

ABSTRACT
Flavivirus nonstructural protein 2B (NS2B) is a transmembrane protein that functions as a cofactor for viral NS3 protease. The cytoplasmic region (amino acids 51 to 95) alone of NS2B is sufficient for NS3 protease activity, whereas the role of transmembrane domains (TMDs) remains obscure. Here, we demonstrate for the first time that flavivirus NS2B plays a critical role in virion assembly. Using Japanese encephalitis virus (JEV) as a model, we performed a systematic mutagenesis at the flavivirus conserved residues within the TMDs of NS2B. As expected, some mutations severely attenuated (L38A and R101A) or completely destroyed (G12L) viral RNA synthesis. Interestingly, two mutations (G37L and P112A) reduced viral RNA synthesis and blocked virion assembly. None of the mutations affected NS2B-NS3 protease activity. Because mutations G37L and P112A affected virion assembly, we selected revertant viruses for these two mutants. For mutant G37L, replacement with G37F, G37H, G37T, or G37S restored virion assembly. For mutant P112A, insertion of K at position K127 (leading to K127KK) of NS2B rescued virion assembly. A biomolecular fluorescent complementation (BiFC) analysis demonstrated that (i) mutation P112A selectively weakened NS2B-NS2A interaction and (ii) the adaptive mutation K127KK restored NS2B-NS2A interaction. Collectively, our results demonstrate that, in addition to being a cofactor for NS3 protease, flavivirus NS2B also functions in viral RNA replication, as well as virion assembly.
IMPORTANCE Many flaviviruses are important human pathogens. Understanding the molecular mechanisms of the viral infection cycle is essential for vaccine and antiviral development. In this study, we demonstrate that the TMDs of JEV NS2B participate in both viral RNA replication and virion assembly. A viral genetic study and a BiFC assay demonstrated that interaction between NS2B and NS2A may participate in modulating viral assembly in the flavivirus life cycle. Compensatory-mutation analysis confirmed that there was a correlation between viral assembly and NS2B-NS2A interaction. TMDs of NS2B may serve as novel antiviral targets to prevent flavivirus infection, and the structure determination of NS2B will help us to understand the functional mechanism of NS2B in viral RNA replication and assembly. The results have uncovered a new function of flavivirus NS2B in virion assembly, possibly through interaction with the NS2A protein.
INTRODUCTION
Flaviviruses are a large group of small, enveloped viruses transmitted by arthropods. Many flaviviruses are important human pathogens, such as Japanese encephalitis virus (JEV), West Nile virus (WNV), tick-borne encephalitis virus (TBEV), dengue virus (DENV), and yellow fever virus (YFV). The flavivirus genome is a positive-sense RNA about 11 kb in length. The single open reading frame encodes a long polyprotein that is co- and posttranslationally processed by cellular proteases and viral protease into three structural proteins (capsid [C], premembrane/membrane [prM/M], and envelope [E]) and seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) (1). The structural proteins form the virus particle, while the nonstructural proteins function in viral RNA replication, virion assembly, and evasion of the host antiviral immune responses (2–5). Among them, NS3 is a multifunctional protein with an N-terminal protease domain and a C-terminal RNA helicase/NTPase domain. The NS3 protease activity requires NS2B as a cofactor (NS2B-NS3pro) and is responsible for cleaving the C terminus of mature capsid protein, as well as the junctions of NS2A/NS2B, NS2B/NS3, NS3/NS4A and NS4B/NS5 (6).
NS2B is a small integral membrane protein (approximately 130 amino acids) with a molecular mass of 14 kDa. It contains a conserved central hydrophilic region (amino acids 51 to 95 in JEV) and three hydrophobic regions that are predicted to be transmembrane domains (TMDs) (7, 8). It has been demonstrated that the central hydrophilic region of NS2B is necessary and sufficient for the activation of NS3 protease, and mutations in this region could affect protease activities and NS3 stability, resulting in defects of viral replication (1, 7, 9–13). Its hydrophobic TMDs are generally considered to help the NS2B-NS3pro complex anchor into the host endoplasmic reticulum (ER) membranes for efficient activation of the NS3 protease (7, 11). Although the TMDs of NS2B are not essential for NS3 protease activity, there is some evidence that the TMDs of NS2B may have additional functions during viral replication (7, 14). Chambers et al. showed that mutations in the predicted TMDs of YFV NS2B had subtle effects on proteolytic processing but decreased viral replication (7). Consistently, we also found that a Met-to-Thr mutation at amino acid position 102 of JEV NS2B (NS2B-M102T) located at the predicted third TMD led to proteolysis-independent impairment of JEV replication (14). It is currently unclear how the NS2B TMDs regulate flaviviral replication.
A recent nuclear magnetic resonance (NMR) study revealed the membrane topology of DENV4 NS2B (15). The hydrophilic cofactor region forms a β-strand structure, while the TMDs contains four short helical segments. Notably, each of the four TMD helices contains a small-XXX-small motif (“small” represents amino acids with small side chains [Gly, Ala, and Ser], and “X” represents any amino acid) that is important for TMD-TMD interactions. Such structural characteristics of NS2B might confer more freedom to induce conformational changes in membranes under different conditions, facilitating interaction with other viral membrane proteins through its TMDs (15). Indeed, using fluorescent resonance energy transfer (FRET)- and biomolecular fluorescent complementation (BiFC)-based imaging methods, Yu et al. showed that WNV NS2B could interact with itself, as well as most other NS proteins, including the other three membrane proteins, NS2A, NS4A, and NS4B (16). These results, together with its known function as an NS3 protease cofactor, suggest that NS2B may play a critical role in bringing the four membrane NS proteins together in the replication complex and in bridging them with non-membrane-associated NS3 and NS5 proteins (16). Although the details of these interactions are still obscure, they strongly suggest that the NS2B protein might perform some functions other than as a protease cofactor through its TMDs.
In this study, mutagenesis analyses of NS2B in the context of JEV reverse-genetic systems were performed. The results showed that all three NS2B TMDs are essential for viral replication, but none of the tested TMD mutations affected trans- or cis-protease activities of NS2B-NS3. Interestingly, NS2B mutations G37L and P112A impaired virion assembly. Revertant and protein-protein interaction analyses showed that the defect in virus assembly caused by the P112A mutation correlated with a weakened interaction between NS2B and NS2A. Remarkably, an adaptive mutation, K127KK, in NS2B could compensate for the defects in virion assembly, as well as NS2A-NS2B interaction. These results indicate that NS2B participates in virion assembly through interaction with NS2A, a protein known to be important for flavivirus assembly (17–20). Overall, our data demonstrate, for the first time, that flavivirus NS2B participates in virion assembly.
MATERIALS AND METHODS
Cell culture and viruses.
Baby hamster kidney (BHK-21) cells and human embryo kidney 293T cells were grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS), 100 units penicillin ml−1, and 100 μg streptomycin ml−1 in 5% CO2 at 37°C. Wild-type (WT) and mutant viruses were generated from the infectious cDNA clone of JEV by electroporation of BHK-21 cells with in vitro-transcribed RNA. The supernatants of transfected cells were harvested at different time points posttransfection and frozen at −80°C.
Membrane topology prediction.
The membrane topology of JEV NS2B was predicted using various Web servers. These servers included HMMTOP, SOSUI, Split, TOPCONS, PSIPRED, DAS, TMHMM2, and Polyphobius.
Plasmid construction.
The infectious clone of a Renilla luciferase (Rluc)-JEV reporter virus, pACYC-Rluc-JEV (whose construction will be reported elsewhere), was used as the backbone to introduce NS2B mutations. Briefly, the NS2B fragments containing different mutations were produced by overlap PCR and then engineered into the pACYC-Rluc-JEV infectious clone at BspEI and BamHI restriction sites. The full-length pACYC-JEV infectious clone and the Renilla luciferase replicon (JEV-Rluc-Rep) carrying the corresponding NS2B mutations were constructed by replacement of the BspEI/BamHI fragment from the pACYC-Rluc-JEV infectious clone at the same sites. For the construction of the NS2B-NS3pro-HA expression plasmids, a fragment of NS2B-NS3 (amino acids 1 to 180) tagged with a hemagglutinin (HA) sequence at its C terminus was amplified using the infectious clone containing the corresponding NS2B mutations as a template and cloned into the pCAGGS vector at the XhoI and ClaI restriction sites. For the construction of BiFC vectors, the plasmid encoding the Venus protein (a variant of yellow fluorescent protein) (21) constructed by our laboratory was used as the backbone. Two separate fragments representing the N terminus (VenusN; amino acids 1 to 173) and the C terminus (VenusC; amino acids 174 to 239) of the full-length protein were amplified by PCR with a GGGSGGGS linker sequence at the 5′ end and inserted into pEGFPN1 at AgeI and NotI sites to replace the original enhanced green fluorescent protein (eGFP) gene of the vector, generating VenusN and VenusC expression vectors, respectively. NS2B fragments with different mutations and other individual NS proteins were amplified by PCR using the corresponding infectious clone as a template and inserted into the VenusN and VenusC vectors at the XhoI/AgeI sites, respectively. The NS2B TMD3 (amino acid positions 98 to 131) sequences were amplified and cloned in frame with eGFP into pEGFPN1 at EcoRI/AgeI restriction sites to generate the NS2B TMD3-eGFP fused-protein expression plasmids.
RNA transcription and transfection.
The plasmids for the infectious clone and replicon were linearized by XhoI, followed by purification by phenol-chloroform extraction. RNAs were then transcribed from the linearized plasmids using a T7 mMESSAGE mMACHINE kit (Ambion) according to the manufacturer's protocols. For transfection, 5 μg of in vitro-transcribed RNA was electroporated into 8 × 106 BHK-21 cells following a protocol described previously (14).
Luciferase assay.
Various numbers of cells transfected with WT and mutant Rluc-JEV RNAs were seeded in 12-well plates. At 2, 12, 24, 48, 72, and 96 h posttransfection (hpt), the supernatants and cells were collected. For Rluc-JEV infection, 100 μl of supernatants harvested at 48 and 72 hpt was used to infect naive BHK-21 cells in 12-well plates that were seeded at a density of 1 × 105 cells per well 1 day prior to infection. After 2 h of incubation, the cells were washed three times with phosphate-buffered saline (PBS) and maintained in Dulbecco's modified Eagle's medium (DMEM) with 2% FBS. Forty-eight hours postinfection (hpi), the cells were lysed and stored at −80°C for the subsequent luciferase assay. Luciferase activity was measured in a microplate reader (Varioskan Flash; Thermo Fisher) by mixing 20 μl lysates with 50 μl substrate (Promega). The JEV-Rluc-replicon transient-transfection assay was performed to detect the replication efficiencies of the mutants using a protocol described previously (14).
Immunofluorescence assay (IFA) and plaque assay.
The cells transfected with the JEV genomic RNA were washed once with PBS and fixed with cold (−20°C) 5% acetone in methanol for 10 min at room temperature. The fixed cells were then washed with PBS three times and incubated with anti-St. Louis encephalitis (SLEV) envelope protein mouse monoclonal antibody (MAb) (Chemicon; diluted 1:250) for 1 h. Following three washes with PBS, the cells were incubated with Texas Red-conjugated goat anti-mouse antibody (ProteinTech; diluted 1:125) for 1 h at room temperature. After washing with PBS three times, the images were acquired and analyzed under a fluorescence microscope. The virus titer was determined by single-layer plaque assay, as described previously (14). Briefly, a series of 1:10-diluted viruses were seeded onto a 24-well plate containing confluent BHK-21 cells. After 4 days of incubation at 37°C with 5% CO2, the cells were fixed and stained with 3.7% formaldehyde containing 1% crystal violet solution. The plaque morphology of the mutant viruses was determined by double-layer plaque assay as described in a previous study (14).
Real-time RT-PCR.
After electroporation, approximately 4 × 105 cells transfected with WT and mutant RNAs were seeded in a 6-well plate. Following incubation at 37°C with 5% CO2 for 2 h, the transfected cells were washed with PBS three times to remove the untransfected RNAs in the culture fluids and then maintained in DMEM containing 2% FBS. At 24, 48, and 72 hpt, the supernatants and cells were harvested. After centrifugation at 500 × g for 5 min, the cell pellet was resuspended in 1 ml PBS. Half of the cell suspensions were centrifuged to pellet the cells, and the cell pellets were added with 500 μl TRIzol and frozen at −80°C to extract the intracellular total RNAs. The other half of the cell suspensions were centrifuged at 500 × g for 5 min, followed by resuspension with 300 μl DMEM containing 2% FBS. After three rounds of freezing and thawing, cellular debris was removed by centrifugation at 17,000 × g for 5 min at 4°C, and the supernatants were tested for intracellular infectious-virus production using a plaque assay. Viral RNAs in the supernatants of the transfected cells and the intracellular total RNAs were extracted using a QIAamp viral-RNA minikit (Qiagen) according to the manufacturer's protocol. The viral-RNA levels were measured using a one-step real-time reverse transcription (RT)-PCR with SYBR green and the primer pair 5′-TACAACATGATGGGAAAGCGAGAGAAAAA-3′ and 5′-GTGTCCCAGCCGGCGGTGTCATCAGC-3′. The number of genomic RNA copies was determined with a standard curve of the in vitro-transcribed RNA derived from the infectious clone.
DNA transfection.
For the proteolytic processing and BiFC assays, a standard calcium phosphate precipitation protocol was used for DNA transfection. Briefly, 293T cells were seeded in 35-mm-diameter dishes or a chamber slide (Nalge Nunc) and grown overnight to 70 to 80% confluence. A mixture of equal amounts of the two plasmids (NS2B-NS3pro-HA expression plasmid plus NS4A-NS4B-FLAG polyprotein expression plasmid, or the VenusN-fused NS2B plus VenusC-fused viral-protein expression plasmids) was transfected into the cells. Cells expressing NS2B-NS3pro and NS4A-NS4B proteins were lysed with 200 μl lysis buffer (20 mM Tris, pH 7.5, 100 mM NaCl, 0.5% n-dodecyl-β-d-maltopyranoside (DDM), and EDTA-free protease inhibitor cocktail) on ice for 20 min. The lysates were clarified by centrifugation at 17,000 × g at 4°C for 30 min and subjected to Western blot assay. For detection of interactions of NS2B with other NS proteins, the fluorescence in transfected cells was visualized at 12 hpt with a Zeiss fluorescence microscope under an oil immersion 60× objective lens (numerical aperture [NA], 1.4) with an excitation wavelength of 495/10 nm (center wavelength [CWL]/bandwidth [BW]) and an emission wavelength of 542/27 nm (CWL/BW). For the NS2B subcellular localization assay, the plasmids encoding NS2B-eGFP fused proteins were transfected into 293T cells using Lipofectamine 2000 (Invitrogen). After 24 hpt, the cells were fixed with cold (−20°C) 5% acetone in methanol for 10 min at room temperature and then subjected to IFA using the anti-eGFP rabbit antibody as described previously (22).
SDS-PAGE and Western blotting.
Cellular lysates were heated at 75°C for 10 min and then separated on 15% SDS-PAGE gels. Proteins were transferred onto a polyvinylidene difluoride (PVDF) membrane using a Trans-Blot Turbo system (Bio-Rad), followed by incubation with 5% skim milk (Bio-Rad) in TBST buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, and 0.1% Tween 20) at room temperature for 1 h. The blots were then subjected to sequential incubation with primary (anti-HA/anti-FLAG mouse monoclonal antibody at 1:2,000 dilution) and secondary (horseradish peroxidase [HRP]-conjugated goat anti-mouse IgG at 1:3,000 dilution) antibodies. Following three washes with TBST buffer, the signals were detected with a chemiluminescence system (ChemiDoc; Bio-Rad).
Statistical analysis.
The Student t test was used to determine if there were significant differences (P < 0.05) for all the tested reporter viruses. The statistical analyses were performed in IBM SPSS Statistics v18.0.
RESULTS
Functional analysis of the hydrophobic regions of JEV NS2B protein using Rluc-JEV.
The aim of this study was to identify new functions of the transmembrane regions of flavivirus NS2B protein. We initially used several bioinformatics algorithms to analyze the membrane topology of JEV NS2B protein. Most programs (SOSUI, Split, TOPCONS, DAS, TMHMM2, and Polyphobius) consistently predicted three TMDs with minor variations in the length and position of each TMD (Fig. 1A). After combining the predicted topology with a recent NMR model of DENV4 NS2B topology (15), we proposed a topology model of JEV NS2B (Fig. 1B). Amino acid sequence alignment of NS2B proteins of different flaviviruses revealed 13 conserved residues located at different TMDs (Fig. 1C): E6, A10, V11, G12, and L21 in TMD1; G37, L38, M39, V44, and G47 in TMD2; and R101, P112, and I115 in TMD3.
FIG 1.

Prediction of the membrane binding domain of JEV NS2B protein and alignment analysis of flavivirus NS2B sequences. (A) Schematic representation of the membrane topology predictions of JEV NS2B by various Web servers. The gray boxes represent the predicted transmembrane segments. The positions of the first and the last amino acid residues in the predicted transmembrane domains are indicated. (B) Model of JEV NS2B transmembrane domains with selected mutations indicated. (C) Multiple-sequence alignment of NS2B proteins from different flaviviruses using ClustalW software (http://embnet.vital-it.ch/software/ClustalW.html). The gray shading indicates the residues selected for mutagenesis in this study. The ellipses represent the hydrophilic regions for which the sequence is not presented. Asterisks indicate positions that have a single fully conserved residue; colons indicate conservation between groups with strongly similar physicochemical properties; dots indicate conservation between groups with weakly similar physicochemical properties.
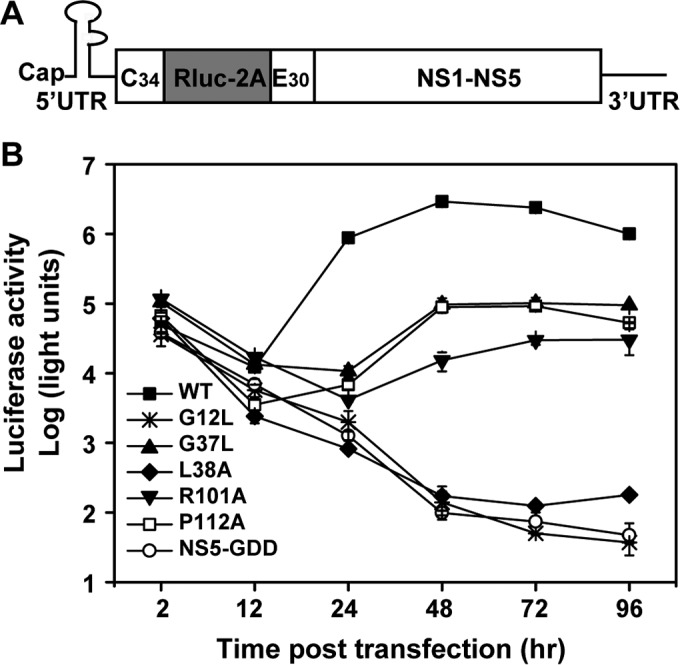
We used a luciferase reporter virus (Rluc-JEV) to probe the function of the flavivirus conserved TMD residues (Fig. 2A). The construction and characterization of the Rluc-JEV reporter virus will be reported elsewhere. In the context of Rluc-JEV, each of the conserved non-Ala and non-Gly residues was replaced with Ala; the small Gly and Ala residues were replaced with large hydrophobic Leu residues. Equal amounts of wild-type and mutant Rluc-JEV RNAs were transfected into BHK-21 cells, and viral replication was monitored by measuring luciferase activities in the transfected cells at different times posttransfection. Similar levels of luciferase activities were detected at 2 and 4 hpt for all transfected RNAs (data not shown), suggesting comparable transfection efficiencies for the RNAs. However, different levels of luciferase signals were detected for the WT and mutant RNAs at 24, 48, and 72 hpt (Fig. 2B). At 48 hpt, mutants E6A, A10L, V11A, L21A, G37L, M39A, V44A, G47L, and I115A showed slight to moderate defects in viral replication, with less than 100-fold decreases in luciferase signals compared with the WT; the L38A, R101A, and P112A mutants exhibited severe defects in replication, with 100- to 1,200-fold decreases in luciferase activity; mutant G12L was lethal, with a luciferase profile similar to that of a negative-control NS5-GDD Rluc-JEV (containing a deletion of a GDD active site of NS5 polymerase). These results demonstrate that mutations at the TMDs could attenuate viral replication.
FIG 2.

Characterization of viral replication and infectivity of NS2B mutants using the Rluc-JEV reporter virus. (A) Schematic diagram of the Rluc-JEV reporter virus. The Rluc reporter gene was inserted between the 5′ untranslated region (UTR) and the capsid gene of the JEV genome. The first 34 amino acids of the capsid coding sequences were maintained in frame preceding the Rluc gene. Foot-and-mouth disease virus (FMDV) 2A cleavage peptides were inserted between the Rluc and capsid genes of the viral genome. (B) Rluc-JEV reporter virus was used to detect the replication efficiencies of WT and NS2B mutant viruses. Equal amounts of WT and mutant Rluc-JEV RNAs were transfected into BHK-21 cells, and luciferase activity was measured at the indicated time points posttransfection. (C) The production of infectious virus particles was detected by infecting naive BHK-21 cells with the supernatants collected at 48 and 72 hpt, and the luciferase activity was detected at 48 hpi. (D) Summary of the phenotypic effects of NS2B mutants. Representative data from three independent experiments are presented. Each experiment was performed in triplicate, and the error bars represent standard deviations.
To determine the effect of each mutation on infectious-virus production, we inoculated naive BHK-21 cells with the supernatants harvested from the transfected cells at 48 or 72 hpt. At 48 h postinfection, the cells were lysed to measure luciferase activities (Fig. 2C). The luciferase activities expressed in the infected cells were used to indicate virus production from the transfected cells. Consistent with the observation in cells transfected with G12L RNA, the luciferase signals were barely detectable in G12L mutant-infected cells. For the mutants that efficiently replicated in the transfected cells (E6A, A10L, V11A, L21A, M39A, V44A, G47L, and I115A), the corresponding infected cells produced similar or slightly lower luciferase signals than the WT-virus-infected cells, indicating that these mutations did not prominently affect virus production. For the mutants that severely inhibited viral replication (L38A and R101A), the infected cells produced luciferase signals significantly lower than that of the WT (t test; P < 0.01), confirming that the two mutations attenuate viral RNA replication. Remarkably, although the G37L mutant RNA replicated more efficiently than L38A and R101A mutants in the transfected cells (Fig. 2B), it produced background levels of luciferase signals in the infected cells (similar to the G12L mutant and the NS5-GDD negative control), suggesting that G37L mutation blocks virus assembly and/or release. A similar phenomenon was also found in the P112A mutant.
Therefore, NS2B mutations could be classified into four groups based on the phenotypes of viral replication and infectivity of the Rluc-JEV reporter virus (Fig. 2D). Group I mutants had mild to moderate defects in viral replication (E6A, A10L, V11A, L21A, M39A, V44A, G47L, and I115A). Group II mutants severely affected viral replication (L38A and R101A). The group III mutant showed a lethal phenotype (G12L). Group IV mutants were attenuated in both viral replication and virus production (G37L and P112A). Overall, our primary data suggest that NS2B TMDs play important roles in both viral RNA replication and particle production during the viral life cycle.
Characterization of NS2B mutants using an authentic JEV infectious cDNA clone.
To validate the results derived from the luciferase reporter JEV, we engineered mutations from groups II, III, and IV into a full-length JEV infectious cDNA clone. Equal amounts of WT and mutant genome length RNAs were electroporated into BHK-21 cells. Viral replication and spreading were first assessed through an IFA using an anti-E protein monoclonal antibody (Fig. 3A). Consistent with the observation with reporter viruses, group II (L38A and R101A) mutant RNAs yielded far fewer IFA-positive cells than the WT, whereas the total number of IFA-positive cells increased throughout the 120-h time course; these results confirm that mutations L38A and R101A affect RNA replication without blocking progeny virus production. Group III (G12L) mutant RNA did not yield any IFA-positive cells, demonstrating the lethality of the mutation. Interestingly, group IV (G37L and P112A) mutant RNAs produced scattered IFA-positive cells whose number did not increase from 24 to 120 hpt, indicating that the mutant RNAs were replicative but failed to produce infectious viruses to initiate new rounds of infection.
FIG 3.

Characterizations of the phenotypes of NS2B mutants in the context of a JEV infectious clone. (A) Immunofluorescence analysis of NS2B mutants. Equal amounts of the NS2B mutant RNAs derived from the full-length infectious clone were transfected into BHK-21 cells, and IFA was performed at the indicated time points. The SLEV envelope protein antibody and Texas Red-conjugated goat anti-mouse IgG were used as primary and secondary antibodies, respectively. (B) Extracellular viral titers of the NS2B mutants at 24, 48, and 72 hpt. The supernatants harvested at the indicated time points were subjected to plaque assay to determine viral titers. (C) Intracellular viral titers of NS2B mutants at 24, 48, and 72 hpt. The transfected cells were lysed at the indicated time points by three rounds of freezing and thawing and used for plaque assays as described in Materials and Methods. (D) Amounts of extracellular RNAs of NS2B mutants. The amounts of viral RNAs in the supernatants at 24, 48, and 72 hpt were determined by quantitative RT-PCR as described in Materials and Methods. (E) Amounts of intracellular RNAs of NS2B mutants. The amounts of viral RNA in the transfected cells at 24, 48, and 72 hpt were monitored by quantitative RT-PCR as described in Materials and Methods. Representative data from three independent experiments are presented. Each experiment was performed in triplicate, and the error bars represent standard deviations.
To quantify viral RNA replication and virion production, we determined the intra- and extracellular viral titers, as well as intra- and extracellular viral-RNA levels, using plaque assay and real-time RT-PCR, respectively. As shown in Fig. 3B to E, the amounts of intra- and extracellular infectivity and viral RNA were reduced for all the mutants in comparison with those of the WT. For group II mutants (L38A and R101A), the levels of intracellular infectivity correlated well with the levels of extracellular infectivity (Fig. 3B and C). The group III G12L mutant displayed a lethal phenotype similar to that of the NS5-GDD negative control. For group IV mutations (G37L and P112A), no intra- or extracellular infectious virus was detected (Fig. 3B and C) and no extracellular viral RNA was detected (Fig. 3D); in contrast, consistent levels of intracellular viral RNAs were observed for both mutants (Fig. 3E). These data demonstrated that the group IV mutations G37L and P112A blocked virion assembly and/or release.
Characterization of NS2B mutations using a JEV replicon.
We used a Renilla luciferase reporter replicon of JEV (JEV-Rluc-replicon) to further validate the mutational effect on viral RNA replication. JEV-Rluc-replicon was constructed by replacing the structural genes with a Renilla luciferase reporter gene in frame (Fig. 4A). Mutations from groups II, III, and IV were individually engineered into JEV-Rluc-replicon. Equal amounts of WT and mutant JEV-Rluc-replicon RNAs were transfected into BHK-21 cells and assayed for luciferase activity. As shown in Fig. 4B, similar levels of luciferase signals were produced for all the replicons at 2 hpt, indicating that none of the mutations affects viral RNA translation. In contrast, after 24 hpt, different replicons produced distinct profiles of luciferase activity. The group III mutant replicon (G12L) exhibited the same luciferase profile as the NS5-GDD lethal replicon. For group II (L38A and R101A) and IV (G37L and P112A) mutants, replication efficiencies were in the following order: WT > G37L ≈ P112A ≥ R101A > L38A. Except for G37L and P112A, the differences in replication efficiency between any two mutants were significant (t test; P < 0.01). It should be noted that the G37L and P112A mutants failed to produce infectious viruses but replicated more efficiently in the replicon system than the R101A and L38A mutants, both of which could generate infectious viruses; these data exclude the possibility that the failure of viral particle production for G37L and P112A mutants was due to defects in RNA replication. Taken together, the results demonstrate that JEV NS2B is required for viral RNA replication and virion assembly processes during the viral life cycle. Additionally, we observed that the L38A mutant displays a severe defect in replication compared to R101A using the replicon (Fig. 4B), but this defect looks less severe using the JEV reporter virus (Fig. 2B). The reason for this discrepancy needs be explored in future studies.
FIG 4.
Replicon analysis of NS2B mutants. (A) Schematic diagram of a JEV replicon with a Renilla gene. JEV-Rluc-replicon was constructed by replacing the structural protein genes with the Rluc gene. (B) Comparison of the replication efficiencies of NS2B mutants and the WT using the JEV-Rluc-replicon system. Equal amounts of replicon RNAs (WT and NS2B mutants) were transfected into BHK-21 cells and assayed for luciferase activity at the indicated time points posttransfection. Representative data from three independent experiments are presented. Each experiment was performed in triplicate, and the error bars represent standard deviations.
The mutations in NS2B TMDs have no apparent effects on NS2B-NS3pro protease activity.
As NS2B acts as the cofactor of NS3 protease, we examined whether the replication and assembly defects caused by NS2B mutations were due to the impairment of NS2B-NS3pro protease activity. The NS2B mutations were individually engineered into an NS2B-NS3pro polyprotein expression plasmid that contains the full-length NS2B, the N-terminal protease domain (1 to 180 amino acids) of NS3, and a C-terminal HA tag (Fig. 5A). A construct expressing the NS2B-NS3-HA polyprotein with a D75A mutation in the protease catalytic site was prepared as a negative control. In addition, an expression plasmid encoding the NS4A-NS4B polyprotein with a C-terminal FLAG tag was constructed as a substrate for NS2B-NS3 in trans (14). After cotransfection of 293T cells with the NS2B-NS3-HA polyprotein and NS4A-NS4B-FLAG expression plasmids, Western blotting was performed to analyze the cleavage efficiencies at the NS2B/NS3 (cis) and NS4A/NS4B (trans) junctions. For cis cleavage, all the mutants and WT NS2B-NS3pro-HA polyproteins generated equivalent levels of NS3pro-HA product (Fig. 5B). As a negative control, no cleavage products were detected for the inactive NS3-D75A mutant. For trans cleavage, equal amounts of product NS4B-FLAG were observed for the WT and NS2B mutants (Fig. 5C). In agreement with a previous study (7), our data indicate that the protease activities of NS2B-NS3pro are not affected by mutations within the NS2B TMDs.
FIG 5.

Effects of NS2B mutations on the proteolytic processing activities of NS2B-NS3pro. (A) Schematic diagram of the NS2B-NS3pro and NS4A-NS4B polyprotein constructs. The white arrow indicates the cleavage site at the NS2B/NS3 junction. The gray arrow indicates the cleavage site at the NS4A/NS4B junction. (B and C) The NS2B mutations were engineered into NS2B-NS3pro constructs and coexpressed with NS4A-NS4B polyprotein in 293T cells, and the cis- and trans-cleavage activities were detected by Western blotting using anti-HA and anti-FLAG antibodies, respectively, at 24 hpt.
The P112A mutation weakens the interaction between NS2B and NS2A.
It has been found, using various imaging-based techniques, that NS2B interacts with the other three membrane proteins, NS2A, NS4A, and NS4B, in addition to the known interaction with NS3 in the context of the in vitro WNV NS protein expression system (16). We developed a Venus-based BiFC system to analyze the interactions of NS2B with other NS proteins of JEV (Fig. 6A). The full-length NS2B was fused in frame with the N-terminal 173 amino acids of the Venus protein (VenusN) through a GGGGSGGGGS linker; other NS proteins were individually fused to the C-terminal fragment (174 to 239 amino acids) of the Venus protein (VenusC) via the same peptide linker. The NS2B-VenusN plasmid was cotransfected into 293T cells with the plasmid expressing the NS-VenusC fusion protein. At 12 hpt, strong fluorescence signals were observed with the pairs NS2B-VenusN plus NS2A-, NS2B-, NS4A-, or NS4B-VenusC (Fig. 6B); the result is consistent with a previous report that WNV NS2B interacts with itself, as well as with NS2A, NS4A, and NS4B (16). No fluorescent signal was detected in the pair of NS2B-VenusN and NS1-VenusC plasmids, indicating no interaction between NS1 and NS2B. Weak fluorescence was observed when NS2B was coexpressed with NS3 or NS5. Collectively, the results suggest that NS2B interacts with transmembrane proteins (NS2A, NS2B, NS4A, and NS4B) more strongly than with the cytoplasmic proteins (NS3 and NS5).
FIG 6.

BiFC analysis of the interactions of NS2B mutants with viral NS proteins in living cells. (A) Schematic illustration of BiFC constructs and the workflow of BiFC analysis used in the study. The Venus protein was divided into two parts. The N-terminal part contained amino acids 1 to 173, and the C-terminal part consisted of residues 174 to 239 of the Venus protein. NS2B was fused with the N-terminal fragment with a GGGGSGGGGS linker, and the viral NS proteins were fused with the C-terminal segment using the same linker sequence. The NS2B-VenusN plasmid and the individual VenusC-fused constructs were cotransfected into 293T cells. (B and C) images were taken at 12 hpt with a Zeiss fluorescence microscope at an excitation wavelength of 495/10 nm (CWL/BW) and an emission wavelength of 542/27 nm (CWL/BW). (B) Visualization of the interactions of WT NS2B with viral NS proteins by BiFC. The complement fluorescence images were clearly visualized in the BiFC pairs, including NS2B-VenusN plus NS2A/NS2B/NS3/NS4A/NS4B/NS5-VenusC. (C) Visualization of the interactions of NS2B mutants (L38A in group II, G12L in group III, and G37L and P112A in group IV) with viral membrane proteins by BiFC. The NS2B mutations were individually engineered into the NS2B-VenusN construct, and the effects of the NS2B mutations on the interactions of NS2B with NS2A, NS2B, NS4A, and NS4B were detected using BiFC analysis. representative data from three independent experiments are presented.
Next, we used the BiFC assay to examine the effects of NS2B mutations on its interactions with NS2A, NS2B, NS4A, and NS4B (Fig. 6C). Mutations G12L, G37L, L38A, and P112A were individually engineered into the NS2B-VenusN and NS2B-VenusC plasmids. After cotransfecting 293T cells with NS2B-VenusN and NS2B-VenusC bearing the same mutations, no significant difference in BiFC fluorescence was observed between the homologous WT NS2B/NS2B pair and mutant pairs, suggesting that mutations do not affect NS2B self-interaction (Fig. 6C, top). When the mutant NS2B-VenusN plasmid was cotransfected with the WT NS2A-, NS4A-, or NS4B-VenusC plasmid, only mutant P112A NS2B-VenusN consistently reduced the fluorescent signal when paired with WT NS2A-VenusC (Fig. 6C, bottom). For the other member, the G37L mutation, the results were not consistent due to unknown reasons, and no specific conclusions could be reached. Overall, the results suggest that mutation P112A in NS2B TMD2 weakens the NS2A-NS2B interaction.
Revertant analysis of group IV mutant viruses.
To investigate the mechanism by which NS2B regulates viral RNA replication and assembly, we performed revertant analysis of group IV mutant viruses (G37L and P112A) by continuously passaging culture fluids on BHK-21 cells. For each mutant virus, 12 independent selections were performed to improve the chances of getting recovered viruses harboring second-site mutations. Nine independent selections were successfully performed for each mutant. All the recovered viruses, G37L (Fig. 7A, top) and P112A (Fig. 7A, bottom), produced visible plaques. To determine the genetic basis of these recovered viruses, we extracted viral RNA from each selection and sequenced the complete genome (Fig. 7B). For the G37L revertants, we recovered only pseudoreversions to G37F (from three selections), G37H (from two selections), G37T (from three selections), and G37S (from one selection). For P112A revertants, all the viruses retained the original P112A mutation but contained one or two Lys insertions immediately after amino acid 127 or 130 in NS2B (Fig. 7B). Specifically, seven independent selections had one Lys insertion at the K127 position (K127KK), one selection had two Lys insertions at the K127 position (K127KKK), and one selection had one Lys insertion at the 130K position (K130KK). Such Lys insertion mutations indicated that the basic residue cluster at the C terminus of NS2B might play important roles in rescuing defects in viral RNA synthesis and/or assembly caused by the NS2B-P112A mutation. No mutation outside the NS2B gene was recovered. These results suggest that the adaptive mutations are responsible for restoring the complete infectious cycle of the G37L and P112A mutants.
FIG 7.

Selection of revertant viruses containing group IV mutations (G37L and P112A). (A) Plaque morphologies of the recovered viruses from different selections of the NS2B G37L and P112A mutants. (B) Compensatory mutation identified from revertant viruses.
Compensatory mutations specifically rescue virus assembly defects of group IV mutants.
To validate the sequence result for revertants, we engineered the adaptive mutations into the Rluc-JEV genome length RNA. For the G37L revertant, we chose adaptive mutation G37F for validation. As shown in Fig. 8A, similar levels of luciferase activities were detected in cells transfected with G37F and G37L Rluc-JEV RNAs, although the luciferase signals from both mutants were lower than that of the WT. In the viral infection assay (Fig. 8B), only the adaptive mutant G37F produced a replicative level of luciferase signal, which was significantly higher than those detected in the original G37L mutant and the negative-control NS5-GDD Rluc-JEV (P < 0.05). These results demonstrate that the G37F mutation could rescue the defect in virion assembly caused by the G37L mutation.
FIG 8.

Genetic analysis of compensatory effects of NS2B-G37F mutation using the Rluc-JEV reporter virus. (A) Equal amounts of WT and mutant Rluc-JEV RNAs were transfected into BHK-21 cells, and luciferase activity was measured at the indicated time points posttransfection. (B) The production of infectious virus particles was detected by infecting naive BHK-21 cells with the supernatants collected at 48 and 72 hpt, and the luciferase activity was detected at 48 hpi. Representative data from two independent experiments are presented. Each experiment was performed in triplicate, and the error bars represent standard deviations.
For the P112A revertant, we chose the adaptive mutation K127KK for functional validation, since it was found in most of the recovered P112A viruses (seven of nine) (Fig. 7B). Adaptive mutation K127KK was engineered into the P112A JEV genome length RNA (without a luciferase reporter). Upon transfection into BHK-21 cells, double-mutant (P112A plus K127KK) JEV RNA produced many more IFA-positive cells than single-mutant (P112A) RNA (Fig. 9A). Plaque assay showed that infectious virus was recovered from the P112A-plus-K127KK double mutant but not from the P112A single mutant (Fig. 9B). Since the P112A mutation attenuated both viral RNA synthesis and virion assembly, we examined the effect of compensatory mutation K127KK on viral RNA replication using a luciferase replicon. Surprisingly, addition of K127KK to the P112A replicon significantly reduced viral RNA replication (Fig. 9C) (t test; P < 0.01). These results clearly demonstrate that the restoration of virus production by the K127KK mutation is not due to the improvement of viral RNA synthesis.
FIG 9.

Genetic analysis of compensatory effects of the NS2B-K127KK mutation using the JEV infectious clone and subgenomic replicon. (A) IFA. BHK-21 cells were electroporated with the genome length RNAs of the WT and NS2B-P112A and NS2B-P112A-plus-K127KK mutants. The transfected cells were analyzed by IFA at the indicated time points using the method described in Materials and Methods. (B) Virus production. The virus titers of the transfected cells at each time point posttransfection were detected by plaque assay. (C) Transient replicon assay. Equal amounts of WT or mutant replicon RNAs were transfected into BHK-21 cells, and the cells were lysed for the luciferase activity assay at the indicated time points. Representative data from three independent experiments are presented. Each experiment was performed in triplicate, and the error bars represent standard deviations.
Analysis of the effect of adaptive mutation K127KK on NS2A-NS2B interaction.
Since the BiFC assay showed that the P112A mutation in NS2B weakened NS2B-NS2A interaction (Fig. 6C), we asked whether the compensatory mutation K127KK rescues virion assembly by restoring the NS2A-NS2B interaction. To this end, the WT or mutant (P112A and P112A plus K127KK) NS2B-VenusN plasmid was cotransfected with a NS2A-VenusC construct. As shown in Fig. 10A, the K127KK mutation in NS2B did not improve the interaction between NS2A and NS2B P112A mutants. However, when the single (P112A) and double (P112A-plus-K127KK) mutations were engineered into TMD3-VenusN containing only the TMD3 fragment of NS2B (representing C-terminal amino acids 98 to 131), the BiFC results (Fig. 10B) showed that (i) the P112A single mutation weakened the NS2B TMD3-NS2A interaction and (ii) P112A-plus-K127KK double mutations restored the NS2B TMD3-NS2A interaction to the WT level. The discrepancy between the complete NS2B and TMD3 alone could be due to steric hindrance of such mutants in the context of complete NS2B.
FIG 10.

BiFC analysis of compensatory effects of the NS2B-K127KK mutation on NS2B-NS2A interaction. (A and B) The full-length NS2B-VenusN (A) or the truncated NS2B TMD3-VenusN (B) plasmid (WT, P112A, and P112A plus K127KK) was cotransfected with NS2A-VenusC into 293T cells, and the effects of the NS2B mutations on NS2B-NS2A interaction were compared by BiFC assay. (C) Schematic diagram of the constructs used in fluorescence microscopy. WT and mutant NS2B TMD3 fragments with single P112A or double P112A-plus-K127KK mutations were cloned into a GFP fusion vector. (D) Comparison of the subcellular localizations of WT and mutant NS2B TMD3 fusion proteins. Representative data from three independent experiments are presented.
Additionally, the subcellular localization of TMD3 of NS2B was determined by fluorescence microscopy using a green fluorescent protein (GFP) fused with WT and mutant (P112A or P112A-plus-K127KK) NS2B TMD3 (Fig. 10C). WT and P112A-plus-K127KK NS2B TMD3 showed dot-like distribution patterns in the cytoplasm, while P112A NS2B TMD3 displayed even distribution in cells. The results demonstrate that the compensatory mutation K127KK rescues the topology of P112A-NS2B on the membrane within the cytoplasm, which may help to recover the interaction between NS2B and NS2A.
DISCUSSION
Flavivirus NS2B is a small integral transmembrane protein containing a conserved central hydrophilic region flanked by three putative hydrophobic transmembrane regions (7, 8). It interacts with the NS3 protease domain through its central hydrophilic region to form an NS2B-NS3pro heterodimer, which is responsible for proteolytic processing of the viral polyprotein. Its transmembrane segments are generally considered dispensable for the activity of NS3 protease. In this study, we found that the TMDs of JEV NS2B are involved in both viral RNA synthesis and virion assembly. Remarkably, it was also demonstrated that the interaction between NS2B and NS2A may play a critical role in the production of infectious particles. Overall, our data provide evidence for the first time that NS2B functions in both viral RNA synthesis and assembly through its TMDs in the flavivirus replication cycle.
To rapidly dissect the effects of each individual mutation on different steps of the JEV replication cycle, we initially assessed viral RNA replication and virus production using the JEV-Rluc reporter virus (Fig. 2B and C). All 13 mutations were classified into four groups based on their phenotypes (Fig. 2). Group I, including most of the mutations (E6A, A10L, V11A, L21A, M39A, V44A, G47L, and I115A), displayed slight or moderate reduction in viral RNA synthesis. Group II, including L38A and R101A, was severely defective in viral RNA replication. Group III included only one mutation, G12L, which was lethal for viral replication. Group IV, which included G37L and P112A, exhibited defects in both viral RNA synthesis and infectious virus production. Notably, mutagenesis analysis in the JEV infectious clone and subgenomic replicon provided further evidence that the virion assembly defect observed with these group IV mutants (G37L and P112A) is not due to their mutational effect on viral RNA synthesis. (i) Both genomic RNAs yielded E protein IFA-positive cells as early as 24 hpt, whereas no significant increase in the number of IFA-positive cells was observed during the whole time course (Fig. 3A), which indicated no efficient virus spread. (ii) Either extracellular viral RNA or extracellular/intracellular viral titers in these mutant genomic-RNA-transfected cells were barely detectable, while the two mutations had minor effects on the accumulation of intracellular viral RNA (Fig. 3B to E). (iii) The replicon assay clearly showed that both G37L and P112A substitutions were still able to replicate efficiently, exhibiting a much higher level of Rluc signals than even group II mutants (L38A and R101A) with a minor impact on virus assembly (Fig. 4B).
Considering that NS2B functions as the cofactor of NS3 protease (7, 10, 23), we compared both cis (NS2B-NS3 self-cleavage) and trans (NS4A-NS4B cleavage) activities of NS2B-NS3pro protease bearing different NS2B TMD mutations using an in vitro cleavage system (Fig. 5B and C). No significant differences in either cis-cleavage or trans-cleavage activities were observed between mutant NS2B-NS3pro proteases and wild-type NS2B-NS3pro protease. This indicates that the defects in viral RNA synthesis and/or assembly observed in NS2B TMD mutants are not caused by impairing NS2B-NS3pro protease activity. In YFV, Chambers et al. also found that substitution or deletion mutations within the hydrophobic domains of NS2B that were lethal to viral replication had subtle effects on the cleavage activity of the NS2B-NS3pro protease (7). These data not only further confirm that NS2B TMDs are dispensable for NS2B-NS3 protease activity (1, 12, 24), but also imply that NS2B might modulate viral RNA synthesis and virus assembly through its TMDs.
It has been reported that the virus budding sites are in close proximity to the pores of replication vesicles. Such spatial distribution may ensure efficient production and delivery of viral RNA for the assembly of infectious virus progeny (25). Therefore, NS proteins that are thought to play important roles in viral RNA replication are always found to be involved in virion assembly. NS2A is the first identified NS protein engaged in both processes. Mutations within NS2A of YFV, DENV-2, and the Kunjin subtype of West Nile virus (KUNV) were found to affect viral RNA synthesis and/or the production of infectious particles (18, 19, 26). Additionally, the second-site mutations in NS3 helicase could compensate for the defect in infectious YFV production caused by NS2A mutations (17, 19), which suggested that the genetic interaction between NS2A and NS3 is critical for virion production (17, 19). In addition to the evidence from the compensatory mutations in the NS3 helicase domain (17, 19), the role of NS3 in virion assembly has also been directly demonstrated by a mutation in YFV NS3 that selectively blocked infectious-virus production (27). During viral RNA synthesis, NS3 interacts with NS2B (7), NS4A (28), NS4B (29), and NS5 (30–32) to exert its enzymatic activities, facilitating viral RNA replication. In addition to being essential for RNA replication (33), NS1 has also been found to modulate infectious-particle production via interaction with the structural proteins (34). In a recent study, it was demonstrated, using FRET- and BiFC-based imaging approaches, that WNV NS2B interacted with many NS proteins, including the known NS3 and the other three membrane proteins (NS2A, NS4A, and NS4B), as well as itself, although the underlying mechanism for these interactions is unknown (16).
In our BiFC assay, we also observed that JEV NS2B was able to interact with most NS proteins, except NS1. It should be noted that the result regarding the interaction between NS2B and NS5 seems to contradict the previous study, in which NS2B was not able to interact with NS5 (16). This discrepancy may be caused by the different imaging approaches used: FRET in the previous study (16) versus BiFC in the current study. In contrast to FRET, which requires two chromophores at a separation radius of approximately 10 nm or less for positive signals, BiFC can occur if the two fluorescent fragments are within 10 nm with appropriate linkers (35). Further experiments may be needed to determine whether there is NS2B-NS5 interaction. Using this system, we did not find that the introduction of group II and III mutations into the full-length NS2B construct affected the interactions of NS2B with these viral proteins. Given that each membrane protein (NS2A, NS2B, NS4A, and NS4B) contains more than one TMD, we cannot rule out the possibility that multiple TMDs of individual membrane proteins are involved in protein interactions, and a single amino acid mutation in the full-length NS2B is not enough to destroy the interactions to a detectable degree in the BiFC assay. A systematic investigation of the effects of these replication-defective mutations on the interactions of NS2B with other viral NS proteins will be conducted in the context of an individual TMD segment of each membrane protein in the future.
In contrast to groups II and III, which dramatically impaired viral RNA replication, one of the group IV mutations affecting both viral RNA synthesis and assembly, P112A, dramatically attenuated the interaction between NS2B and NS2A (Fig. 6C). However, no compensatory mutation occurred in NS2A from the recovered viruses of the NS2B-P112A mutant. For the P112A mutant, all of the recovered viruses acquired a 1- or 2-lysine-residue insertion mutation adjacent to the C-terminal codon K127 or K130 in NS2B itself (seven strains with K127KK, one strain with K127KKK, and one strain with K130KK) (Fig. 7B). Genetic analysis of the compensatory mutation (K127KK) revealed that the adaptive mutation specifically rescued the virus production defect of a P112A mutant, since the combined mutant replicon K127KK plus P112A exhibited an even lower capability for viral RNA synthesis than P112A alone (Fig. 9C). Interestingly, it was also found that the K127KK compensatory mutation recovered interactions between NS2B TMD3 and NS2A caused by the P112A mutation (Fig. 10). It is therefore tempting to speculate that the physical interaction between NS2B and NS2A may contribute to virion assembly.
Based on the NMR structure of DENV4 NS2B reported recently (15), the conserved residue P112 is located at the turn between α3 (residues N90 to G105 in DENV4 NS2B) and α4 (residues P112 to T125 in DENV4 NS2B) helix segments in the third hydrophobic region. It was proposed that P112 might be responsible for conformational changes and the correct position of the C terminus of α4 in membranes. In contrast to proline, alanine is a strong helix-favoring residue. Thus, it is very possible that Pro-to-Ala substitution at this position affects the NS2B topology structure in the membrane, resulting in the impairment of its function, as well as interactions with other viral proteins. In support of this hypothesis, we did observe dramatic alterations of the subcellular localization of NS2B induced by the P112A mutation in the context of truncated NS2B TMD3 (Fig. 10D). The mutant GFP-fused NS2B TMD3 protein carrying the P112A mutation displayed even cytoplasmic accumulation, in contrast to the distinct dot-like distribution of the WT. At the same time, the addition of the K127KK compensatory mutation restored NS2B subcellular distribution caused by the P112A mutation. Thus, we speculated that the correct orientation of the NS2B α4 helix in the membrane may be a prerequisite for NS2B to interact with NS2A, which is important for virus assembly, and the insertion of the positive polar residue lysine at the C terminus of TMD3 may compensate for the topology of the P112A mutant protein in the membrane to a certain degree. This may also provide an explanation for why the compensatory mutations of P112A occurred exclusively in NS2B itself instead of in NS2A.
As for the other member of group IV, G37L, situated in the second TMD of NS2B, only a variety of pseudorevertants, such as G37F (three strains), G37H (two strains), G37T (three strains), and G37S (one strain), were obtained from the recovered viruses. Although genetic analysis showed that the pseudoreversion G37F was able to specifically restore the defect in assembly of G37L, which was similar to the observation in the P112A mutant, we did not observe apparent alterations of interaction between NS2B and other NS proteins. This indicates that the G37L mutation impairs virus assembly through a mechanism that is different from that for the P112A mutation.
In summary, our study provides evidence for the first time that the TMDs of JEV NS2B participate in both viral RNA replication and virion assembly. Using a BiFC-based imaging method, we observed that the interaction between NS2B and NS2A was weakened by a virus assembly-deficient mutant, NS2B-P112A, which could be recovered by its compensatory mutation, NS2B-K127KK. These data imply an essential role for the interaction between NS2B and NS2A during virus particle assembly. Although it is still hard to propose a clear mechanistic model for NS2B TMDs in viral replication and assembly based on the limited information from our data, it clearly indicates that NS2B TMDs may serve as a potential target for antiviral drug development.
ACKNOWLEDGMENTS
We thank the Core Facility and Technical Support, Wuhan Institute of Virology, and the Wuhan Key Laboratory on Emerging Infectious Diseases and Biosafety for helpful support during the course of the work.
Funding Statement
This work was funded by National Basic Research Program of China (973 Program) (2012CB518904).
REFERENCES
- 1.Sampath A, Padmanabhan R. 2009. Molecular targets for flavivirus drug discovery. Antivir Res 81:6–15. doi: 10.1016/j.antiviral.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brinton MA. 2014. Replication cycle and molecular biology of the West Nile virus. Viruses 6:13–53. doi: 10.3390/v6010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morrison J, Aguirre S, Fernandez-Sesma A. 2012. Innate immunity evasion by dengue virus. Viruses 4:397–413. doi: 10.3390/v4030397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nikonov A, Molder T, Sikut R, Kiiver K, Mannik A, Toots U, Lulla A, Lulla V, Utt A, Merits A, Ustav M. 2013. RIG-I and MDA-5 detection of viral RNA-dependent RNA polymerase activity restricts positive-strand RNA virus replication. PLoS Pathog 9:e1003610. doi: 10.1371/journal.ppat.1003610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shi PY. 2014. Structural biology. Unraveling a flavivirus enigma. Science 343:849–850. doi: 10.1126/science.1251249. [DOI] [PubMed] [Google Scholar]
- 6.Falgout B, Pethel M, Zhang YM, Lai CJ. 1991. Both nonstructural proteins NS2B and NS3 are required for the proteolytic processing of dengue virus nonstructural proteins. J Virol 65:2467–2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chambers TJ, Nestorowicz A, Amberg SM, Rice CM. 1993. Mutagenesis of the yellow fever virus NS2B protein: effects on proteolytic processing, NS2B-NS3 complex formation, and viral replication. J Virol 67:6797–6807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang Q, Chen AS, Li Q, Kang C. 2011. Expression, purification, and initial structural characterization of nonstructural protein 2B, an integral membrane protein of Dengue-2 virus, in detergent micelles. Protein Expr Purif 80:169–175. doi: 10.1016/j.pep.2011.08.008. [DOI] [PubMed] [Google Scholar]
- 9.Arias CF, Preugschat F, Strauss JH. 1993. Dengue 2 virus NS2B and NS3 form a stable complex that can cleave NS3 within the helicase domain. Virology 193:888–899. doi: 10.1006/viro.1993.1198. [DOI] [PubMed] [Google Scholar]
- 10.Chambers TJ, Nestorowicz A, Rice CM. 1995. Mutagenesis of the yellow fever virus NS2B/3 cleavage site: determinants of cleavage site specificity and effects on polyprotein processing and viral replication. J Virol 69:1600–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clum S, Ebner KE, Padmanabhan R. 1997. Cotranslational membrane insertion of the serine proteinase precursor NS2B-NS3(Pro) of dengue virus type 2 is required for efficient in vitro processing and is mediated through the hydrophobic regions of NS2B. J Biol Chem 272:30715–30723. doi: 10.1074/jbc.272.49.30715. [DOI] [PubMed] [Google Scholar]
- 12.Droll DA, Krishna Murthy HM, Chambers TJ. 2000. Yellow fever virus NS2B-NS3 protease: charged-to-alanine mutagenesis and deletion analysis define regions important for protease complex formation and function. Virology 275:335–347. doi: 10.1006/viro.2000.0488. [DOI] [PubMed] [Google Scholar]
- 13.Zuo Z, Liew OW, Chen G, Chong PC, Lee SH, Chen K, Jiang H, Puah CM, Zhu W. 2009. Mechanism of NS2B-mediated activation of NS3pro in dengue virus: molecular dynamics simulations and bioassays. J Virol 83:1060–1070. doi: 10.1128/JVI.01325-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li XD, Li XF, Ye HQ, Deng CL, Ye Q, Shan C, Shang BD, Xu LL, Li SH, Cao SB, Yuan ZM, Shi PY, Qin CF, Zhang B. 2014. Recovery of a chemically synthesized Japanese encephalitis virus reveals two critical adaptive mutations in NS2B and NS4A. J Gen Virol 95:806–815. doi: 10.1099/vir.0.061838-0. [DOI] [PubMed] [Google Scholar]
- 15.Li Y, Li Q, Wong YL, Liew LS, Kang C. 2015. Membrane topology of NS2B of dengue virus revealed by NMR spectroscopy. Biochim Biophys Acta 1848:2244–2252. doi: 10.1016/j.bbamem.2015.06.010. [DOI] [PubMed] [Google Scholar]
- 16.Yu L, Takeda K, Markoff L. 2013. Protein-protein interactions among West Nile non-structural proteins and transmembrane complex formation in mammalian cells. Virology 446:365–377. doi: 10.1016/j.virol.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 17.Kummerer BM, Rice CM. 2002. Mutations in the yellow fever virus nonstructural protein NS2A selectively block production of infectious particles. J Virol 76:4773–4784. doi: 10.1128/JVI.76.10.4773-4784.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leung JY, Pijlman GP, Kondratieva N, Hyde J, Mackenzie JM, Khromykh AA. 2008. Role of nonstructural protein NS2A in flavivirus assembly. J Virol 82:4731–4741. doi: 10.1128/JVI.00002-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vossmann S, Wieseler J, Kerber R, Kummerer BM. 2015. A basic cluster in the N terminus of yellow fever virus NS2A contributes to infectious particle production. J Virol 89:4951–4965. doi: 10.1128/JVI.03351-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie X, Zou J, Puttikhunt C, Yuan Z, Shi PY. 2015. Two distinct sets of NS2A molecules are responsible for dengue virus RNA synthesis and virion assembly. J Virol 89:1298–1313. doi: 10.1128/JVI.02882-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A. 2002. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat Biotechnol 20:87–90. doi: 10.1038/nbt0102-87. [DOI] [PubMed] [Google Scholar]
- 22.Li XD, Ye HQ, Deng CL, Liu SQ, Zhang HL, Shang BD, Shi PY, Yuan ZM, Zhang B. 2015. Genetic interaction between NS4A and NS4B for replication of Japanese encephalitis virus. J Gen Virol 96:1264–1275. doi: 10.1099/vir.0.000044. [DOI] [PubMed] [Google Scholar]
- 23.Chambers TJ, Droll DA, Tang Y, Liang Y, Ganesh VK, Murthy KH, Nickells M. 2005. Yellow fever virus NS2B-NS3 protease: characterization of charged-to-alanine mutant and revertant viruses and analysis of polyprotein-cleavage activities. J Gen Virol 86:1403–1413. doi: 10.1099/vir.0.80427-0. [DOI] [PubMed] [Google Scholar]
- 24.Leung D, Schroder K, White H, Fang NX, Stoermer MJ, Abbenante G, Martin JL, Young PR, Fairlie DP. 2001. Activity of recombinant dengue 2 virus NS3 protease in the presence of a truncated NS2B co-factor, small peptide substrates, and inhibitors. J Biol Chem 276:45762–45771. doi: 10.1074/jbc.M107360200. [DOI] [PubMed] [Google Scholar]
- 25.Welsch S, Miller S, Romero-Brey I, Merz A, Bleck CK, Walther P, Fuller SD, Antony C, Krijnse-Locker J, Bartenschlager R. 2009. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe 5:365–375. doi: 10.1016/j.chom.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu RH, Tsai MH, Chao DY, Yueh A. 2015. Scanning mutagenesis studies reveal a potential intramolecular interaction within the C-terminal half of dengue virus NS2A involved in viral RNA replication and virus assembly and secretion. J Virol 89:4281–4295. doi: 10.1128/JVI.03011-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patkar CG, Kuhn RJ. 2008. Yellow fever virus NS3 plays an essential role in virus assembly independent of its known enzymatic functions. J Virol 82:3342–3352. doi: 10.1128/JVI.02447-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shiryaev SA, Chernov AV, Aleshin AE, Shiryaeva TN, Strongin AY. 2009. NS4A regulates the ATPase activity of the NS3 helicase: a novel cofactor role of the non-structural protein NS4A from West Nile virus. J Gen Virol 90:2081–2085. doi: 10.1099/vir.0.012864-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Umareddy I, Chao A, Sampath A, Gu F, Vasudevan SG. 2006. Dengue virus NS4B interacts with NS3 and dissociates it from single-stranded RNA. J Gen Virol 87:2605–2614. doi: 10.1099/vir.0.81844-0. [DOI] [PubMed] [Google Scholar]
- 30.Johansson M, Brooks AJ, Jans DA, Vasudevan SG. 2001. A small region of the dengue virus-encoded RNA-dependent RNA polymerase, NS5, confers interaction with both the nuclear transport receptor importin-beta and the viral helicase, NS3. J Gen Virol 82:735–745. doi: 10.1099/0022-1317-82-4-735. [DOI] [PubMed] [Google Scholar]
- 31.Kapoor M, Zhang L, Ramachandra M, Kusukawa J, Ebner KE, Padmanabhan R. 1995. Association between NS3 and NS5 proteins of dengue virus type 2 in the putative RNA replicase is linked to differential phosphorylation of NS5. J Biol Chem 270:19100–19106. doi: 10.1074/jbc.270.32.19100. [DOI] [PubMed] [Google Scholar]
- 32.Yon C, Teramoto T, Mueller N, Phelan J, Ganesh VK, Murthy KH, Padmanabhan R. 2005. Modulation of the nucleoside triphosphatase/RNA helicase and 5′-RNA triphosphatase activities of Dengue virus type 2 nonstructural protein 3 (NS3) by interaction with NS5, the RNA-dependent RNA polymerase. J Biol Chem 280:27412–27419. doi: 10.1074/jbc.M501393200. [DOI] [PubMed] [Google Scholar]
- 33.Youn S, Li T, McCune BT, Edeling MA, Fremont DH, Cristea IM, Diamond MS. 2012. Evidence for a genetic and physical interaction between nonstructural proteins NS1 and NS4B that modulates replication of West Nile virus. J Virol 86:7360–7371. doi: 10.1128/JVI.00157-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scaturro P, Cortese M, Chatel-Chaix L, Fischl W, Bartenschlager R. 2015. Dengue virus non-structural protein 1 modulates infectious particle production via interaction with the structural proteins. PLoS Pathog 11:e1005277. doi: 10.1371/journal.ppat.1005277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kerppola TK. 2008. Bimolecular fluorescence complementation (BiFC) analysis as a probe of protein interactions in living cells. Annu Rev Biophys 37:465–487. doi: 10.1146/annurev.biophys.37.032807.125842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miller S, Kastner S, Krijnse-Locker J, Buhler S, Bartenschlager R. 2007. The non-structural protein 4A of dengue virus is an integral membrane protein inducing membrane alterations in a 2K-regulated manner. J Biol Chem 282:8873–8882. doi: 10.1074/jbc.M609919200. [DOI] [PubMed] [Google Scholar]