

Abstract
Anti-neutrophil cytoplasm antibody (ANCA)-associated vasculitis (AAV) is a group of rare autoimmune diseases. Although the aetiology of AAV is uncertain, it is likely that genetic and environmental factors contribute. We report the unusual case of two brothers presenting with AAV with differing clinical pictures and differing ANCA specificity. There is a recently identified difference in genetic risk factors associated with ANCA specificity, making it surprising that first-degree relatives develop AAV with differing clinical and serological features. Our report illustrates the complex aetiology of AAV and suggests that further research on the interaction of genetic and environmental factors is needed.
Keywords: ANCA, familial, vasculitis
Background
Anti-neutrophil cytoplasm antibody (ANCA)-associated vasculitis (AAV) is the term used to describe a group of systemic diseases characterized by small vessel vasculitis [1]. There are three clinical syndromes: granulomatosis with polyangitis (GPA); microscopic polyangitis (MPA); and eosinophilic granulomatosis with polyangitis (EGPA). Within a Caucasian population, GPA is usually associated with antibodies against proteinase-3 (PR3), MPA is usually associated with antibodies against myeloperoxidase (MPO) and EGPA is often associated with anti-MPO antibodies but is sometimes ANCA negative. Renal limited vasculitis is also recognized with either antibody specificity. These are rare conditions with an annual incidence of around 20 cases per million within the UK [2]. They are often associated with significant morbidity and mortality despite progress in immunosuppressive regimens designed to maximize efficiency and limit toxicity [3]. The aetiology of AAV remains uncertain; however, it is likely that both genetic predisposition and environmental triggers are important. There is also mounting evidence for the pathogenicity of ANCA [4]. We report the very unusual occurrence of two brothers with AAV, but with differing clinical syndromes and ANCA types.
Case reports
Patient 1
A 49-year-old Caucasian man presented as an emergency with a 1 day history of severe epistaxis. He had a 3-month history of nasal congestion, left ear and supraorbital pain, and lethargy. On examination he had bilateral uveitis and episcleritis and a faint purpuric rash over his groin. Flexible nasendoscopy demonstrated copious dried blood and secretions. Urine dipstick showed 4+ blood and 4+ protein.
Investigations showed preserved renal function, creatinine 91 µmol/L and mild anaemia, haemoglobin 12 g/dL. Inflammatory markers were raised, C-reactive protein 368 mg/L and erythrocyte sedimentation rate 119 mm/h. Computed tomography (CT) chest demonstrated diffuse ground glass infiltrates throughout both lung fields (Figure 1). CT sinuses showed diffuse mucosal thickening of the maxillary sinuses. A diagnosis of probable vasculitis was made and the patient was started on 30 mg/day of oral prednisolone. He was found to be C-ANCA positive with anti-PR3 titre of 431 IU (normal range <25). He developed haemoptysis with a fall in his haemoglobin to 10.5 g/dL, and acute kidney injury with peak creatinine 185 µmol/L. Nasal swabs were negative for methicillin-resistant staphylococcal aureus but were not tested for methicillin-sensitive staphylococcal aureus.
Fig. 1.

CT chest image from Patient 1 showing bilateral diffuse ground glass infiltrates.
The diagnosis of GPA was made and he was transferred to our centre and treated by our current protocol for vasculitis with pulmonary haemorrhage. He received 60 mg/day oral prednisolone, 1 g IV cyclophosphamide and ten 4 L plasma exchanges. Renal biopsy was not performed because of the urgency of starting treatment. Following discharge he received five further doses of IV cyclophosphamide (total 4 g) and 2× 1 g IV rituximab (again in keeping with our current protocol). Prednisolone was weaned gradually and stopped by 9 months as he became Cushingoid. He was maintained on azathioprine alone at a dose of 150 mg once daily. He had some minor infective complications with recurrent dental infections due to underlying dental caries which resolved following tooth extraction. Two and a half years later he is in complete remission with a creatinine of 82 µmol/L, although he remains C-ANCA positive with anti-PR3 titre of 70 IU.
Patient 2
Six months later the patient's brother presented at the age of 53 years with 6 weeks of worsening lethargy, myalgia and arthralgia. He had a dry cough but no haemoptysis, and weight loss of 10 kg. Urine dipstick showed 3+ blood and 1+ protein. Investigations showed acute kidney injury, creatinine 338 µmol/L (no baseline available but 213 µmol/L when checked by his general practitioner 2 days previously). He was P-ANCA positive with anti-MPO titre of 357 IU (normal range <25). CT chest was unremarkable except for a single pulmonary nodule which did not meet criteria for further investigation. Renal biopsy showed pauci-immune crescentic glomerulonephritis and necrotizing vasculitis (Figure 2).
Fig. 2.
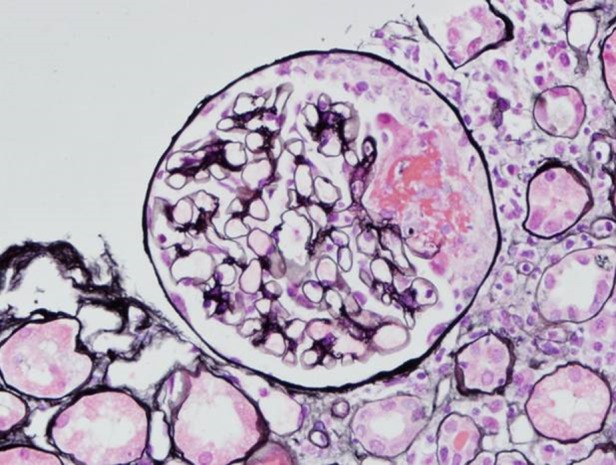
Renal biopsy from Patient 2. Silver stain showing glomerulus with cellular crescent formation (courtesy of Professor Terry Cook).
The diagnosis of MPA was made, and he was treated with 60 mg/day oral prednisolone, 1 g IV rituximab and 750 mg IV cyclophosphamide. Following discharge he received a further 1 g IV rituximab and five further doses of IV cyclophosphamide (total 3.5 g) and was maintained on low-dose oral prednisolone (5 mg/day) and azathioprine 75 mg/day. His treatment was by our current protocol for treatment of patients with vasculitis but not requiring dialysis and without pulmonary haemorrhage. His treatment course was uneventful with no infectious complications. He remains in remission 2 years later with stable renal function (creatinine 140 µmol/L) although still P-ANCA positive with anti-MPO titre of 40 IU.
Discussion
We describe the occurrence of two different ANCA-associated vasculitides, GPA and MPA, in two brothers who were brought up together in West London. They presented within 6 months of each other with distinct clinical pictures and differing ANCA specificity. One brother had renal, pulmonary, and ear, nose and throat (ENT) involvement with positive PR3-ANCA typical of GPA, and the other brother had positive MPO-ANCA and clinical features in keeping with MPA. They had no significant family history, in particular no history of vasculitis or autoimmune disease. The brothers have a shared HLA haplotype of A*3, B*44, Cw7, DR*11, DQ*7, DQB1*03/05, which to our knowledge has not been reported to be associated with AAV.
There are few previous reports of familial AAV. The majority describe familial clusters of patients presenting separated in time, but with similar clinical phenotypes and the same ANCA type. We previously reported three members of an Indo-Asian family with GPA who all presented with ENT and renal involvement [5]. Hay et al. [6] reported on two siblings with GPA, both with renal and pulmonary involvement, and Sewell and Hamilton [7] reported a mother and daughter with GPA, both with ENT and renal involvement. There are even fewer reported cases of first-degree relatives presenting with small vessel vasculitis with different ANCA types. A family reported by Gomes et al. [8] includes a father and daughter who both presented with renal disease; one was anti-PR3 positive and the other anti-MPO positive. Manganelli et al. [9] reported two brothers with EGPA (ANCA negative) and GPA (anti-PR3 positive).
AAV is known to have important genetic associations. A genome-wide association study [10] identified four single nucleotide polymorphisms (SNPs) that associated with AAV. GPA/PR3-ANCA associated with SNPs within HLA-DP, SERPINA1 (which encodes α1-antitrypsin) and PRTN3 (which encodes proteinase 3), whereas a SNP within HLA-DQ associated with MPA/MPO-ANCA. Further analysis showed that these SNPs were more strongly associated with ANCA type than with clinical phenotype, suggesting that these diseases should perhaps be re-classified as PR3-ANCA or MPO-ANCA vasculitis, rather than GPA or MPA.
Environmental factors are also likely to be important in the pathogenesis of AAV and a variety of toxins, infections and drugs have been implicated [3]. Silica is the environmental toxin for which there is the greatest evidence, with a population case–control study showing high lifetime silica exposure associated with AAV [11]. Brener et al. [12] reported the case of two brothers with pulmonary and renal vasculitis (anti-MPO positive); both brothers had been exposed to silica but four other unexposed siblings did not have vasculitis. Neither of our patients reported exposure to any environmental factor which could have predisposed them to developing AAV.
Given the identification of a difference in genetic risk factors associated with different AAV phenotypes and ANCA specificity it is perhaps surprising that first-degree relatives develop AAV with differing clinical and serological features. It may be that there was some unrecognized environmental exposure, such as a toxin or infection, within their shared upbringing. Our report illustrates the complex aetiology of AAV and suggests that further research on the interaction of genetic and environmental factors is needed.
Conflict of interest statement
The authors have no conflicts of interests to declare.
Acknowledgements
We acknowledge support from the NIHR Imperial Biomedical Research Centre, and thank our colleagues who have looked after these patients. We are grateful to Dr Paul Brookes, who performed the tissue typing.
References
- 1.Jennette JC, Falk RJ, Bacon PA. 2012 revised International Chapel Hill consensus conference nomenclature of vasculitides. Arthritis Rheum 2013; 65: 1–11 [DOI] [PubMed] [Google Scholar]
- 2.Watts RA, Mooney J, Skinner J et al. The contrasting epidemiology of granulomatosis with polyangitis (Wegener's) and microscopic polyangitis. Rheumatology 2012; 15: 926–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hamour S, Salama AD, Pusey CD. Management of ANCA-associated vasculitis: current trends and future prospects. Ther Clin Risk Manag 2010; 6: 253–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jennette JC, Falk RJ, Hu P et al. Pathogenesis of antineutrophil cytoplasmic autoantibody-associated small-vessel vasculitis. Annu Rev Pathol 2013; 8: 139–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tanna A, Salama AD, Brookes P et al. Familial granulomatosis with polyangitis: three cases of this rare disorder in one Indoasian family carrying an identical HLA DPB1 allele. BMJ Case Rep 2012; doi:10.1136/bcr.01.2012.5502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hay EM, Beaman M, Ralston AJ et al. Wegener's granulomatosis occurring in siblings. Br J Rheumatol 1991; 30: 144–145 [DOI] [PubMed] [Google Scholar]
- 7.Sewell RF, Hamilton DV. Time associated Wegener's granulomatosis in two members of a family. Nephrol Dial Transplant 1992; 7: 82. [PubMed] [Google Scholar]
- 8.Gomes AM, Nery F, Almeida C et al. Familial clusters of ANCA small-vessel vasculitis. NDT Plus 2009; 2: 34–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Manganelli P, Giacosa R, Fietta P et al. Familial vasculitides: Churg-Strauss syndrome and Wegener's granulomatosis in 2 first-degree relatives. J Rheumatol 2003; 30: 618–621 [PubMed] [Google Scholar]
- 10.Lyons PA, Rayner TF, Trivedi S et al. Genetically distinct subsets within ANCA-associated vasculitis. N Engl J Med 2012; 367: 214–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hogan SL, Cooper GS, Savitz DA et al. Association of silica exposure with anti-neutrophil cytoplasmic autoantibody small-vessel vasculitis: a population-based, case-control study. Clin J Am Soc Nephrol 2007; 2: 290–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brener Z, Cohen L, Goldberg SJ et al. ANCA-associated vasculitis in Greek siblings with chronic exposure to silica. Am J Kidney Dis 2001; 38: E28. [DOI] [PubMed] [Google Scholar]