

ABSTRACT
The coxsackievirus and adenovirus receptor (CAR) is a member of the immunoglobulin superfamily (IgSF) and functions as a receptor for coxsackie B viruses (CVBs). The extracellular portion of CAR comprises two glycosylated immunoglobulin-like domains, D1 and D2. CAR-D1 binds to the virus and is essential for virus infection; however, it is not known whether D2 is also important for infection, and the role of glycosylation has not been explored. To understand the function of these structural components in CAR-mediated CVB3 infection, we generated a panel of human (h) CAR deletion and substitution mutants and analyzed their functionality as CVB receptors, examining both virus binding and replication. Lack of glycosylation of the CAR-D1 or -D2 domains did not adversely affect CVB3 binding or infection, indicating that the glycosylation of CAR is not required for its receptor functions. Deletion of the D2 domain reduced CVB3 binding, with a proportionate reduction in the efficiency of virus infection. Replacement of D2 with the homologous D2 domain from chicken CAR, or with the heterologous type C2 immunoglobulin-like domain from IgSF11, another IgSF member, fully restored receptor function; however, replacement of CAR-D2 with domains from CD155 or CD80 restored function only in part. These data indicate that glycosylation of the extracellular domain of hCAR plays no role in CVB3 receptor function and that CAR-D2 is not specifically required. The D2 domain may function largely as a spacer permitting virus access to D1; however, the data may also suggest that D2 affects virus binding by influencing the conformation of D1.
IMPORTANCE An important step in virus infection is the initial interaction of the virus with its cellular receptor. Although the role in infection of the extracellular CAR-D1, cytoplasmic, and transmembrane domains have been analyzed extensively, nothing is known about the function of CAR-D2 and the extracellular glycosylation of CAR. Our data indicate that glycosylation of the extracellular CAR domain has only minor importance for the function of CAR as CVB3 receptor and that the D2 domain is not essential per se but contributes to receptor function by promoting the exposure of the D1 domain on the cell surface. These results contribute to our understanding of the coxsackievirus-receptor interactions.
INTRODUCTION
Coxsackie B viruses (CVBs) initiate infection of their host cells by interaction with the coxsackievirus and adenovirus receptor (CAR). An additional cell surface protein, decay-accelerating factor (DAF), promotes binding to the cell surface but is not sufficient for infection (1–3). CAR is a member of the immunoglobulin superfamily (IgSF) and is composed of two extracellular immunoglobulin-like domains, D1 (amino acid [aa] 20 to 139) and D2 (aa 142 to 229), as well as a typical hydrophobic transmembrane domain (TMD; aa 236 to 258) and an internal cytoplasmic domain (ICD; aa 259 to 365) (4). The extracellular immunoglobulin-like domains vary in their secondary structure by different β-strand folding. Whereas D1 shows a typical V-type fold structure, D2 has a C2-type immunoglobulin fold (5, 6). Several studies indicate a primary role for the D1 domain in CAR interactions with CVB3 (7) and adenovirus (8), as well as in CAR/CAR homophilic interactions (9–11). Although the isolated D1 domain, produced in Escherichia coli, binds adenovirus efficiently (8), the same D1 domain was found to bind poorly to CVB3 (7), suggesting a possible supporting role for the D2 domain during CAR/CVB3 interaction.
Numerous picornavirus receptors are members of the IgSF: intercellular adhesion molecule-1 (ICAM-1) is a receptor for coxsackievirus A21 (CAV21) and the major group of human rhinoviruses (HRVs), and CD155 serves as the poliovirus receptor (PVR). In each case, virus interacts with the membrane-distal D1 domain (12–14). However, studies with PV, HRV, and hepatitis A virus (HAV) have shown that deletion of the membrane-proximal extracellular domains decreases virus binding to the receptor, as well as infection of the cells (13, 15–17). Replacement of these proximal domains with homologous domains from other species restored normal receptor function (18, 19), but replacement with heterologous protein domains did not (16, 20). Thus, domains that are located membrane proximal to the virus-binding D1 domain are important to maintain virus receptor properties, but the mechanisms that are involved are not well understood.
CAR functions not only as the main receptor for CVB but also as an attachment receptor for subgroup A and C to F adenoviruses (2, 21). Experiments with adenovirus 5 revealed significantly attenuated binding and infection through CAR lacking the D2 domain, suggesting that the D2 may contribute to of the D1 domain with the adenovirus fiber-knob (22). Moreover, additional experiments showed that glycosylation of CAR's two extracellular domains influences adenovirus infection (23). In general, posttranslational modifications such as glycosylation play an important role in protein folding and conformation (22) and, as a result, many physiological activities such as migration, cell adhesion, and receptor/ligand binding are affected by glycosylation (23). CAR has two N-glycosylation sites, one on each of its extracellular domains (N106 in D1 and N201 in D2). Glycosylation of ICAM-1 is not required for rhinovirus binding (19, 24) or for poliovirus interaction with PVR (25). In contrast, the binding and infection efficiency of hepatitis A virus (HAV) depends on glycosylation of the HAV cellular receptor havcr-1, and deglycosylation results in decreased susceptibility of the cells to HAV infection (26). These data suggest either that glycosylation is needed to maintain the conformation of the havcr-1-D1 domain, which is required for virus binding and uptake, or that the N-glycan itself forms part of the binding site (26).
The role of CAR glycosylation in CVB binding and infection has not been defined. Further, although CAR-D1 is known to be make direct contact with virus (7), and the TMD and ICD are known to be dispensable for infection (27); the function of the D2 domain is not certain. In the present study, we intended to clarify the role of CAR glycosylation, and the specific role of D2, on CVB3 binding and infection. We found that elimination of CAR's N-linked glycosylation sites by mutagenesis and inhibition of glycosylation by tunicamycin treatment had no effect on CAR-mediated infection by CVB3. Further, experiments with domain deletion and substitution mutants provided evidence that CAR-D2, although it is not specifically required for binding or infection, nonetheless contributes to CAR's interaction with the virus. The D2 domain may act as a spacer that facilitates virus access to D1, and/or it may promote binding by influencing the conformation of the D1 domain.
MATERIALS AND METHODS
Cells and viruses.
CVB3 (Nancy strain; VR-30) was obtained from ATCC and propagated in HeLa cells. Virus expansion, virus labeling, and plaque assays were performed on low-passage-number HeLa cells. HeLa cells were maintained in minimal essential medium (Gibco/Life Technologies, Grand Island, NY) supplemented with 5% fetal bovine serum (FBS), 1% penicillin-streptomycin, and 1× nonessential amino acids (Gibco). Chinese hamster ovary (CHO-K1) cells and CHO-DAF cells (28) were cultivated in F-12 medium containing 1% penicillin-streptomycin and 10% FBS.
Construction of various CAR and soluble CAR expression plasmids.
To generate plasmid phCAR, hCAR encoding cDNA was amplified from pZS2/hCARs (29) and inserted via EcoRI/SalI into the plasmid pscAAV-MCS (30). To generate phCARD1 DNA fragments containing the hCAR-D1 domain (aa 1 to 139 of hCAR, where aa 1 is the initial methionine of the leader sequence) and hCAR, the TMD and ICD were amplified from phCAR and fused by overlap PCR. The fused fragment was digested with EcoRI and SalI and inserted into pscAAV-MCS. The plasmids phCAR-CD80 and phCAR-CD155 were generated as follows. Total RNA was isolated from HeLa cells and reverse transcribed using SuperScript II reverse transcriptase (Invitrogen, Karlsruhe, Germany). The CD80-D2 (NCBI accession no. NM_005191; aa 140 to 232) and CD155-D2 (accession no. NM_006505; aa 141 to 242) domains were amplified and fused to the hCAR-D1 (behind aa 141) domain by PCR and then inserted into plasmid pscAAV-MCS as described above. To generate plasmid phCAR-chCAR, a PCR fragment containing the hCAR-D1 domain (aa 1 to 139) was amplified from pZS2/hCARs, and a PCR fragment containing the D2 domain of chicken CAR (chCAR; aa 144 to 240) was amplified from a chCAR cDNA (kindly provided by Fritz G. Rathjen; Max Delbrück Center for Molecular Medicine, Berlin, Germany; NCBI accession no. XM_004938354). The fragments were fused by overlap PCR and inserted into phCAR, which leads to replacement of the extracellular domain of hCAR by the chimeric hCAR-D1/chCAR-D2 domain. To construct pchCAR-hCAR, the D1 domain of chCAR (aa 1 to 143) was amplified from chCAR cDNA and fused to the D2 and TM/IC domains of hCAR by overlap PCR, and the chimeric chCAR-D1/hCAR-D2 domain was inserted into phCAR as described above. For phCAR-IgSF11, the D2 domain of IgSF11 (aa 142 to 240) was amplified from IgSF11 cDNA (accession no. BC034411 [Thermo Scientific]) and fused to the hCAR-D1 domain by overlap PCR. The chimeric hCAR-D1/IgSF11-D2 domain was inserted into phCAR as described above. The plasmids phCARN106Q, phCARN201Q, and phCARN106/201Q containing the hCAR glycosylation mutants hCARN106Q, hCARN201Q, and hCARN106/201Q, respectively, were generated by site-directed mutagenesis of phCAR following the protocol described previously (31).
To generate the sCAR-Fc (i.e., the fusion protein of the extracellular domain of CAR and the carboxyl terminus of human IgG1 Fc domain) expression plasmid psCAR-Fc, the sCAR-Fc cDNA was amplified from the DNA of the adenoviral vector AdG12 (32) by PCR with Pfu polymerase (PfuUltra high-fidelity DNA polymerase; Agilent Technologies, Boeblingen, Germany) using primers linked with EcoRI sites and ligated into the EcoRI-digested pscAAV-MCS (30). The sCAR-Fc variants with deleted D2 domain were generated by PCR. Therefore, we used the sense primer, including the EcoRI restriction site, and BamHI-linked antisense primers that bound to CAR aa 139. The PCR fragment was inserted into BamHI/EcoRI digested psCAR-Fc upstream and in frame with the carboxy terminus of human IgG-Fc. The plasmid was termed psCARD1-Fc.
The plasmids psCARN106Q-Fc, psCARN201Q-Fc, and psCARN106/201Q-Fc containing the coding sequence for the soluble versions of the hCAR glycosylation mutants (sCARN106Q-Fc, sCARN201Q-Fc, and sCARN106/201Q-Fc) were generated as follows. The extracellular domains of the hCAR glycosylation mutants were amplified by PCR from phCARN106Q, phCARN201Q, and phCARN106/201Q using EcoRI- and BamHI-linked primers and inserted into EcoRI/BamHI-digested psCAR-Fc, upstream and in frame with the carboxy terminus of human IgG-Fc. The correctness of all plasmids was proven by sequencing.
Transfection of CHO-K1 cells and CHO-DAF cells.
CHO-K1 and CHO-DAF cells were transfected using polyethylenimine (Polyethylenimine Max; Polyciences, Warrington, PA) 24 h after seeding at a density of 70 to 90%. At 4 h posttransfection (p.t.), the medium was replaced with fresh medium containing 10% FBS.
Western blotting.
CAR-transfected CHO-K1 cells were scraped at 48 h p.t. from six-well plates and lysed. For each sample, 20 μg of protein was separated on a 4 to 20% Bis-Tris gel (Bio-Rad, Hercules, CA) and transferred onto a polyvinylidene difluoride membrane. To detect CAR and GAPDH (glyceraldehyde-3-phosphate dehydrogenase), the membrane was incubated, after blocking with 5% dry milk/PBS-T (phosphate-buffered saline with 0.1% Tween 20), with affinity-purified rabbit anti-CAR antibody at a dilution of 1:1,000 at room temperature for 1 h. After three washes with PBS-T, the membrane was incubated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit antibody (GE Healthcare) at a dilution of 1:5,000 in 5% dry milk/PBS-T for 1 h. After an extensive washing, CAR expression was detected by measuring the chemiluminescence using SuperSignal West Dura extended-duration substrate (Thermo Scientific). For the loading control, the membrane was stripped with Restore Western Blot stripping buffer (Thermo Scientific), reblocked, and incubated with directly HRP-conjugated rabbit-anti-GAPDH antibody at a dilution of 1:200 overnight at 4°C and then developed as described above.
Flow cytometry to detect receptor expression on the cell surface.
Transfected CHO-K1 cells were detached at 48 h p.t. with PBS–2.5 mM EDTA and then washed with PBS. The cells were stained on ice with an affinity-purified rabbit-anti-CAR antibody (33) for 1 h at a dilution of 1:1,000, followed by secondary goat anti-rabbit antibody conjugated to fluorescein isothiocyanate (FITC) at a 1:500 dilution in 10% goat serum (GE Healthcare, Pittsburgh, PA). After the final washing step, the cells were resuspended in PBS and analyzed by flow cytometry using a BD Accuri C6 (BD Biosciences, San Jose, CA). The mean fluorescence was calculated by determining the geometric mean of CAR-expressing cells minus the geometric mean of a negative control sample.
Immunofluorescence microscopy.
CHO-K1 cells were seeded on eight-well chamber slides coated with poly-l-lysine (BD Biosciences) at a density of 5 × 104 cells/well and then transfected with wild-type hCAR- or chimeric CAR-expressing plasmids 24 h later. To analyze protein localization, the cells were fixed at 48 h p.t. with 4% formalin, and the CAR proteins were stained with an affinity-purified rabbit anti-CAR antibody (33) for 1 h at a dilution of 1:1,000, and a secondary goat anti-rabbit antibody conjugated to Texas Red (GE Healthcare) was added after three washing steps for an additional 1 h at a dilution of 1:500 containing 10% goat serum. The cells were embedded with Vectashield mounting medium containing DAPI (4′,6′-diamidino-2-phenylindole; Vector Laboratories, Burlingame, CA). To detect newly synthesized CVB3 antigen, transfected CHO-K1 cells were incubated with CVB3 (at a multiplicity of infection [MOI] of 10) and fixed at 10 h p.i. with a 3:1 mixture of ice-cold methanol-acetone; the CVB3 was then stained with mouse anti-enterovirus VP1 monoclonal antibody (NCL-Entero; Leica Biosystems, Buffalo Grove, IL) at a 1:500 dilution for 1 h and goat anti-mouse secondary antibody conjugated to FITC (GE Healthcare).
Radiolabeling of CVB3 and purification of mature particles.
Confluent HeLa cells, growing in a 75-cm2 flask, were washed twice with methionine-cysteine and serum-free medium (Dulbecco modified Eagle medium [DMEM] plus 20 mM HEPES) and incubated with 2 ml of CVB3 (MOI of 40) for 45 min at room temperature with continuous rocking. After the virus dilution was aspirated, the cells were washed once with CO2-equilibrated radiolabeling medium (methionine-cysteine-free DMEM, 1× penicillin-streptomycin, 1× l-glutamine, and 5% dialyzed FBS) and then incubated with 10 ml of radiolabeling medium at 37°C. After 3 h, the medium was replaced with fresh radiolabeling medium containing 400 μCi of [35S]methionine-cysteine, and the incubation was continued overnight. The cells were completely lysed by three freeze-thaw cycles following incubation on ice after the addition of 10% Triton X-100/PBS (final concentration, 1%). Virus-containing supernatant was cleared from cell debris by centrifugation (20 min, 12,000 × g, 16°C) and further incubated with 0.1 volume of 10% sodium dodecyl sulfate. Radiolabeled virus was then pelleted through a 30% sucrose cushion in acetate buffer (1 M NaCl, 20 mM Tris-acetate [pH 7]) at 240,000 × g for 90 min at 16°C and then thoroughly resuspended in 150 μl of heparan sulfate (HS) buffer. Mature virus particles (160S) were separated from procapsid, A-particles (135S), and empty capsids (80S) by ultracentrifugation on a 15 to 30% continuous sucrose gradient at 240,000 × g for 60 min, followed by fractionation (200 μl per fraction from top to bottom). Next, 5 μl of each fraction were measured by scintillation counting to identify the radioactive peak. Peak radioactive fractions corresponding to mature virions were then pooled and stored at −80°C.
Virus binding to transfected cells.
Radiolabeled CVB3 (20,000 cpm/well) was bound to transfected CHO-K1 cells in 24-well plates for 1 h at room temperature with continuous rocking. Unbound virus was removed by three washing steps with PBS, the cells were lysed with detergent (Solvable; Perkin-Elmer, Waltham, MA), and the cell-bound radioactivity was assessed using a scintillation counter.
Production of the sCAR-Fc variants.
To produce the different sCAR-Fc variants, the HeLa cells were transfected with polyethylenimine at a density of 90%. The medium was replaced with fresh medium containing 1% FBS at 16 h p.t., and supernatants containing sCAR-Fc proteins were harvested after an additional 3 days. The concentrations of the various sCAR-Fc mutants were measured by an enzyme-linked immunosorbent assay detecting human IgG-Fc (Bethyl Laboratories, Montgomery, TX).
Virus binding to soluble receptors immobilized on beads.
Portions (10 μl) of Dynabeads Protein G (Life Technologies) and 0.5 μg of each chimeric Fc-linked protein were diluted in 10 μl of immunoprecipitation buffer (20 mM Tris-Cl [pH 7.4], 135 mM NaCl, 1% Triton X-100, 10% glycerol) and 80 μl of DMEM with 1% FBS, followed by incubation for 1 h at 4°C. The beads were washed once and incubated with radiolabeled CVB3 (10,000 cpm) in DMEM containing 1% FBS and 10% immunoprecipitation buffer for an additional 1 h at 4°C. The beads were washed five times with immunoprecipitation buffer and suspended in 50 μl of PBS, and the amount of bound virus was quantified by scintillation counting.
Virus infection and plaque assay.
CHO-K1 and CHO-DAF cells were incubated at 48 h after plasmid transfection with CVB3 for 1 h at room temperature. After incubation, the virus was removed, and the cells were washed twice with PBS to remove unbound virus. Thereafter, medium was added to the cells, and the cells were frozen after additional incubation for 0, 24, or 48 h at 37°C. After three freeze-thaw cycles probes were centrifuged to remove cell debris, and the infectious virus particles were quantified by plaque assay as described previously (30). In experiments with tunicamycin, CHO-K1 cells were incubated with 0.25 μg of tunicamycin (Sigma-Aldrich, catalog no. T7765)/ml for 4 h before transfection with hCAR expression plasmids, and treatment was continued for an additional 36 h before infection.
Statistical analysis.
For statistical analysis, Prism software 5.0 (GraphPad, San Diego, CA) was used. The results are expressed as means ± the standard deviations (SD). To compare two groups, a Student t test was performed. Asterisks are used in the legends and figures indicate statistically significant differences (*, P < 0.05; **, P < 0.01).
RESULTS
CAR-mediated CVB3 infection does not require glycosylation of the CAR-D1 or -D2 domains.
To determine whether glycosylation is important for CAR interactions with CVB3, we examined the receptor function of CAR mutants lacking the sites for N-linked glycosylation of either D1 (hCARN106Q) or D2 (hCARN201Q) or lacking both glycosylation sites (hCARN106/201Q) (Fig. 1A).
FIG 1.

Expression of hCAR glycosylation mutants in CHO-K1 cells. (A) Schematic illustration of wild-type hCAR and hCAR glycosylation mutants. (B) Protein expression in transiently transfected CHO-K1 cells analyzed by Western blotting with polyclonal anti-CAR rabbit antibody. Mutants lacking glycosylation of the D1 domain (hCARN106Q), the D2 domain (hCARN201Q), or both domains (hCARN106/201Q) show increased mobility compared to wild-type hCAR. CHO-K1 cells exposed to transfection reagent without plasmid DNA (mock) served as a negative control. All lanes are from a single exposure of a single immunoblot; the line indicates where irrelevant lanes were deleted. Lane M, MagicMark XP Western protein standard. (C) Localization of wild-type hCAR and hCAR mutants detected with anti-CAR rabbit antibody (red). DAPI was used for nuclear staining (blue). Both wild-type hCAR and hCAR glycosylation mutants were concentrated at cell-cell junctions (arrows). Magnification, ×400.
Mutated proteins were expressed by transient transfection of the CAR-negative cell line CHO-K1 and analyzed by Western blotting, flow cytometry, and immunocytochemistry. When analyzed by Western blotting (Fig. 1B), fully glycosylated wild-type CAR migrated at 46 kDa, as previously reported (1). The resulting protein bands from the CAR mutants reflect the loss of glycosylation in a stepwise manner: single glycosylation-deficient mutants hCARN106Q and hCARN201Q showed an equal protein size, smaller than hCAR but larger than the double glycosylation-deficient variant hCARN106/201Q, which migrated at 40 kDa, a finding consistent with the CAR amino acid sequence (34). Analysis by flow cytometry confirmed that all four CAR variants were expressed at the surfaces of similar numbers of transfected CHO-K1 cells and in similar amounts (Table 1). As shown in Fig. 1C, immunofluorescence analysis indicated that, like wild-type hCAR, the glycosylation-deficient mutants were located at sites of cell-cell contact.
TABLE 1.
Surface expression of wild-type and mutant CAR moleculesa
| CAR molecule | Description | Mean ± SD |
|
|---|---|---|---|
| CAR-positive cells (% total) | MFI (×103) | ||
| Wild type | hCAR | 36.3 ± 7.2 | 48.4 ± 3.9 |
| Glycosylation mutants | hCARN106Q | 34.9 ± 3.9 | 48.0 ± 4.1 |
| hCARN201Q | 33.8 ± 2.8 | 59.3 ± 9.0 | |
| hCARN106/201Q | 37.6 ± 7.5 | 55.5 ± 11.2 | |
| D2 domain chimeras | hCARD1 | 39.9 ± 6.2 | 51.7 ± 8.0 |
| hCAR-CD155 | 36.2 ± 7.3 | 71.4 ± 7.0 | |
| hCAR-CD80 | 36.4 ± 1.6 | 64.9 ± 9.4 | |
| hCAR-IgSF11 | 39.7 ± 6.4 | 45.4 ± 16.1 | |
| hCAR/chCAR chimeras | chCAR-hCAR | 34.7 ± 2.3 | 40.3 ± 12.9 |
| hCAR-chCAR | 36.0 ± 8.2 | 39.5 ± 12.8 | |
Transfected CHO-K1 cells were stained with polyclonal rabbit anti-CAR antibody and FITC-conjugated anti-rabbit secondary antibody 48 h after transfection. The percentages of CAR-positive cells and the geometric mean fluorescence intensity (MFI) of the total population were determined using stained mock-transfected CHO-K1 cells as a negative control. Both the percentages and the geometric means of CAR-positive CHO-K1 cells at 48 h after transfection are indicated.
To determine whether glycosylation-deficient mutants functioned as receptors for CVB3, we measured their capacity to bind virus and mediate infection. We used two different approaches to quantify the binding of CVB3 to the hCAR glycosylation mutants and compared them to fully glycosylated hCAR. First, CHO-K1 cells were transiently transfected and incubated 48 h later with radioactive 35S-labeled CVB3 at room temperature. Unbound virus was washed away, and cell-bound virus was quantified by scintillation counting (Fig. 2A). In contrast to results previously reported for adenovirus, CVB3 bound equally well to the mutants and to wild-type CAR. To measure virus interaction with a precisely controlled quantity of receptor protein, in the absence of other membrane components, we also measured virus binding to soluble (s) CAR proteins bound to protein G-beads. For these experiments, the extracellular domains of the three CAR glycosylation mutants were fused to the constant domain (Fc) of the human IgG; we used fully glycosylated sCAR-Fc (32) as a positive control and soluble Fc (sFc) as negative control to evaluate subsequent binding ability to CVB3. In accordance with our previous results, the efficiency of virus binding to sCAR-Fc proteins was not affected by different glycosylation levels (Fig. 2B), whereas no virus bound to the negative control. These experiments indicate that glycosylation of the CAR extracellular part (D1 and D2 domain) does not alter CVB3 binding.
FIG 2.
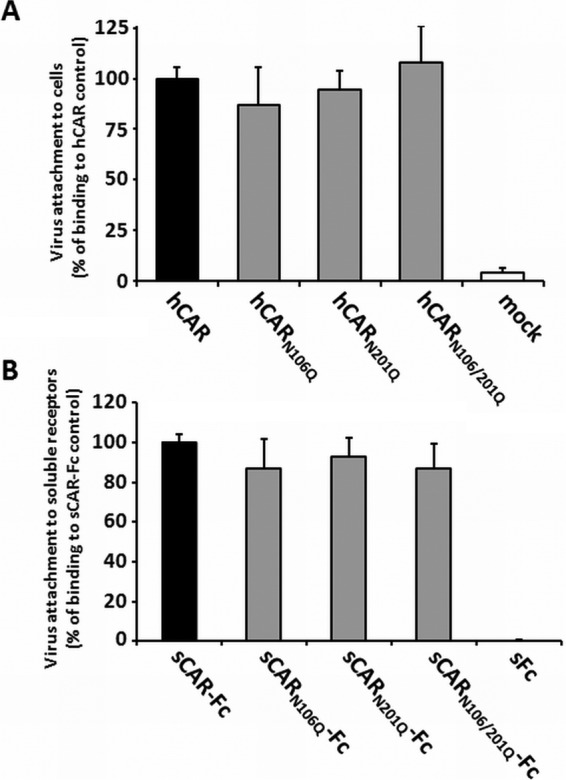
Loss of hCAR glycosylation has no influence on CVB3 attachment. (A) Virus binding to CHO-K1 cells transfected with hCAR glycosylation variants was determined as described in Materials and Methods. CHO-K1 cells exposed to transfection reagent without plasmid DNA (mock) served as a negative control. (B) Different glycosylated CAR variants were expressed as soluble receptors fused to human IgG-Fc, and equal amounts were bound to protein G-coated magnetic beads. Soluble Fc was used as negative control. Bead-coupled soluble receptor proteins were incubated with 35S-labeled CVB3 (10,000 cpm) for 1 h at 4°C. After washing, bound virus was measured by scintillation counting.
To measure infection, we incubated transfected CHO-K1 cells with virus (MOI of 5) and quantified viral replication by the detection of infectious viral particles by plaque assay at 0 and 24 h postinfection. As shown in Fig. 3A and in accordance with the previous binding data, deglycosylation of the D1 domain, the D2 domain, or both domains had no adverse affect on CVB3 infection and replication; in fact, the virus titers were somewhat higher in the cells expressing hCARN201Q, although not in the double deglycosylated CAR variant (hCARN106/201Q). These results were confirmed in an additional time-dependent infection experiment over a period of 48 h postinfection (Fig. 3B). To confirm by another approach that glycosylation was not required for infection, we used tunicamycin to inhibit glycosylation in CAR-transfected CHO-K1 cells. In cells treated with 0.25 μg of tunicamycin/ml, Western blotting revealed no glycosylated CAR (46 kDa) but only the 40-kDa unglycosylated protein (Fig. 3C); nonetheless, virus replicated to equal titers in tunicamycin-treated and control cells (Fig. 3D).
FIG 3.

Loss of hCAR glycosylation does not impair CVB3 replication. (A) Infection at an MOI of 5. CHO-K1 cells were transfected with different hCAR glycosylation variants, cultured for 48 h, incubated with CVB3 (5 PFU/cell) for 1 h at room temperature, washed twice to remove unbound virus, and incubated again for 0 h or 24 h at 37°C. Virus titers were determined by plaque assay. Mock-transfected cells (no DNA) were included as a control. (B) Time-dependent replication of CVB3 in CHO-K1 cells expressing hCAR or the various glycosylation variants. Transfected cells were exposed to CVB3 (MOI of 0.1), and virus titers were measured at 0, 24, and 48 h after infection by plaque assay. (C) Inhibition of glycosylation by tunicamycin. CHO-K1 cells were treated without or with tunicamycin (0.05 to 0.25 μg/ml) for 4 h before transfection with the hCAR expression plasmid, and treatment was continued for 36 h after transfection. A Western blot shows a dose-dependent reduction in the apparent molecular mass of hCAR from 46 kDa (mock treatment) to 40 kDa (predicted). Lane M, MagicMark XP Western protein standard. (D) The glycosylation of hCAR has no influence on viral infection and replication. CHO-K1 cell were transfected with hCAR expression plasmid and treated with or without tunicamycin at 0.25 μg/ml as described for panel C and then infected with CVB3 (MOI of 0.1) at 36 h p.t., and the virus titers were determined by plaque assay 24 h later. The results are shown as means and the SD for three independent experiments each performed in duplicate. (E) CAR-dependent infection in cells that also express DAF. Stably transfected CHO cells expressing hDAF (CHO-hDAF) were transfected with different hCAR glycosylation variants and infected with CVB3 (MOI of 1) 48 h later. Virus titers were determined by plaque assay at 0 and 24 h after infection. *, P < 0.05 (compared to hCAR control value).
Because DAF, in addition to CAR, functions as an attachment receptor for CVB1, -3, and -5, we examined the effect of CAR glycosylation in cells that also express DAF. CHO cells stably expressing DAF (CHO-DAF cells) were transfected with wild-type CAR or with glycosylation-deficient mutants and then assessed for susceptibility to infection by CVB3 (Fig. 3E). Although viral replication in these cells depended on the presence of CAR, the replication efficiency was higher in cells expressing both DAF and CAR than in cells expressing CAR alone (compare these results to those in Fig. 3A). The findings support the idea that virus attachment to DAF facilitates CAR-dependent virus infection. However, consistent with our previous observations, replication was independent of CAR glycosylation. In addition to DAF and CAR, a third molecule, HS, has been shown to serve as a receptor for a single CVB3 isolate, CVB3-PD (35). We found that soluble heparin had no effect on infection by the CVB3 Nancy strain used in our studies, whereas CVB3-PD was completely inhibited (data not shown). Taken together, these experiments indicate that glycosylation is not necessary for CAR localization to the cell surface and, further, that glycosylation of D1 and D2 is not important for CAR's function as a CVB3 receptor.
Deletion of the D2 domain reduces virus attachment but does not prevent infection by CVB3.
Like CVB3 (7), adenovirus binds directly to the CAR-D1 domain (8); nonetheless, the D2 domain has also been reported to be important for adenovirus binding and infection (31). To test the importance of the D2 domain for CAR function, we generated a D2 deletion mutant in which the D1 domain was joined directly to the TMD and ICD (Fig. 4A). As determined by flow cytometry, hCARD1 was expressed on the cell surface at levels approximately equal to those observed for hCAR (Table 1). However, immunofluorescence microscopy revealed that hCARD1, unlike wild-type hCAR, did not concentrate at intercellular junctions (Fig. 4B).
FIG 4.
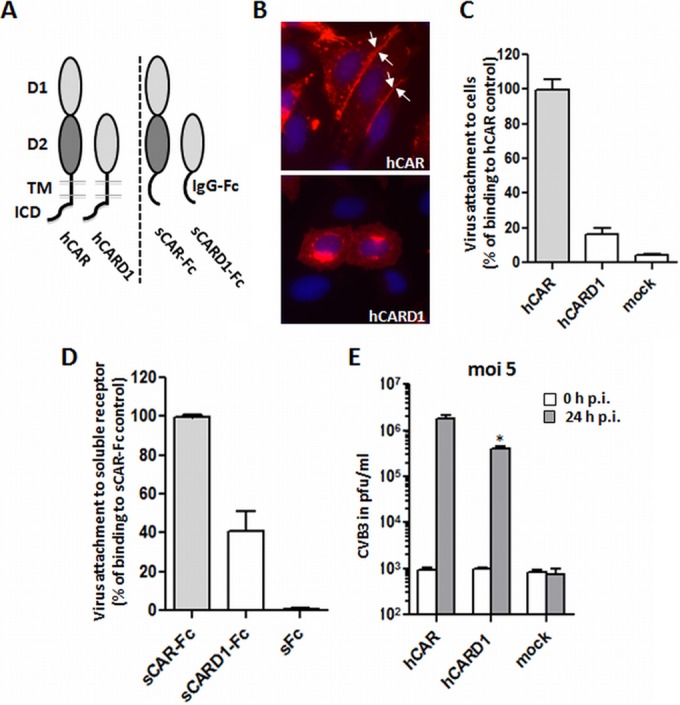
Deletion of CAR-D2 domain reduces, but does not prevent, virus attachment and infection. (A) Schematic illustration of wild-type hCAR and a D2 deletion mutant (hCARD1), expressed as cell surface receptors (left) and as dimeric soluble Fc fusion proteins (shown as monomers, right). hCAR consists of extracellular domains D1 and D2, a transmembrane domain (TMD), and an intracellular domain (ICD). In the deletion mutant hCARD1, the D1 domain is directly fused to the hCAR TMD. In the wild-type soluble fusion protein sCAR-Fc, the entire CAR ectodomain is fused to the immunoglobulin G Fc region; in the deletion mutant sCARD1-Fc, D1 alone is fused to Fc. (B) CAR localization in transfected CHO-K1 was analyzed by immunofluorescence using polyclonal anti-CAR antibodies. For the wild-type hCAR, the image was taken from Fig. 1. Wild-type hCAR was concentrated at cell-cell junctions; in contrast, hCARD1 showed little or no localization to junctions. (C) Binding of 35S-labeled CVB3 to CHO-K1 cells expressing hCAR or hCARD1, measured as described in Materials and Methods. The results are shown as means and the SD for three independent experiments. (D) Binding of 35S-labeled CVB3 to sCAR-Fc and the D2 deletion sCARD1-Fc mutant. Equal amounts of each soluble fusion protein (or sFc as a negative control) were bound to protein G-coupled magnetic beads, and virus binding was measured as described in Materials and Methods. (E) Viral replication in cells transfected with wild-type hCAR or hCARD1. Transfected CHO-K1 cells were incubated with CVB3 at an MOI of 5 for 1 h at room temperature, washed twice to remove unbound virus, and then incubated for 24 h at 37°C. Virus titers were determined by plaque assay at 0 and 24 h after infection. *, P < 0.05 (compared to hCAR control value).
Despite its expression at the cell surface, the deletion mutant hCARD1 bound radiolabeled CVB3 with distinctly less efficiency (16%) than did wild-type hCAR (Fig. 4C). This suggests either that the D2 domain contributes directly to binding or that the D2 domain facilitates virus binding to D1, possibly by permitting it to project farther from the cell membrane. We also examined binding to hCARD1 expressed as a fusion protein (Fig. 4D). Deletion of the D2 domain from sCAR-Fc reduced CVB3 binding (Fig. 4D), but the reduction was less marked than that observed with transfected cells (40% rather than 16%); one possible explanation for this is that the addition of the IgG Fc region renders the D1 domain more accessible to virus.
The expression of hCARD1 permitted infection of transfected CHO-K1 cells, as determined by the production of progeny virus at 24 h (Fig. 4E) and by the synthesis of intracellular viral antigens (not shown), but the viral titers were reduced by 80% compared to those obtained from cells expressing full-length hCAR. The reduction in virus replication was proportional to the 84% decrease we observed for virus binding (Fig. 4C). Taken together, these results indicate that hCARD1 without a D2 domain can function as a CVB3 receptor, although in an inefficient manner. Further, the results suggest that, although the D2 domain is needed for optimal virus binding, it is not essential for postattachment events in infection, such as virus internalization or uncoating.
Complete recovery of CAR function after substitution of the D2 domain by other IgSF-D2 domains.
We suspected that D2 might function simply as a spacer, permitting the virus-binding D1 domain of CAR to project above the cell surface. If this is the case, we expected that replacement of the D2 domain with other sequences—as long as they did not perturb the D1 structure—would restore the capacity of the receptor to bind virus and mediate infection. We therefore produced chimeras in which the hCAR-D2 domain was replaced by C2-type immunoglobulin-like domains from members of the IgSF such as CD80, CD155 (PVR), IgSF11, and chicken CAR (chCAR) (Fig. 5A). Among these domains, chCAR-D2 shows the greatest sequence homology to hCAR-D2 (45%), although chCAR does not function as a CVB3 receptor (36). IgSF11-D2 has 31% homology, CD155-D2 has 21% homology, and CD80-D2 has no sequence similarities to hCAR-D2.
FIG 5.

Complete recovery of CAR function after substitution of the D2 domain by other D2 domains of the IgSF. (A) Schematic illustration of various CAR proteins: left, wild-type hCAR and wild-type chCAR; center, chimeric chCAR/hCAR receptors containing the chCAR-D1 domain fused to the hCAR-D2 domain (chCAR-hCAR) or the hCAR-D1 domain fused to chCAR-D2 (hCAR-chCAR); and right, hCAR-D1 domains fused to the D2 domains of CD80, CD155, or IgSF11. (B) Localization of CAR chimeras expressed in CHO-K1 cells. Cells transfected with CAR chimeras were stained with polyclonal anti-CAR antibodies and examined by immunofluorescence microscopy. Typical localization to cell-cell junctions was observed for wild-type hCAR, the hCAR/chCAR chimeras, and hCAR-IgSF11 (white arrows), whereas the other substitution mutants (hCAR-CD80 and hCAR-CD155) showed little or no localization to the junctions. (C) Binding of with 35S-labeled CVB3 to CHO-K1 cells expressing wild-type CAR or chimeric receptors, measured as described in Materials and Methods. (D) Viral replication in cells expressing chimeric receptors (MOI of 5). Transfected CHO-K1 cells were incubated with CVB3 for 1 h at room temperature, washed twice to remove unbound virus, and then incubated at 37°C. Viral titers were determined by plaque assay at 0 and 24 h. (E) CHO-K1 cells stably expressing hDAF (CHO-hDAF) were transfected with wild-type or chimeric hCAR receptors (or mock transfected). At 48 h after transfection, the cells were exposed to CVB3 at the indicated MOIs, washed, and incubated at 37°C for 24 h. Virus titers were determined by plaque assay. The results are shown as means and the SD for triplicate samples. ns, not significant; *, P < 0.05; **, P < 0.01 (compared to hCAR control value).
FACS analysis confirmed that all the chimeric receptor constructs were expressed in similar amounts on the surface of transfected CHO-K1 cells (Table 1). Interestingly, although all of the chimeras were recognized by a polyclonal anti-CAR antibody, the monoclonal antibody RmcB did not recognize hCAR chimeras bearing the D2 domains of CD80, CD155, or IgSF11; nor did RmcB recognize a chimera in which the hCAR-D1 domain was replaced by chCAR-D1 (data not shown). Only wild-type hCAR and the hCAR-chCAR chimera in which the hCAR-D2 domain was replaced with chCAR-D2 were detected by both antibodies. Immunofluorescence revealed differences in localization of the chimeric CAR variants in transfected CHO-K1 cells. Like hCARD1, the hCAR-CD80 and hCAR-CD155 chimeras showed a diffuse distribution with little or no accumulation at cell-cell contact sites. In contrast, hCAR-IgSF11 and hCAR-chCAR, as well as chCAR-hCAR, localized to cell-cell junctions (Fig. 5B, white arrows).
We then examined binding of radiolabeled CVB3 to transiently transfected CHO-K1 expressing the different CAR chimeras. In general, virus bound more efficiently to constructs including any of the D2 domains (Fig. 5C) than to D2-deleted hCAR (hCARD1) (Fig. 4C). CVB3 binding to hCAR-IgSF11 and hCAR-chCAR was as efficient as virus binding to wild-type hCAR itself, whereas binding to hCAR-CD80 and hCAR-CD155 was moderately less efficient, with decreases of about 53% (hCAR-CD80) and 15% (hCAR-CD155), respectively, compared to hCAR. The results demonstrate that, with regard to virus binding, at least some heterologous D2 domains can completely replace the function of hCARD2.
To determine whether heterologous domains could restore the full capacity of hCARD1 to mediate CVB3 infection, we measured virus replication in transfected cells (Fig. 5D). The virus titers in CHO-K1 cells expressing hCAR-chCAR and hCAR-IgSF11 24 h after CVB3 infection were the same as those in cells expressing wild-type hCAR. This indicates that these chimeric proteins were as active as wild-type CAR in mediating virus infection. In contrast, CVB3 infection was distinctly reduced in cells expressing hCAR-CD80 or hCAR-CD155. Interestingly, although CVB3 binding to hCAR-CD155-expressing cells was more efficient (85%) than binding to hCAR-CD80-expressing cells (47%) (Fig. 5C), the virus titers were nearly 10-fold higher in cells expressing hCAR-CD80 than in cells expressing hCAR-CD155 (Fig. 5D). The coexpression of hDAF (CHO-DAF cells) as an alternative attachment receptor increased viral replication, but the relative efficacies of the different CAR chimeras did not change in the presence of DAF (Fig. 5E). Thus, although DAF may enhance viral attachment, it does not compensate for decreases in CAR function that occur when the D2 domain is eliminated.
Although hCAR-chCAR appeared to function as efficiently as wild-type hCAR, we observed neither virus binding (Fig. 5C) nor viral replication in CHO-K1 cells transfected with chCAR and chCAR-hCAR (Fig. 5D and E). This confirms the previously described inability of chCAR to mediate CVB3 infection (36) and is consistent with strong evidence that D1 is directly involved in virus binding (7).
Taken together, these results with chimeras demonstrate that the hCAR-D2 domain is not per se necessary for CVB3 binding or infection. Receptor function is reduced after deletion of the hCAR-D2 domain, but it can be fully restored by substitution with homologous or heterologous C2 type immunoglobulin-like domains of proteins from other IgSF members.
DISCUSSION
Many picornaviruses use receptor proteins that belong to the IgSF (1, 13, 26, 37). In each case, the virus binding site is located within the amino-terminal D1 domain of the receptor (7, 13, 26). Earlier work demonstrated that the CAR transmembrane and cytoplasmic domains are not required for CVB3 infection (7, 27), but the role of D2 had not been investigated. Further, although extracellular glycosylation is known to be important for other CAR-mediated functions, including homotypic adhesion and adenovirus infection (38), the role of glycosylation in CVB3 infection had not been examined. We found here that glycosylation of the extracellular portion of CAR does not contribute to CVB3 infection. Further, we found that, although the CAR-D2 domain is not specifically required, the presence of either CAR-D2 or D2 domains from some other IgSF members facilitated both virus binding and infection. We believe that CAR-D2 acts primarily as a spacer, enhancing virus access to the D1 domain—either because a longer stalk allows D1 to project beyond the cellular glycocalyx or because it facilitates virus interaction with more than one receptor at a time. Although D2 appears to facilitate virus attachment, the data suggest that it has no important role in postattachment events such as internalization and uncoating.
It is important to note that our experiments were all performed under conditions in which virions, which are multivalent, may have the opportunity to interact simultaneously with multiple CAR molecules, either at the cell surface or immobilized on beads. We did not attempt to measure the affinity of a single receptor molecule with a single binding site on the virion's surface, and our experiments do not exclude the possibility that the specific binding affinities (as opposed to the effective avidities) of the mutated receptors we studied might differ from those of wild-type CAR. Nonetheless, the results indicate that—under the conditions in which CVB3 naturally encounters receptors at the cell surface—neither deglycosylated CAR nor CAR in which D2 had been replaced by some other immunoglobulin-like domains differed from wild-type CAR in their capacity to mediate virus attachment and infection.
Studies of other picornavirus receptors have indicated variable roles for glycosylation in receptor function. For example, glycosylation of havcr-1 is needed to maintain the required conformation for binding and uptake of HAV. In contrast, glycosylation appears to inhibit PV binding to PVR (16, 26). CAR has two glycosylation sites, one on each extracellular domain (38). He et al. (7) observed that a CAR-D1 domain, produced in bacteria and therefore lacking glycosylation, bound poorly to CVB3 (7), and other authors (38) have suggested that this might indicate a role for glycosylation. However, we found that deglycosylation of the D1 and/or D2 domains of CAR had no adverse effect on CVB3 binding or infection. If anything, infection was more efficient in the absence of D1 glycosylation.
Based on structural evidence (7), CVB3 binds to the CAR-D1 domain, but it seems that additional domains of IgSF receptors may be important for infection by other picornaviruses. A soluble ICAM-1 variant consisting only of the D1 and D2 domains inhibits HRV3 infection less effectively than a soluble ICAM with all five domains (39), and binding of HRV14 to membrane-bound ICAM-1 is decreased after the deletion of D3, D4, and D5 (40). We found that CVB3 bound less efficiently when CAR-D2 was deleted and that binding was partially or even fully restored when CAR-D2 was replaced by domains from some other members of the IgSF. This suggests that D2 serves in large part as a spacer, permitting the optimal exposure of CAR-D1 to the virus at the cell surface. Our results are consistent with earlier studies of the PVR and ICAM-1, which demonstrated that D1 was sufficient for receptor function, although the efficiency of binding and infection was increased by the addition of some, but not all, heterologous immunoglobulin-like domains (18–20, 41).
We found that only the D2 domains from IgSF11 and chCAR—the two domains most similar to hCAR-D2—restored binding to wild-type levels. Although we cannot exclude the possibility that some shared D2 sequences participate in virus binding, the sequence identity among these domains is quite low (31% for CAR and IgSF11), and most of the identical residues are remote from D1 in the available crystal structure (data not shown) (10). We believe it more likely that replacement of CAR-D2 with D2 domains of CD55 or CD80 affected the structure or orientation of CAR-D1. In this regard, it is interesting that chimeras with these D2 domains were not localized to cell-cell contacts, perhaps because they interfered with D1-D1 homophilic interactions. An influence of CAR-D2 on proper D1 conformation may also be suggested by the results of He et al. (7), who reported that an unglycosylated CAR-D1 domain fragment bound poorly to CVB3, whereas a glycosylated D1/D2 fragment bound much better. In the light of our data, which show that glycosylation itself is not important for CAR binding to CVB3, the data obtained by He et al. may support the idea that D2 promotes virus binding by influencing the conformation of D1.
The results reported here add to our understanding of how CAR's structure contributes to its function as a coxsackievirus receptor. They may also impact the application of the virus receptor trap sCAR-Fc as an antiviral treatment. The antiviral action of sCAR-Fc against CVB infection in vitro and in vivo has been extensively examined by us and by other groups (32, 42–44). Large-scale production of sCAR-Fc in animal cells is inefficient and costly, and the production of glycosylated sCAR-Fc is a limiting factor for clinical applications. Although protein production in prokaryotes is quite efficient, prokaryotes do not perform many important posttranslational modifications, including N-linked glycosylation (23, 45). Our observation that glycosylation is not required for CAR interaction with CVB3 opens the possibility of large-scale soluble receptor production in prokaryotes, an important step toward using soluble CAR proteins as therapeutics in CVB infections.
ACKNOWLEDGMENTS
We thank Fritz G. Rathjen for kindly providing the chCAR cDNA plasmid.
This study was supported by the Deutsche Forschungsgemeinschaft through personal research grant PI 1125/1-1 to S.P. and through project grand Fe785/2-2 to H.F., by National Institutes of Health grant R01 AI52281 to J.B., and by the Plotkin Endowed Chair at the Children's Hospital of Philadelphia.
REFERENCES
- 1.Bergelson JM, Cunningham JA, Droguett G, Kurt-Jones EA, Krithivas A, Hong JS, Horwitz MS, Crowell RL, Finberg RW. 1997. Isolation of a common receptor for coxsackie B viruses and adenoviruses 2 and 5. Science 275:1320–1323. doi: 10.1126/science.275.5304.1320. [DOI] [PubMed] [Google Scholar]
- 2.Tomko RP, Xu R, Philipson L. 1997. HCAR and MCAR: the human and mouse cellular receptors for subgroup C adenoviruses and group B coxsackieviruses. Proc Natl Acad Sci U S A 94:3352–3356. doi: 10.1073/pnas.94.7.3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shafren DR, Bates RC, Agrez MV, Herd RL, Burns GF, Barry RD. 1995. Coxsackieviruses B1, B3, and B5 use decay accelerating factor as a receptor for cell attachment. J Virol 69:3873–3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carson SD. 2001. Receptor for the group B coxsackieviruses and adenoviruses: CAR. Rev Med Virol 11:219–226. doi: 10.1002/rmv.318. [DOI] [PubMed] [Google Scholar]
- 5.Jiang S, Caffrey M. 2007. Solution structure of the coxsackievirus and adenovirus receptor domain 2. Protein Sci 16:539–542. doi: 10.1110/ps.062643507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang S, Jacobs A, Laue TM, Caffrey M. 2004. Solution structure of the coxsackievirus and adenovirus receptor domain 1. Biochemistry 43:1847–1853. doi: 10.1021/bi035490x. [DOI] [PubMed] [Google Scholar]
- 7.He Y, Chipman PR, Howitt J, Bator CM, Whitt MA, Baker TS, Kuhn RJ, Anderson CW, Freimuth P, Rossmann MG. 2001. Interaction of coxsackievirus B3 with the full-length coxsackievirus-adenovirus receptor. Nat Struct Biol 8:874–878. doi: 10.1038/nsb1001-874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bewley MC. 1999. Structural analysis of the mechanism of adenovirus binding to its human cellular receptor, CAR. Science 286:1579–1583. doi: 10.1126/science.286.5444.1579. [DOI] [PubMed] [Google Scholar]
- 9.van Raaij MJ, Chouin E, van der Zandt H, Bergelson JM, Cusack S. 2000. Dimeric structure of the coxsackievirus and adenovirus receptor D1 domain at 1.7 Å resolution. Structure 8:1147–1155. doi: 10.1016/S0969-2126(00)00528-1. [DOI] [PubMed] [Google Scholar]
- 10.Patzke C, Max KEA, Behlke J, Schreiber J, Schmidt H, Dorner AA, Kröger S, Henning M, Otto A, Heinemann U, Rathjen FG. 2010. The coxsackievirus-adenovirus receptor reveals complex homophilic and heterophilic interactions on neural cells. J Neurosci 30:2897–2910. doi: 10.1523/JNEUROSCI.5725-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coyne CB, Bergelson JM. 2005. CAR: a virus receptor within the tight junction. Adv Drug Deliv Rev 57:869–882. doi: 10.1016/j.addr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 12.Rossmann MG, He Y, Kuhn RJ. 2002. Picornavirus-receptor interactions. Trends Microbiol 10:324–331. doi: 10.1016/S0966-842X(02)02383-1. [DOI] [PubMed] [Google Scholar]
- 13.Koike S, Ise I, Nomoto A. 1991. Functional domains of the poliovirus receptor. Proc Natl Acad Sci U S A 88:4104–4108. doi: 10.1073/pnas.88.10.4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rossmann MG. 1994. Viral cell recognition and entry. Protein Sci 3:1712–1725. doi: 10.1002/pro.5560031010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Silberstein E, Xing L, van de Beek W, Lu J, Cheng H, Kaplan GG. 2003. Alteration of hepatitis A virus (HAV) particles by a soluble form of HAV cellular receptor 1 containing the immunoglobulin-and mucin-like regions. J Virol 77:8765–8774. doi: 10.1128/JVI.77.16.8765-8774.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bernhardt G, Harber J, Zibert A, deCrombrugghe M, Wimmer E. 1994. The poliovirus receptor: identification of domains and amino acid residues critical for virus binding. Virology 203:344–356. doi: 10.1006/viro.1994.1493. [DOI] [PubMed] [Google Scholar]
- 17.Lineberger DW, Uncapher CR, Graham DJ, Colonno RJ. 1992. Domains 1 and 2 of ICAM-1 are sufficient to bind human rhinoviruses. Virus Res 24:173–186. doi: 10.1016/0168-1702(92)90005-T. [DOI] [PubMed] [Google Scholar]
- 18.Morrison ME, Racaniello VR. 1992. Molecular cloning and expression of a murine homolog of the human poliovirus receptor gene. J Virol 66:2807–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McClelland A, deBear J, Yost SC, Meyer AM, Marlor CW, Greve JM. 1991. Identification of monoclonal antibody epitopes and critical residues for rhinovirus binding in domain 1 of intercellular adhesion molecule 1. Proc Natl Acad Sci U S A 88:7993–7997. doi: 10.1073/pnas.88.18.7993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Selinka HC, Zibert A, Wimmer E. 1992. A chimeric poliovirus/CD4 receptor confers susceptibility to poliovirus on mouse cells. J Virol 66:2523–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roelvink PW, Lizonova A, Lee JG, Li Y, Bergelson JM, Finberg RW, Brough DE, Kovesdi I, Wickham TJ. 1998. The coxsackievirus-adenovirus receptor protein can function as a cellular attachment protein for adenovirus serotypes from subgroups A, C, D, E, and F. J Virol 72:7909–7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mitra N, Sinha S, Ramya TNC, Surolia A. 2006. N-linked oligosaccharides as outfitters for glycoprotein folding, form, and function. Trends Biochem Sci 31:156–163. doi: 10.1016/j.tibs.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 23.Ohtsubo K, Marth JD. 2006. Glycosylation in cellular mechanisms of health and disease. Cell 126:855–867. doi: 10.1016/j.cell.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 24.Jiménez D, Roda-Navarro P, Springer TA, Casasnovas JM. 2005. Contribution of N-linked glycans to the conformation and function of intercellular adhesion molecules (ICAMs). J Biol Chem 280:5854–5861. doi: 10.1074/jbc.M412104200. [DOI] [PubMed] [Google Scholar]
- 25.Zibert A, Wimmer E. 1992. N glycosylation of the virus binding domain is not essential for function of the human poliovirus receptor. J Virol 66:7368–7373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thompson P, Lu J, Kaplan GG. 1998. The Cys-rich region of hepatitis A virus cellular receptor 1 is required for binding of hepatitis A virus and protective monoclonal antibody 190/4. J Virol 72:3751–3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang X, Bergelson JM. 1999. Coxsackievirus and adenovirus receptor cytoplasmic and transmembrane domains are not essential for coxsackievirus and adenovirus infection. J Virol 73:2559–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Milstone AM, Petrella J, Sanchez MD, Mahmud M, Whitbeck JC, Bergelson JM. 2005. Interaction with coxsackievirus and adenovirus receptor, but not with decay-accelerating factor (DAF), induces A-particle formation in a DAF-binding coxsackievirus B3 isolate. J Virol 79:655–660. doi: 10.1128/JVI.79.1.655-660.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fechner H, Wang X, Wang H, Jansen A, Pauschinger M, Scherübl H, Bergelson JM, Schultheiss HP, Poller W. 2000. Trans-complementation of vector replication versus Coxsackie-adenovirus-receptor overexpression to improve transgene expression in poorly permissive cancer cells. Gene Ther 7:1954–1968. doi: 10.1038/sj.gt.3301321. [DOI] [PubMed] [Google Scholar]
- 30.Fechner H, Sipo I, Westermann D, Pinkert S, Wang X, Suckau L, Kurreck J, Zeichhardt H, Müller O, Vetter R, Erdmann V, Tschope C, Poller W. 2008. Cardiac-targeted RNA interference mediated by an AAV9 vector improves cardiac function in coxsackievirus B3 cardiomyopathy. J Mol Med 86:987–997. doi: 10.1007/s00109-008-0363-x. [DOI] [PubMed] [Google Scholar]
- 31.Excoffon KJD, Traver GL, Zabner J. 2005. The role of the extracellular domain in the biology of the coxsackievirus and adenovirus receptor. Am J Respir Cell Mol Biol 32:498–503. doi: 10.1165/rcmb.2005-0031OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pinkert S, Westermann D, Wang X, Klingel K, Dörner A, Savvatis K, Grössl T, Krohn S, Tschöpe C, Zeichhardt H, Kotsch K, Weitmann K, Hoffmann W, Schultheiss H-P, Spiller OB, Poller W, Fechner H. 2009. Prevention of cardiac dysfunction in acute coxsackievirus B3 cardiomyopathy by inducible expression of a soluble coxsackievirus-adenovirus receptor. Circulation 120:2358–2366. doi: 10.1161/CIRCULATIONAHA.108.845339. [DOI] [PubMed] [Google Scholar]
- 33.Cohen CJ, Xiang ZQ, Gao G-P, Ertl HCJ, Wilson JM, Bergelson JM. 2002. Chimpanzee adenovirus CV-68 adapted as a gene delivery vector interacts with the coxsackievirus and adenovirus receptor. J Gen Virol 83:151–155. doi: 10.1099/0022-1317-83-1-151. [DOI] [PubMed] [Google Scholar]
- 34.Freimuth P, Philipson L, Carson SD. 2008. The coxsackievirus and adenovirus receptor. Curr Top Microbiol Immunol 323:67–87. [DOI] [PubMed] [Google Scholar]
- 35.Zautner AE, Jahn B, Hammerschmidt E, Wutzler P, Schmidtke M. 2006. N- and 6-O-sulfated heparan sulfates mediate internalization of coxsackievirus B3 variant PD into CHO-K1 cells. J Virol 80:6629–6636. doi: 10.1128/JVI.01988-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Freiberg F, Sauter M, Pinkert S, Govindarajan T, Kaldrack J, Thakkar M, Fechner H, Klingel K, Gotthardt M. 2014. Interspecies differences in virus uptake versus cardiac function of the coxsackievirus and adenovirus receptor (CAR). J Virol 88:7345–7356. doi: 10.1128/JVI.00104-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Register RB, Uncapher CR, Naylor AM, Lineberger DW, Colonno RJ. 1991. Human-murine chimeras of ICAM-1 identify amino acid residues critical for rhinovirus and antibody binding. J Virol 65:6589–6596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Excoffon KJDA, Gansemer N, Traver G, Zabner J. 2007. Functional effects of coxsackievirus and adenovirus receptor glycosylation on homophilic adhesion and adenoviral infection. J Virol 81:5573–5578. doi: 10.1128/JVI.02562-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Greve JM, Forte CP, Marlor CW, Meyer AM, Hoover-Litty H, Wunderlich D, McClelland A. 1991. Mechanisms of receptor-mediated rhinovirus neutralization defined by two soluble forms of ICAM-1. J Virol 65:6015–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Staunton DE, Dustin ML, Erickson HP, Springer TA. 1990. The arrangement of the immunoglobulin-like domains of ICAM-1 and the binding sites for LFA-1 and rhinovirus. Cell 61:243–254. doi: 10.1016/0092-8674(90)90805-O. [DOI] [PubMed] [Google Scholar]
- 41.Selinka HC, Zibert A, Wimmer E. 1991. Poliovirus can enter and infect mammalian cells by way of an intercellular adhesion molecule 1 pathway. Proc Natl Acad Sci U S A 88:3598–3602. doi: 10.1073/pnas.88.9.3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yanagawa B, Spiller OB, Proctor DG, Choy J, Luo H, Zhang HM, Suarez A, Yang D, McManus BM. 2004. Soluble recombinant coxsackievirus and adenovirus receptor abrogates coxsackievirus b3-mediated pancreatitis and myocarditis in mice. J Infect Dis 189:1431–1439. doi: 10.1086/382598. [DOI] [PubMed] [Google Scholar]
- 43.Lim B, Choi J, Nam J, Gil C, Shin J. 2006. Virus receptor trap neutralizes coxsackievirus in experimental murine viral myocarditis. Cell 71:517–526. [DOI] [PubMed] [Google Scholar]
- 44.Werk D, Pinkert S, Heim A, Zeichhardt H, Grunert H-P, Poller W, Erdmann VA, Fechner H, Kurreck J. 2009. Combination of soluble coxsackievirus-adenovirus receptor and anti-coxsackievirus siRNAs exerts synergistic antiviral activity against coxsackievirus B3. Antiviral Res 83:298–306. doi: 10.1016/j.antiviral.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 45.Gopal GJ, Kumar A. 2013. Strategies for the production of recombinant protein in Escherichia coli. Protein J 32:419–425. doi: 10.1007/s10930-013-9502-5. [DOI] [PubMed] [Google Scholar]