

Abstract
A 40-year-old Japanese man who had a medical history of hypokalemic periodic paralysis 4 months prior was hospitalized to undergo a cholecystectomy. Hypokalemia, nephrocalcinosis and alkaluria suggesting distal renal tubular acidosis (dRTA) were detected, but metabolic acidosis was not evident. An ammonium chloride/furosemide–fludrocortisone/bicarbonate loading test demonstrated a remarkable disability in urinary H+ excretion. A novel heterozygous mutation in the ATP6V0A4 gene encoding the vacuolar H+-ATPase (V-ATPase) a4 subunit p.S544L was detected. Among cases of V-ATPase a4 mutations, this is the first case in which a heterozygous mutation developed to an incomplete or latent form of dRTA.
Keywords: ATP6V0A4, distal renal tubular acidosis (dRTA), heterozygous mutation, V-ATPase a4 subunit
Introduction
Distal renal tubular acidosis (dRTA) is characterized by an inability to secrete H+ in α-intercalated cells. The clinical manifestations of the disease consist of hyperchloremic metabolic acidosis, hypokalemia, hypercalciuria, hypocitraturia, urinary concentrating ability defect, recurrent urinary stone formation, nephrocalcinosis and bone demineralization. With regard to the genetic cause of dRTA, several currently known mutations have been identified in α-intercalated cells, including anion exchanger 1 (AE1), the B1 or a4 subunits of vacuolar H+-ATPase (V-ATPase) and cytosolic carbonic anhydrase 2 (CA2) [1–3]. In cases of dRTA caused by V-ATPase mutations, the autosomal recessive form that manifests the disease in infancy is common [1].
We herein describe an adult patient with incomplete dRTA caused by a novel heterogeneous mutation of V-ATPase a4. Among cases of dRTA due to V-ATPase a4 mutations, this is the first case that developed to an incomplete or late-onset form of the disease.
Case report
A 40-year-old Japanese man was admitted to our hospital to undergo a laparoscopic cholecystectomy. An unknown hypokalemia [potassium (K) 2.7 mmol/L] was found and he was referred to our department presurgery. He had been hospitalized in another hospital for periodic paralysis due to unknown severe hypokalemia (K 1.7 mmol/L) 4 months prior. He and his father, who had died of a stroke, had been diagnosed with nephrocalcinosis in their 20s. None of his three siblings had any medical history. His physical and laboratory findings were as follows: height, 182 cm; weight, 81.5 kg; blood pressure, 120/78 mmHg. No abnormalities were observed in a physical examination. His audibility test was normal. In a blood examination, his serum creatinine level was elevated at 1.44 mg/dL (127.3 μmol/L). His serum K level was low, at 2.7 mmol/L, while his sodium (Na), chlorine (Cl), calcium (Ca), phosphorus (P) and magnesium (Mg) levels were all normal, at 144 mmol/L, 108 mmol/L, 8.7 mg/dL, 2.7 mg/dL and 2.6 mg/dL, respectively. The venous bicarbonate () level was at the lower limit of the normal range, at 22.8 mmol/L. His blood anion gap was normal at 13.2. His complete blood counts were all normal. The levels of immunoglobulins and complements were all within normal limits, and he tested negative for antinuclear, anti-SS-A and anti-SS-B antibodies. The plasma renin activity (normal: 0.1–2) and plasma aldosterone concentration (normal: 35.7–240) were 0.7 ng/mL/h and 141.1 pg/mL, respectively. The spot urinary pH was 7.5 and the fractional excretion of K was inappropriately normal, at 10.3%, even in a hypokalemic state. The urinary Ca/creatinine ratio (mol) was 0.70. The urinary anion gap was positive at 30.9 (=Na 67 mmol/L + K 13.9 mmol/L – Cl [minus sign] 50 mmol/L). Abdominal CT revealed medullary nephrocalcinosis in both kidneys. A bone mineral density test showed normal levels. We suspected dRTA and started a prescription of potassium aspartate and sodium/potassium citrate during the perioperative period. The level of serum K was corrected to within the normal range at 3.6 mmol/L. Black-pigmented stones in the patient's gallbladder were detected after a successful laparoscopic cholecystectomy.
After suspension of the prescription for 7 days, the hypokalemia became advanced (K 3.0 mmol/L), while metabolic acidosis was not evident ( 29.3 mmol/L). We performed ammonium chloride (NH4Cl), furosemide–fludrocortisone and HCO3− loading tests (Figures 1–3 ). The NH4Cl loading test demonstrated that the patient had dysfunction in both urinary acidification and net acid excretion under the acid loading condition (Figure 1A–C). In the furosemide–fludrocortisone loading test, the potential to secrete H+ into the lumina at distal nephrons with an enhanced negative charge was extremely weak (Figure 2A). In this test, instead of H+, the patient's K secretion increased sharply compared with a healthy control (Figure 2B). In the bicarbonate loading test, the patient's basal H+ secretion dysfunction was also demonstrated (Figure 3A); conversely, the tubular maximal reabsorption rate of () was elevated compared with the control (Figure 3B).
Fig. 1.

Results of the NH4Cl loading test. In the NH4Cl loading test, 2 mg/kg per dose of NH4Cl in powder form was orally administered to the patient. Urine samples were immediately sealed with liquid paraffin to minimize the ammonia gas loss and pH change. The urinary pH was immediately measured using a glass electrode pH meter and the urinary excretion of both H+ (E-H+) and were defined as follows: E-H+: urinary : urinary . Urinary H+ and urinary were calculated by the following formula: urinary H+ (umol/L) = 103× 10−UPH, UpH = urinary pH; urinary = (measured-Uosm (osm/L) − uGlu/18 − uUN/2.8)/2 − (uNa+ uK), uGlu = urinary glucose (mg/dL), uUN = urinary urea nitrogen (mg/dL), uNa = urinary Na (mmol/L), uK = urinary K (mmol/L). The tests were also performed on an age- and sex-matched healthy volunteer as a control in the same way. (A) The patient's UpH was much higher than that of the control. After loading, in contrast to that of the control, urinary acidification was not evident in the patient. (B) To correct any influences derived from a difference in the urinary dilution/concentration between the two, E-H+, which denotes the relative amount of isolated H+, was calculated. The differences in the E-H+ level and in its change after loading were more prominent than those for UpH. The ratio of the patient's E-H+ to that of the control during the test was in the range of 0.023–0.062. (C) mainly reflects the net acid excretion in the urine. The patient's was smaller to that of the control. The ratio of the patient's to that of the control during the test was in the range of 0.37–0.67. Note that the patient/control ratio of was more than 10 times larger than that of E-H+.
Fig. 3.

Results of the HCO3− loading test. In the HCO3− loading test, 0.08 g/kg per dose of in powder form was orally administered to the patient five times hourly during the test. Urine samples were immediately sealed with liquid paraffin to minimize the carbon dioxide (CO2) gas loss and pH change. All blood and urine samples was immediately measured using a gas analyzer or a glass electrode pH meter. (U-B) CO2 and net excretion were calculated by the following formula: (U-B) CO2 (mmHg) = uCO2 – bCO2 [uCO2= urinary CO2 (mmHg), bCO2= blood CO2 (mmHg)]; (mmol/100 mL/GFR) = 0.1 × (+ uCO2× 0.0301) × sCr/uCr (GFR = glomerular filtration rate); = 0.0301 × uCO2 × 10UpH–UPK′, UPK′ = 6.33–0.5 × √(uNa/1000 + uK/1000 + ) [sCr = serum creatinine (mg/dL), uCr = urinary creatinine (mg/dL)]. (A) (U-B) CO2 denotes the states of basal H+ secretion in distal nephrons. In the control, an increase of the (U-B) CO2 level, which indicates a basal state of H+ secretion accompanied by a dose-dependent accumulation of filtrate overflowing into the lumens of distal nephrons, was observed in the first half period of the test, while the levels of (U-B) CO2 were kept down, which was assumed to be an ‘adaptive change’ (i.e. suppression of H+ secretion) against advanced metabolic alkalosis in the second half period. In the patient, however, (U-B) CO2 remained almost unchanged at extremely low levels during the test. (B) The level of rose abruptly when the plasma level exceeded . In the test, the level of the patient's was relatively high, at approximately 32 mmol/L, compared with that of the control (28 mmol/L). Under a level of lower than that of , the patient's was slightly detected (at approximately 1 mEq/min/GFR) while that of the control was not detected at all. The reason for this is explained by the difference in the acid/alkaline state of the urine samples between the two subjects. In an acid state, is easily converted to CO2 gas and lost from the samples, while this does not occur in an alkaline state.
Fig. 2.

Results of the furosemide–fludrocortisone loading test. In the furosemide–fludrocortisone loading test, 40 mg of furosemide and 1 mg of fludrocortisone per dose were orally administered to the patient. UpH was immediately measured using a glass electrode pH meter and E-H+ and E-K (defined as urinary K/g·Cr) were calculated. (A) In both the patient and the control, the level of E-H+ reached its peak at 3 h after loading and then decreased. The level of the patient's E-H+ during the test was extremely low compared with that of the control. (B) The patient's E-K transiently decreased at 1 h after loading, but then increased sharply compared with that of the control. Both the maximum and final level of the patient's E-K during the test were higher than that of the control.
We performed comprehensive genetic testing for known causal genes of RTA in the patient using next-generation sequencing (NGS) technology based on the capturing method (Agilent Technologies). The genes SLC4A1 (encoding AE1), ATP6V1B1, ATP6V0A1, ATP6V0A4 (encoding the B1, a1 and a4 subunits of V-ATPase, respectively), SLC4A4 (encoding NBCe1-A), SLC34A1 (encoding NaPi-2a) and CA2 were all examined simultaneously. The detected mutations were narrowed down to those that might be disease-causing mutations according to the following two filters: one for novel variants that have not been reported previously or for variants with minor allele frequencies (MAFs) of up to 1% in the four single nucleotide polymorphism (SNP) databases [Human Genetic Variation Database (Japanese) [4], 1000 Genomes [5], ESP6500 [6], Exome Aggregation Consortium [7]] and a second one for variants except for synonymous mutations if the variants are in the coding region. Consequently, only one mutation, which is a novel heterozygous mutation in ATP6V0A4 (exon 15, c.C1631T, p.S544L) was detected. As shown in Figure 4, the detected variants were validated with conventional Sanger sequencing and the result was consistent with that of the NGS diagnosis. Moreover, to confirm whether a large structural variant such as a large deletion or insertion exists as the second mutation, we performed the copy number variation (CNV) analysis for all the targeted genes by using CONTRA (Supplementary Ref S1). Briefly, this is CNV detection software using NGS data, and it calls copy number gains and losses for each target region by comparing the normalized sequencing depth from control samples. We used the sequence data from seven individuals as controls and did not find any large deletions or insertions in the patient. The results of CNV detection extracting the ATP6V0A4 region are shown in Supplementary Table S1. There were no regions where relative sequencing depth was significantly changed compared with the controls, indicating that this patient did not have large deletions in ATP6V0A4, thus he was believed to be a heterozygous carrier of the ATP6V0A4 mutation. In addition, it was confirmed at the same time that other tested genes (SLC4A1, SLC4A4, ATPV1B1, ATPV0A1, etc.) did not have any CNVs.
Fig. 4.
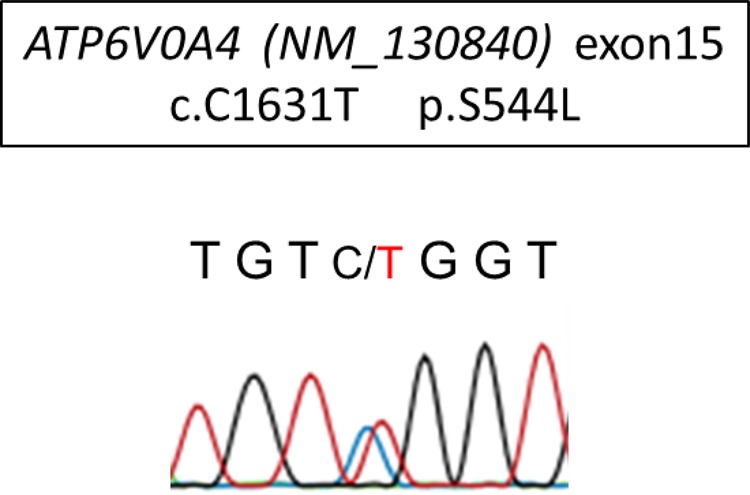
Results of Sanger sequencing of the identified mutation in ATP6V0A4. The mutation C1631T located on exon15 was consistent with that of multiplex genetic testing.
Discussion
As for the genetic causes of dRTA, the culprit protein can be detected in AE1, V-ATPase or CA2, expressed in α-intercalated cells. The late-onset or incomplete/mild type of dRTA is common in autosomal dominant, heterozygous mutations of AE1 [1–3]. The clinical manifestation in the present case resembled cases caused by heterozygous AE1 mutations responsible for the following attributes: late onset, subclinical metabolic acidosis and complicated gallbladder stones. In dRTA from mutant AE1, an aberrant AE1 expressed on the cell membranes of erythrocytes can be a cause of hemolysis and gallbladder stones [1]; however, the relationship between dRTA and gallbladder stones in the present case remains unclear because neither hemolysis nor AE1 mutations were evident.
In cases of dRTA caused by mutant V-ATPase in either the B1 or a4 subunit, >40 gene mutations encoding each of those subunits have been reported to date [8–12]. The B1 subunit consists of a cytoplasmic V1 domain that hydrolyzes ATP, while the a4 transmembrane V0 domain translocates H+ [13]. Cases of dRTA caused by mutant B1 have been recognized to develop deafness early in the disease course, while those caused by mutant a4 do not develop deafness or exhibit deafness with a highly variable phenotype range [13–16]. In relation to the critical form of dRTA, hardly any difference between B1 and a4 in clinical manifestation was observed. However, according to a report from Turkey, polyuria and nephrocalcinosis were more common in dRTA patients with deafness compared with patients without deafness. This may suggest the difference between B1 and a4 in dRTA [17]. The difference in both the distribution and roles played by these two subunits may influence the clinical manifestation; however, the information derived from long-term observation was inconclusive.
In the critical form of mutant V-ATPase, the autosomal recessive form, showing an early onset (developing in infancy) and severe metabolic acidosis, was common until quite recently. However, incomplete dRTA presenting as impaired urinary acidification and/or recurrent renal stones from a heterozygous mutation or polymorphism of the B1 subunit has been reported [18, 19]. Zhang et al. [18] investigated a family of dRTA patients with a B1 subunit gene mutation (p.F468fsX487) and demonstrated impairment of urinary acid acidification and net acid excretion in heterozygotes with normal plasma HCO3−. They also showed an aberrant distribution and expression of mutant B1 in the renal papilla of a heterozygote with severe kidney stones, and demonstrated insufficient V-ATPase activity of the heterologous expression of wild and mutant B1 using transfected mammalian cells and yeast. Most recently, Dhayat et al. [19] investigated a total of 555 recurrent stone formers and showed that the impairment of urinary acidification in acute NH4Cl loading, suggesting incomplete dRTA, was significantly observed in a population that was a heterozygous for the p.E161K SNP in subunit B1. In the present case, there was no variant in ATP6V1B1 (c.481G, p.161E). In regard to the a4 subunit of V-ATPase, to the best of our knowledge, the clinical features of a heterozygous a4 mutant in humans have not been well described to date, but in wild/ATP6V0a4-deletion hybrid mice, hyperchloremic metabolic acidosis under the condition of chronic acid loading has been proven [20]. It is clinically important that the incomplete dRTA from a novel heterozygous V-ATPase a4 subunit mutation in the present case presents as severe hypokalemia and chronic renal insufficiency. None of the six heterozygotes in a mutant B1 subunit described in a recent report [18] had this clinical feature. The loading tests for the present case clearly explained the mechanism of the clinical features, namely a urinary H+ excretion dysfunction with compensatory changes in the proximal tubules (i.e. enhanced production, elevation) and urinary K loss instead of H+. No examinations of the patient's siblings have been done, but these changes may not be specific to the mutation of the present case, as the common mechanisms may be at work in any type of incomplete dRTA caused by a heterozygous mutant V-ATPase.
In terms of the NGS analysis, especially in CNV, it has been well known that detecting relatively large structural variants is still a developing issue in NGS analysis. Moreover, particularly in cases of targeted resequencing like this one, it becomes more difficult to detect CNVs since the sequenced regions are very limited compared with the whole genome (Supplementary Ref S2) and thus detection sensitivity may decrease. Therefore, even though the CNV detection approach was done for NGS data, there remains the possibility that we could not detect such variants perfectly. This could be one of the limitations of this genetic analysis.
Supplementary data
Supplementary data are available online at http://ndt.oxfordjournals.org.
Conflict of interest statement
None declared.
Supplementary Material
References
- 1.Batlle D, Haque SK. Genetic causes and mechanisms of distal renal tubular acidosis. Nephrol Dial Transplant 2012; 27: 3691–3704 [DOI] [PubMed] [Google Scholar]
- 2.Rodríguez Soriano J. Renal tubular acidosis: the clinical entity. J Am Soc Nephrol 2002; 13: 2160–2170 [DOI] [PubMed] [Google Scholar]
- 3.Both T, Zietse R, Hoorn EJ et al. Everything you need to know about distal renal tubular acidosis in autoimmune disease. Rheumatol Int 2014; 34: 1037–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Narahara M, Higasa K, Nakamura S et al. Large-scale East-Asian eQTL mapping reveals novel candidate genes for LD mapping and the genomic landscape of transcriptional effects of sequence variants. PLoS One 2014; 9: e100924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature 2010; 467: 1061–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Exome Variant Server, NHLBI GO Exome Sequencing Project, Seattle, WA. http://evs.gs.washington.edu/EVS/ (December 2014, date last accessed).
- 7.Exome Aggregation Consortium (ExAC), Cambridge, MA. http://exac.broadinstitute.org (July 2015, date last accessed)
- 8.Miura K, Sekine T, Takahashi K et al. Mutational analyses of the ATP6V1B1 and ATP6V0A4 genes in patients with primary distal renal tubular acidosis. Nephrol Dial Transplant 2013; 28: 2123–2130 [DOI] [PubMed] [Google Scholar]
- 9.Elhayek D, Perez de Nanclares G, Chouchane S et al. Molecular diagnosis of distal renal tubular acidosis in Tunisian patients: proposed algorithm for Northern Africa populations for the ATP6V1B1, ATP6V0A4 and SCL4A1 genes. BMC Med Genet 2013; 14: 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nagara M, Voskarides K, Nouira S et al. Molecular investigation of distal renal tubular acidosis in Tunisia, evidence for founder mutations. Genet Test Mol Biomarkers 2014; 18: 741–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pereira PC, Melo FM, De Marco LA et al. Whole-exome sequencing as a diagnostic tool for distal renal tubular acidosis. J Pediatr (Rio J) 2015; 91:583–589 [DOI] [PubMed] [Google Scholar]
- 12.Gómez J, Gil-Peña H, Santos F et al. Primary distal renal tubular acidosis: novel findings in patients studied by next generation sequencing. Pediatr Res 2015; doi:10.1038/pr.2015.243 [DOI] [PubMed] [Google Scholar]
- 13.Breton S, Brown D. Regulation of luminal acidification by the V-ATPase. Physiology (Bethesda) 2013; 28: 318–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stover EH, Borthwick KJ, Bavalia C et al. Novel ATP6V1B1 and ATP6V0A4 mutations in autosomal recessive distal renal tubular acidosis with new evidence for hearing loss. J Med Genet 2002; 39: 796–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li X, Chai Y, Tao Z et al. Novel mutations in ATP6V0A4 are associated with atypical progressive sensorineural hearing loss in a Chinese patient with distal renal tubular acidosis. Int J Pediatr Otorhinolaryngol 2012; 76: 152–154 [DOI] [PubMed] [Google Scholar]
- 16.Gao Y, Xu Y, Li Q et al. Mutation analysis and audiologic assessment in six Chinese children with primary distal renal tubular acidosis. Ren Fail 2014; 36: 1226–1232 [DOI] [PubMed] [Google Scholar]
- 17.Topaloglu R, Baskın E, Bahat E et al. Hereditary renal tubular disorders in Turkey: demographic, clinical, and laboratory features. Clin Exp Nephrol 2011; 15: 108–113 [DOI] [PubMed] [Google Scholar]
- 18.Zhang J, Fuster DG, Cameron MA et al. Incomplete distal renal tubular acidosis from a heterozygous mutation of the V-ATPase B1 subunit. Am J Physiol Renal Physiol 2014; 307: F1063–F1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dhayat NA, Schaller A, Albano G et al. The vacuolar H+-ATPase B1 subunit polymorphism p.E161K associates with impaired urinary acidification in recurrent stone formers. J Am Soc Nephrol 2015. ; doi:10.1681/ASN.2015040367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Norgett EE, Golder ZJ, Lorente-Cánovas B et al. Atp6v0a4 knockout mouse is a model of distal renal tubular acidosis with hearing loss, with additional extrarenal phenotype. Proc Natl Acad Sci USA 2012; 109: 13775–13780 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.