of Schur et al. and Goh et al. (46, 47). The two models for the RSV Gag lattice were developed by entirely independent approaches that yielded essentially the same protein packing, which bears similarity to that of immature M-PMV virions. The findings presented here provide important biological support for these models and are also consistent with previous studies showing that deletions across the RSV FL region allow assembly and budding but disturb the size distribution of released particles (59).
All-atom MD simulations provide both a high-resolution look at the extensive NTD-NTD interfaces of the RSV Gag lattice and a rationale for the assembly-disruptive effects of mutations at key positions. The major NTD-NTD interhexameric dimer interface involves much of the FL, with additional contributions from α2 and α7 (Fig. 7D and 10A). In addition, the loop between helices α5 and α6 participates in trimeric interactions between NTD hexamers (46). Throughout the FL region, the R86, D87, R89, E99, and K107 residues participate in an extensive network of electrostatic and hydrogen bonds across NTD-NTD interfaces as well as intramolecular interactions within the FL.
D87 sits within a cluster of charged residues (R86-D87-R89) at the end of helix α4 and the beginning of the large α4-α5 loop and is involved in hydrogen and electrostatic interactions both within and between monomers (Fig. 1 and 7A). Disruption of the interactions involving D87 may explain the suboptimal particle release and disordered morphology in vivo as well as the aggregation of the mutant Gag proteins upon purification observed with the D87A and D87R mutations. Specifically, loss of the intramolecular D87-R89 salt bridge due to D87A increases flexibility within the large loop (residues 95 to 100 [Fig. 7B]) and also allows R89, which is normally constrained in the WT particle by its interaction with D87, to make rogue inter- and intramolecular interactions with nearby residues (see Table S1 in the supplemental material).
Similarly positioned D83 and R85 residues occur in the MLV CA, and an X-ray crystal structure for an MLV CA hexameric lattice showed that these make salt bridge interactions around a 3-fold interhexameric interface. Although this arrangement has not been confirmed in immature virions, charge-neutralizing mutations in this region of the MLV Gag protein led to its accumulation at the cell membrane and destroyed particle release (16), consistent with the RSV mutant phenotypes described in this present study.
Playing a distinctly different role than that of D87, K107 lies at the beginning of the α5-α6 loop and, in the MD model of an assembled Gag particle, at the point of 3-fold lattice symmetry where the three lysines coordinate a chloride ion (46). The alanine substitution may introduce variability into hexamer packing by preventing these local electrostatic interactions, thereby dysregulating normal lattice curvature and producing the particle heterogeneity observed in K107A by EM and gradient analysis. This effect appears unlikely, however, to fully explain the magnitude of the infectivity defect in K107A.
Role of the FL in host factor interactions?
The above analyses do not preclude possible effects of some FL mutations on other aspects of the virus life cycle, which could include alterations of protein targeting, membrane binding, or interactions with host factors. It is possible that infectivity defects in some FL mutants, most notably those with mutations involving E99, may be due in part to disturbance of interactions between CA and an unknown cytosolic protein or proteins. E99 lies within the large α4 -α5 loop in RSV CA, which is among the most mobile regions in monomeric CA (87). This mobility is reflected in the considerable structural heterogeneity of these loops at various positions in the lattice model (not illustrated here) and the prominent peak in the RMSF plot, centered around residue 97 (Fig. 7B). E99 interacts with R125 (about 50% of the time in the MD simulations), which anchors the loop to helix α7 and potentially provides stability to the region. Loss of this interaction may contribute to the strong temperature sensitivity seen in the E99A mutant (Fig. 2C). Overall, however, the assembly of the RSV E99 mutants appears normal. The E99A Gag assembly behavior was largely unaffected in vivo and in vitro, as were CA assembly and capsid stability (data not shown). We note that the structurally analogous loops in HIV and MLV have been implicated in binding of the restriction factors TRIM5a and Fv-1, respectively, and cyclophilin A in HIV. Thus, it is possible that some or all of the infectivity deficits seen in the E99A and E99Q viruses reflect altered interactions with as-yet-to-be discovered host factors. This scenario is also consistent with our failure to isolate intragenic suppressors of these two mutations (see Results).
Contribution of D87 to mature core formation.
The phenotype associated with the charge-conserving D87E mutation indicates a strong influence of this region on core formation during maturation. Gag function was normal in D87E virus as indicated by both in vivo analyses and in vitro Gag assembly. However, the released particle cores exhibited a detergent-sensitive phenotype comparable to that caused by potent maturation-preventing CTD mutations in RSV (54, 69). Soluble D87E CA protein showed no deficit in assembly activity in vitro consistent with the surface-exposed location of the D87 in the capsid model (Fig. 10B). Instead, the in vivo core maturation defect is more likely due to a failure of the Gag interfaces to properly separate upon PR cleavage or otherwise rearrange properly to allow capsid formation. This is strongly supported by the properties of the spontaneous A134V suppressor, which corrected both the infectivity and core stability defects caused by D87E and in in silico simulations counteracted long-range disturbances of intermolecular bonds involving the lower half of helix α7 (see Table S1 in the supplemental material).
Such long-range effects of core maturation suppressors have been a recurring theme in studies of RSV assembly. Alterations of highly conserved residues in the CTD major homology region sequence compromise the ability of mature CA to form the CTD-CTD dimer interface. This effect can be suppressed by any one of several secondary mutations scattered across the CA sequence, including the NTD (67, 69, 85). Also, a crippling β-hairpin substitution, K5R, was optimally suppressed by a combination of two mutations, R86C and C192R, in the FL and the CTD dimer interface, respectively (63). In this study, network analysis of the MD Gag lattice model identified a pathway of communication connecting the beginning of the FL with the lower half of helix 7. The strength of the dynamic coupling between these opposite ends of the NTD was modulated by the D87E and A134V substitutions in a fashion that correlated with their effects on infectivity and stable core formation, arguing for relevance to the maturation pathway.
D87 contributes to a local network of electrostatic interactions that includes residues near the N terminus of CA (Fig. 7A) (46, 47). Schur and coauthors (47) suggested that cleavages upstream of CA help to regulate maturation by disrupting the interaction of p10 with the CA NTD. The analyses presented here suggest an additional possibility for future consideration: that cleavage at the p10-CA junction and/or folding of the β-hairpin can trigger capsid formation via allosteric signaling across the NTD, promoting the switching of interactions involving the C-terminal half of helix α7 that was previously noted by Goh et al. (46). Similarly, dynamic allostery has been observed in assembled complexes of HIV CA, which demonstrate distal structural alterations when CypA binds (88).
Overall, this study supports a role for FL residues in allowing appropriate NTD packing, leading to the release of well-formed immature virions, and in the proper management of the many possible electrostatic interactions that are critical during maturation to allow the CA domains to separate, rearrange, and transition to the final, infectious retroviral capsid. Evolution has selected for an appropriate balance of charge across the surface of CA and the capsid that permits the competing functions in Gag assembly, core maturation, and the early postentry events to be accommodated in support of effective virus replication.
Supplementary Material
ACKNOWLEDGMENTS
Many people assisted us in the conduct of this research. We thank John Flanagan for his guidance and assistance with protein purification. John Briggs, Volker Vogt, and Florian Schur were very generous in providing consultations and sharing their lattice model prior to publication. Susan Hafenstein contributed helpful discussions and critical reading of the manuscript. We are especially grateful to Roland Myers at the Penn State College of Medicine Imaging Core, who contributed invaluable assistance to 2 decades of studies from this lab prior to his retirement, and to Nate Sheaffer and Joseph Bednarzyk in the Flow Cytometry Core for their essential technical assistance.
The core facilities of Penn State College of Medicine were funded in part by a grant from the Pennsylvania Department of Health using Tobacco CURE Funds. Molecular dynamics simulations were performed on the Blue Waters Supercomputers, financed by the National Science Foundation (OCI-0725070 and ACI-1238993) under Petascale Computational Resource (PRAC) grant ACI-1440026.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00378-16.
REFERENCES
- 1.Tang S, Murakami T, Cheng N, Steven AC, Freed EO, Levin JG. 2003. Human immunodeficiency virus type 1 N-terminal capsid mutants containing cores with abnormally high levels of capsid protein and virtually no reverse transcriptase. J Virol 77:12592–12602. doi: 10.1128/JVI.77.23.12592-12602.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tang S, Murakami T, Agresta BE, Campbell S, Freed EO, Levin JG. 2001. Human immunodeficiency virus type 1 N-terminal capsid mutants that exhibit aberrant core morphology and are blocked in initiation of reverse transcription in infected cells. J Virol 75:9357–9366. doi: 10.1128/JVI.75.19.9357-9366.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.von Schwedler UK, Stray KM, Garrus JE, Sundquist WI. 2003. Functional surfaces of the human immunodeficiency virus type 1 capsid protein. J Virol 77:5439–5450. doi: 10.1128/JVI.77.9.5439-5450.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kanamoto T, Kashiwada Y, Kanbara K, Gotoh K, Yoshimori M, Goto T, Sano K, Nakashima H. 2001. Anti-human immunodeficiency virus activity of YK-FH312 (a betulinic acid derivative), a novel compound blocking viral maturation. Antimicrob Agents Chemother 45:1225–1230. doi: 10.1128/AAC.45.4.1225-1230.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li F, Goila-Gaur R, Salzwedel K, Kilgore NR, Reddick M, Matallana C, Castillo A, Zoumplis D, Martin DE, Orenstein JM, Allaway GP, Freed EO, Wild CT. 2003. PA-457: a potent HIV inhibitor that disrupts core condensation by targeting a late step in Gag processing. Proc Natl Acad Sci U S A 100:13555–13560. doi: 10.1073/pnas.2234683100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou J, Yuan X, Dismuke D, Forshey BM, Lundquist C, Lee KH, Aiken C, Chen CH. 2004. Small-molecule inhibition of human immunodeficiency virus type 1 replication by specific targeting of the final step of virion maturation. J Virol 78:922–929. doi: 10.1128/JVI.78.2.922-929.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li F, Zoumplis D, Matallana C, Kilgore NR, Reddick M, Yunus AS, Adamson CS, Salzwedel K, Martin DE, Allaway GP, Freed EO, Wild CT. 2006. Determinants of activity of the HIV-1 maturation inhibitor PA-457. Virology 356:217–224. doi: 10.1016/j.virol.2006.07.023. [DOI] [PubMed] [Google Scholar]
- 8.Adamson CS, Ablan SD, Boeras I, Goila-Gaur R, Soheilian F, Nagashima K, Li F, Salzwedel K, Sakalian M, Wild CT, Freed EO. 2006. In vitro resistance to the human immunodeficiency virus type 1 maturation inhibitor PA-457 (bevirimat). J Virol 80:10957–10971. doi: 10.1128/JVI.01369-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kelly BN, Kyere S, Kinde I, Tang C, Howard BR, Robinson H, Sundquist WI, Summers MF, Hill CP. 2007. Structure of the antiviral assembly inhibitor CAP-1 complex with the HIV-1 CA protein. J Mol Biol 373:355–366. doi: 10.1016/j.jmb.2007.07.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salzwedel K, Martin DE, Sakalian M. 2010. Maturation inhibitors: a new therapeutic class targets the virus structure. AIDS Rev 9:162–172. [PubMed] [Google Scholar]
- 11.Gamble TR, Vajdos FF, Yoo S, Worthylake DK, Houseweart M, Sundquist WI, Hill CP. 1996. Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell 87:1285–1294. doi: 10.1016/S0092-8674(00)81823-1. [DOI] [PubMed] [Google Scholar]
- 12.Jin Z, Jin L, Peterson DL, Lawson CL. 1999. Model for lentivirus capsid core assembly based on crystal dimers of EIAV p26. J Mol Biol 286:83–93. doi: 10.1006/jmbi.1998.2443. [DOI] [PubMed] [Google Scholar]
- 13.Cornilescu CC, Bouamr F, Yao X, Carter C, Tjandra N. 2001. Structural analysis of the N-terminal domain of the human T-cell leukemia virus capsid protein. J Mol Biol 306:783–797. doi: 10.1006/jmbi.2000.4395. [DOI] [PubMed] [Google Scholar]
- 14.Tang C, Ndassa Y, Summers MF. 2002. Structure of the N-terminal 283-residue fragment of the immature HIV-1 Gag polyprotein. Nat Struct Biol 9:537–543. [DOI] [PubMed] [Google Scholar]
- 15.Mortuza GB, Haire LF, Stevens A, Smerdon SJ, Stoye JP, Taylor IA. 2004. High-resolution structure of a retroviral capsid hexameric amino-terminal domain. Nature 431:481–485. doi: 10.1038/nature02915. [DOI] [PubMed] [Google Scholar]
- 16.Mortuza GB, Dodding MP, Goldstone DC, Haire LF, Stoye JP, Taylor IA. 2008. Structure of B-MLV capsid amino-terminal domain reveals key features of viral tropism, gag assembly and core formation. J Mol Biol 376:1493–1508. doi: 10.1016/j.jmb.2007.12.043. [DOI] [PubMed] [Google Scholar]
- 17.Mortuza GB, Goldstone DC, Pashley C, Haire LF, Palmarini M, Taylor WR, Stoye JP, Taylor IA. 2009. Structure of the capsid amino-terminal domain from the betaretrovirus, Jaagsiekte sheep retrovirus. J Mol Biol 386:1179–1192. doi: 10.1016/j.jmb.2008.10.066. [DOI] [PubMed] [Google Scholar]
- 18.Macek P, Chmelík J, Køížová I, Kadeøávek P, Padrta P, [caron]Zídek L, Wildová M, Hadravová R, Chaloupková R, Pichová I, Ruml T, Rumlová M, Sklenáø V. 2009. NMR structure of the N-terminal domain of capsid protein from the Mason-Pfizer monkey virus. J Mol Biol 392:100–114. doi: 10.1016/j.jmb.2009.06.029. [DOI] [PubMed] [Google Scholar]
- 19.Shin R, Tzou Y-M, Krishna NR. 2011. Structure of a monomeric mutant of the HIV-1 capsid protein. Biochemistry 50:9457–9467. doi: 10.1021/bi2011493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cardone G, Purdy JG, Cheng N, Craven RC, Steven AC. 2009. Visualization of a missing link in retrovirus capsid assembly. Nature 457:694–698. doi: 10.1038/nature07724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pornillos O, Ganser-Pornillos BK, Kelly BN, Hua Y, Whitby FG, Stout CD, Sundquist WI, Hill CP, Yeager M. 2009. X-ray structures of the hexameric building block of the HIV capsid. Cell 137:1282–1292. doi: 10.1016/j.cell.2009.04.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wolf D, Goff SP. 2008. Host restriction factors blocking retroviral replication. Annu Rev Genet 42:143–163. doi: 10.1146/annurev.genet.42.110807.091704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mascarenhas AP, Musier-Forsyth K. 2009. The capsid protein of human immunodeficiency virus: interactions of HIV-1 capsid with host protein factors. FEBS J 276:6118–6127. doi: 10.1111/j.1742-4658.2009.07315.x. [DOI] [PubMed] [Google Scholar]
- 24.Owens CM, Song B, Perron MJ, Yang PC, Stremlau M, Sodroski J. 2004. Binding and susceptibility to postentry restriction factors in monkey cells are specified by distinct regions of the human immunodeficiency virus type 1 capsid. J Virol 78:5423–5437. doi: 10.1128/JVI.78.10.5423-5437.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ganser-Pornillos BK, von Schwedler UK, Stray KM, Aiken C, Sundquist WI. 2004. Assembly properties of the human immunodeficiency virus type 1 CA protein. J Virol 78:2545–2552. doi: 10.1128/JVI.78.5.2545-2552.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ohkura S, Goldstone DC, Yap MW, Holden-Dye K, Taylor IA, Stoye JP. 2011. Novel escape mutants suggest an extensive TRIM5α binding site spanning the entire outer surface of the murine leukemia virus capsid protein. PLoS Pathog 7:e1002011. doi: 10.1371/journal.ppat.1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee K, Ambrose Z, Martin TD, Oztop I, Mulky A, Julias JG, Vandegraaff N, Baumann JG, Wang R, Yuen W, Takemura T, Shelton K, Taniuchi I, Li Y, Sodroski J, Littman DR, Coffin JM, Hughes SH, Unutmaz D, Engelman A, KewalRamani VN. 2010. Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe 7:221–233. doi: 10.1016/j.chom.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maillard PV, Zoete V, Michielin O, Trono D. 2011. Homology-based identification of capsid determinants that protect HIV1 from human TRIM5α restriction. J Biol Chem 286:8128–8140. doi: 10.1074/jbc.M110.187609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Price AJ, Fletcher AJ, Schaller T, Elliott T, Lee K, KewalRamani VN, Chin JW, Towers GJ, James LC. 2012. CPSF6 defines a conserved capsid interface that modulates HIV-1 replication. PLoS Pathog 8:e1002896. doi: 10.1371/journal.ppat.1002896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stevens A, Bock M, Ellis S, LeTissier P, Bishop KN, Yap MW, Taylor W, Stoye JP. 2004. Retroviral capsid determinants of Fv1 NB and NR tropism. J Virol 78:9592–9598. doi: 10.1128/JVI.78.18.9592-9598.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schaller T, Ocwieja KE, Rasaiyaah J, Price AJ, Brady TL, Roth SL, Hué S, Fletcher AJ, Lee K, KewalRamani VN, Noursadeghi M, Jenner RG, James LC, Bushman FD, Towers GJ. 2011. HIV-1 capsid-cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathog 7:e1002439. doi: 10.1371/journal.ppat.1002439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ambrose Z, Lee K, Ndjomou J, Xu H, Oztop I, Matous J, Takemura T, Unutmaz D, Engelman A, Hughes SH, KewalRamani VN. 2012. Human immunodeficiency virus type 1 capsid mutation N74D alters cyclophilin A dependence and impairs macrophage infection. J Virol 86:4708–4714. doi: 10.1128/JVI.05887-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hatziioannou T, Cowan S, Bieniasz PD. 2004. Capsid-dependent and -independent postentry restriction of primate lentivirus tropism in rodent cells. J Virol 78:1006–1011. doi: 10.1128/JVI.78.2.1006-1011.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi J, Aiken C. 2006. Saturation of TRIM5 alpha-mediated restriction of HIV-1 infection depends on the stability of the incoming viral capsid. Virology 350:493–500. doi: 10.1016/j.virol.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 35.Perron MJ, Stremlau M, Lee M, Javanbakht H, Song B, Sodroski J. 2007. The human TRIM5alpha restriction factor mediates accelerated uncoating of the N-tropic murine leukemia virus capsid. J Virol 81:2138–2148. doi: 10.1128/JVI.02318-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee K, Mulky A, Yuen W, Martin TD, Meyerson NR, Choi L, Yu H, Sawyer SL, Kewalramani VN. 2012. HIV-1 capsid-targeting domain of cleavage and polyadenylation specificity factor 6. J Virol 86:3851–3860. doi: 10.1128/JVI.06607-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hori T, Takeuchi H, Saito H, Sakuma R, Inagaki Y, Yamaoka S. 2013. A carboxy-terminally truncated human CPSF6 lacking residues encoded by exon 6 inhibits HIV-1 cDNA synthesis and promotes capsid disassembly. J Virol 87:7726–7736. doi: 10.1128/JVI.00124-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.De Iaco A, Santoni F, Vannier A, Guipponi M, Antonarakis S, Luban J. 2013. TNPO3 protects HIV-1 replication from CPSF6-mediated capsid stabilization in the host cell cytoplasm. Retrovirology 10:20. doi: 10.1186/1742-4690-10-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fricke T, Valle-Casuso JC, White TE, Brandariz-Nuñez A, Bosche WJ, Reszka N, Gorelick R, Diaz-Griffero F. 2013. The ability of TNPO3-depleted cells to inhibit HIV-1 infection requires CPSF6. Retrovirology 10:46. doi: 10.1186/1742-4690-10-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Henning MS, Dubose BN, Burse MJ, Aiken C, Yamashita M. 2014. In vivo functions of CPSF6 for HIV-1 as revealed by HIV-1 capsid evolution in HLA-B27-positive subjects. PLoS Pathog 10:e1003868. doi: 10.1371/journal.ppat.1003868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wright ER, Schooler JB, Ding HJ, Kieffer C, Fillmore C, Sundquist WI, Jensen GJ. 2007. Electron cryotomography of immature HIV-1 virions reveals the structure of the CA and SP1 Gag shells. EMBO J 26:2218–2226. doi: 10.1038/sj.emboj.7601664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Marco A, Davey NE, Ulbrich P, Phillips JM, Lux V, Riches JD, Fuzik T, Ruml T, Kräusslich H-G, Vogt VM, Briggs JaG. 2010. Conserved and variable features of Gag structure and arrangement in immature retrovirus particles. J Virol 84:11729–11736. doi: 10.1128/JVI.01423-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Briggs JA, Riches JD, Glass B, Bartonova V, Zanetti G, Krausslich HG. 2009. Structure and assembly of immature HIV. Proc Natl Acad Sci U S A 106:11090–11095. doi: 10.1073/pnas.0903535106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bharat TAM, Davey NE, Ulbrich P, Riches JD, de Marco A, Rumlova M, Sachse C, Ruml T, Briggs JAG. 2012. Structure of the immature retroviral capsid at 8 Å resolution by cryo-electron microscopy. Nature 487:385–389. doi: 10.1038/nature11169. [DOI] [PubMed] [Google Scholar]
- 45.Schur FKM, Hagen WJH, Rumlová M, Ruml T, Müller B, Kräusslich H-G, Briggs JA, G. 2015. Structure of the immature HIV-1 capsid in intact virus particles at 8.8 Å resolution. Nature 517:505–508. doi: 10.1038/nature13838. [DOI] [PubMed] [Google Scholar]
- 46.Goh BC, Perilla JR, England MR, Heyrana KJ, Craven RC, Schulten K. 2015. Atomic modeling of an immature retroviral lattice using molecular dynamics and mutagenesis. Structure 23:1414–1425. doi: 10.1016/j.str.2015.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schur FKM, Dick RA, Hagen WJH, Vogt VM, Briggs JAG. 2015. The structure of immature-like Rous sarcoma virus Gag particles reveals a structural role for the p10 domain in assembly. J Virol 89:10294–10302. doi: 10.1128/JVI.01502-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ganser BK, Li S, Klishko VY, Finch JT, Sundquist WI. 1999. Assembly and analysis of conical models for the HIV-1 core. Science 283:80–83. doi: 10.1126/science.283.5398.80. [DOI] [PubMed] [Google Scholar]
- 49.Pornillos O, Ganser-Pornillos BK, Yeager M. 2011. Atomic-level modelling of the HIV capsid. Nature 469:424–427. doi: 10.1038/nature09640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ganser-Pornillos BK, Cheng A, Yeager M. 2007. Structure of full-length HIV-1 CA: a model for the mature capsid lattice. Cell 131:70–79. doi: 10.1016/j.cell.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 51.Zhao G, Perilla JR, Yufenyuy EL, Meng X, Chen B, Ning J, Ahn J, Gronenborn AM, Schulten K, Aiken C, Zhang P. 2013. Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature 497:643–646. doi: 10.1038/nature12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Meng X, Zhao G, Yufenyuy E, Ke D, Ning J, DeLucia M, Ahn J, Gronenborn AM, Aiken C, Zhang P. 2012. Protease cleavage leads to formation of mature trimer interface in HIV-1 capsid. PLoS Pathog 8:e1002886. doi: 10.1371/journal.ppat.1002886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Craven RC, Leure-duPree AE, Weldon RA, Wills JW, Weldon RA Jr. 1995. Genetic analysis of the major homology region of the Rous sarcoma virus Gag protein. J Virol 69:4213–4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cairns TM, Craven RC. 2001. Viral DNA synthesis defects in assembly-competent Rous sarcoma virus CA mutants. J Virol 75:242–250. doi: 10.1128/JVI.75.1.242-250.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mammano F, Ohagen A, Hoglund S, Gottlinger HG. 1994. Role of the major homology region of human immunodeficiency virus type 1 in virion morphogenesis. J Virol 68:4927–4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Strambio-de-Castillia C, Hunter E. 1992. Mutational analysis of the major homology region of Mason-Pfizer monkey virus by use of saturation mutagenesis. J Virol 66:7021–7032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Accola MA, Strack B, Gottlinger HG. 2000. Efficient particle production by minimal Gag constructs which retain the carboxy-terminal domain of human immunodeficiency virus type 1 capsid-p2 and a late assembly domain. J Virol 74:5395–5402. doi: 10.1128/JVI.74.12.5395-5402.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Keller PW, Johnson MC, Vogt VM. 2008. Mutations in the spacer peptide and adjoining sequences in Rous sarcoma virus Gag lead to tubular budding. J Virol 82:6788–6797. doi: 10.1128/JVI.00213-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Krishna NK, Campbell S, Vogt VM, Wills JW. 1998. Genetic determinants of Rous sarcoma virus particle size. J Virol 72:564–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Auerbach MR, Shu C, Kaplan A, Singh IR. 2003. Functional characterization of a portion of the Moloney murine leukemia virus gag gene by genetic footprinting. Proc Natl Acad Sci U S A 100:11678–11683. doi: 10.1073/pnas.2034020100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Auerbach MR, Brown KR, Singh IR. 2007. Mutational analysis of the N-terminal domain of Moloney murine leukemia virus capsid protein. J Virol 81:12337–12347. doi: 10.1128/JVI.01286-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Scholz I, Arvidson B, Huseby D, Barklis E. 2005. Virus particle core defects caused by mutations in the human immunodeficiency virus capsid N-terminal domain. J Virol 79:1470–1479. doi: 10.1128/JVI.79.3.1470-1479.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Spidel JL, Wilson CB, Craven RC, Wills JW. 2007. Genetic studies of the beta-hairpin loop of Rous sarcoma virus capsid protein. J Virol 81:1288–1296. doi: 10.1128/JVI.01551-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Callahan EM, Wills JW. 2003. Link between genome packaging and rate of budding for Rous sarcoma virus. J Virol 77:9388–9398. doi: 10.1128/JVI.77.17.9388-9398.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ma YM, Vogt VM. 2002. Rous sarcoma virus Gag protein-oligonucleotide interaction suggests a critical role for protein dimer formation in assembly. J Virol 76:5452–5462. doi: 10.1128/JVI.76.11.5452-5462.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ma YM, Vogt VM. 2004. Nucleic acid binding-induced Gag dimerization in the assembly of Rous sarcoma virus particles in vitro. J Virol 78:52–60. doi: 10.1128/JVI.78.1.52-60.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lokhandwala PM, Nguyen TL, Bowzard JB, Craven RC. 2008. Cooperative role of the MHR and the CA dimerization helix in the maturation of the functional retrovirus capsid. Virology 376:191–198. doi: 10.1016/j.virol.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang GP, Bushman FD. 2006. A statistical method for comparing viral growth curves. J Virol Methods 135:118–123. doi: 10.1016/j.jviromet.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 69.Bowzard JB, Wills JW, Craven RC. 2001. Second-site suppressors of Rous sarcoma virus CA mutations: evidence for interdomain interactions. J Virol 75:6850–6856. doi: 10.1128/JVI.75.15.6850-6856.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Garnier L, Ratner L, Rovinski B, Cao SX, Wills JW. 1998. Particle size determinants in the human immunodeficiency virus type 1 Gag protein. J Virol 72:4667–4677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Garnier L, Parent LJ, Rovinski B, Cao SX, Wills JW. 1999. Identification of retroviral late domains as determinants of particle size. J Virol 73:2309–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Purdy JG, Flanagan JM, Ropson IJ, Rennoll-Bankert KE, Craven RC. 2008. Critical role of conserved hydrophobic residues within the major homology region in mature retroviral capsid assembly. J Virol 82:5951–5961. doi: 10.1128/JVI.00214-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Craven RC, Leure-duPree AE, Erdie CR, Wilson CB, Wills JW. 1993. Necessity of the spacer peptide between CA and NC in the Rous sarcoma virus gag protein. J Virol 67:6246–6252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Humphrey W, Dalke A, Schulten K. 1996. VMD: Visual Molecular Dynamics. J Mol Graph 14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 75.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K. 2005. Scalable molecular dynamics with NAMD. J Comput Chem 26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dolinsky TJ, Czodrowski P, Li H, Nielsen JE, Jensen JH, Klebe G, Baker NA. 2007. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res 35:W522–W525. doi: 10.1093/nar/gkm276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sethi A, Eargle J, Black AA, Luthey-Schulten Z. 2009. Dynamical networks in tRNA:protein complexes. Proc Natl Acad Sci U S A 106:6620–6625. doi: 10.1073/pnas.0810961106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Glykos NM. 2006. Software news and updates carma: a molecular dynamics analysis program. J Comput Chem 27:1765–1768. doi: 10.1002/jcc.20482. [DOI] [PubMed] [Google Scholar]
- 79.Eargle J, Luthey-Schulten Z. 2012. NetworkView: 3D display and analysis of protein-RNA interaction networks. Bioinformatics 28:3000–3001. doi: 10.1093/bioinformatics/bts546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Van Wart AT, Durrant J, Votapka L, Amaro RE. 2014. Weighted implementation of suboptimal paths (WISP): an optimized algorithm and tool for dynamical network analysis. J Chem Theory Comput 10:511–517. doi: 10.1021/ct4008603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Weldon RA Jr, Wills JW. 1993. Characterization of a small (25-kilodalton) derivative of the Rous sarcoma virus Gag protein competent for particle release. J Virol 67:5550–5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Briggs JA, Johnson MC, Simon MN, Fuller SD, Vogt VM. 2006. Cryo-electron microscopy reveals conserved and divergent features of gag packing in immature particles of Rous sarcoma virus and human immunodeficiency virus. J Mol Biol 355:157–168. doi: 10.1016/j.jmb.2005.10.025. [DOI] [PubMed] [Google Scholar]
- 83.Nandhagopal N, Simpson AA, Johnson MC, Francisco AB, Schatz GW, Rossmann MG, Vogt VM. 2004. Dimeric rous sarcoma virus capsid protein structure relevant to immature Gag assembly. J Mol Biol 335:275–282. doi: 10.1016/j.jmb.2003.10.034. [DOI] [PubMed] [Google Scholar]
- 84.Dalessio PM, Craven RC, Lokhandwala PM, Ropson IJ. 2013. Lethal mutations in the major homology region and their suppressors act by modulating the dimerization of the rous sarcoma virus capsid protein C-terminal domain. Proteins 81:316–325. doi: 10.1002/prot.24188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Butan C, Lokhandwala PM, Purdy JG, Cardone G, Craven RC, Steven AC. 2010. Suppression of a morphogenic mutant in Rous sarcoma virus capsid protein by a second-site mutation: a cryoelectron tomography study. J Virol 84:6377–6386. doi: 10.1128/JVI.00207-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rihn SJ, Wilson SJ, Loman NJ, Alim M, Bakker SE, Bhella D, Gifford RJ, Rixon FJ, Bieniasz PD. 2013. Extreme genetic fragility of the HIV-1 capsid. PLoS Pathog 9:e1003461. doi: 10.1371/journal.ppat.1003461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Campos-Olivas R, Newman JL, Summers MF. 2000. Solution structure and dynamics of the Rous sarcoma virus capsid protein and comparison with capsid proteins of other retroviruses. J Mol Biol 296:633–649. doi: 10.1006/jmbi.1999.3475. [DOI] [PubMed] [Google Scholar]
- 88.Lu M, Hou G, Zhang H, Suiter CL, Ahn J, Byeon I-JL, Perilla JR, Langmead CJ, Hung I, Gor'kov PL, Gan Z, Brey W, Aiken C, Zhang P, Schulten K, Gronenborn AM, Polenova T. 2015. Dynamic allostery governs cyclophilin A–HIV capsid interplay. Proc Natl Acad Sci U S A 112:14617–14622. doi: 10.1073/pnas.1516920112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.