

Abstract
Immune-checkpoint inhibitors are emerging as revolutionary drugs for certain malignancies. However, blocking the co-inhibitory signals may lead to immune-related adverse events, mainly in the spectrum of autoimmune diseases including colitis, endocrinopathies and nephritis. Here, we report a case of a 75-year-old man with metastatic malignant melanoma treated with a combination of nivolumab (anti-PD1-antibody) and ipilimumab (anti-CTLA-4 antibody) who developed systemic rash along with severe acute tubulointerstitial nephritis after two doses of combination therapy. Kidney biopsy and peripheral blood immune profile revealed highly proliferative and cytotoxic T cell features. Herein, we discuss the pathophysiology and management of immune checkpoint blockade-related adverse events.
Keywords: acute kidney injury, immune-checkpoint blockade, interstitial nephritis
Background
Immune-checkpoint inhibitors are increasingly used as anti-cancer agents as more efficacies have been proved in multiple cancer species, such as melanoma, non-small-cell lung carcinoma and renal cell carcinoma [1–4]. On the other hand, the therapy comes with a price: immune-related adverse events (irAE) occur in up to 60% of treated patients, usually mild to moderate in grade. The pathophysiology of irAE is considered similar to that of autoimmune diseases, wherein activated lymphocytes target self-antigens [5]. Here we report a case of a patient with metastatic melanoma treated with immune-checkpoint inhibitor combination therapy, who developed acute kidney injury (AKI), systemic maculopapular rash and fever.
Case report
Clinical history and initial laboratory data
A 75-year-old Caucasian male with essential hypertension and recurrent metastatic melanoma presented with systemic morbilliform rash and AKI. The patient was initially diagnosed with invasive cutaneous melanoma of the left nasolabial fold [American Joint Committee of Cancer (AJCC) Stage IIc; T4b, N0, M0. BRAF V600/NRAS-wild-type] and underwent local excision with clear margins. Seven months later, he presented with significant weight loss and a palpable mass in his left submandibular area. PET-CT revealed an FDG-avid left submandibular lymph node and a liver lesion without any brain metastasis [AJCC Stage IV, M1c; Eastern Cooperative Oncology Group (ECOG) performance status 0]. Biopsy confirmed recurrence of malignant melanoma and combination therapy of nivolumab (1 mg/kg) and ipilimumab (3 mg/kg) was initiated. Routine blood test after his second cycle of treatment revealed a creatinine of 3.96 mg/dL, compared with a baseline of 0.91 mg/dL 3 weeks prior, indicating AKI (Table 1). Urinalysis revealed 2+ protein (albumin/creatinine ratio 42.4 mg/gCre), RBC 8/hpf and WBC 9/hpf. Urine sediments showed granular casts and WBC casts (Figure 1A and B). There was no history suggestive of dehydration or exposure to nephrotoxic agents such as antibiotics, contrast or analgesics. His only medications were nivolumab and ipilimumab. He was allergic to sulfa and ciprofloxacin.
Table 1.
Laboratory parameters
| Time since last dose of nivolumab/ipilimumab |
||||
|---|---|---|---|---|
| Baseline | 3 weeks | 5 weeks | (Reference range) | |
| Sodium (mEq/L) | 139 | 141 | 137 | 136–145 |
| Potassium (mEq/L) | 4.2 | 4.1 | 4.1 | 3.4–5.0 |
| Chloride (mEq/L) | 108 | 114 | 101 | 98–107 |
| Bicarbonate (mmol/L) | 24 | 18 | 16 | 22–31 |
| BUN (mg/dL) | 13 | 65 | 73 | 6–23 |
| Cre (mg/dL) | 0.91 | 3.96 | 4.64 | 0.50–1.20 |
| Total bilirubin (mg/dL) | 0.7 | 0.6 | 0.7 | 0.0–1.0 |
| LDH (U/L) | 188 | 189 | 536 | 135–225 |
| Total protein (g/dL) | 9.3 | 5.1 | 6.4–8.3 | |
| Albumin (g/dL) | 3.2 | 2.6 | 3.5–5.2 | |
| ALT (U/L) | 25 | 33 | 10–50 | |
| AST (U/L) | 22 | 31 | 10–50 | |
| Alkaline phosphatase (U/L) | 96 | 67 | 35–130 | |
| WBC (1000/µL) | 7.05 | 7.52 | 7.18 | 4.0–10.0 |
| Hemoglobin (g/dL) | 15.6 | 11.9 | 10.5 | 13.5–18.0 |
| Platelets (1000/µL) | 250 | 252 | 61 | 150–450 |
| ANA | 1:40 (diffuse, speckled) | |||
| dsDNA Ab (IU) | 25 | 0–25 | ||
| Histone Ab (U) | 0.7 | <1.0 | ||
| C3 (mg/dL) | 120 | 90–180 | ||
| C4 (mg/dL) | 31 | 10–40 | ||
| PPD | Negative | |||
| Cortisol (in AM, µg/dL) | 21.3 | 6–24 | ||
| TSH (µIU/mL) | 1.35 | 0.09 | 0.50–5.70 | |
| FT4 (ng/dL) | 1.1 | 1.1 | 0.9–1.7 | |
| THBR | 1.17 | 0.80–1.20 | ||
| TPO Ab (IU/mL) | <10.0 | 0.0–33.9 | ||
ANA, anti-nuclear antibody; PPD, purified protein derivative; TSH, thyroid-stimulating hormone; FT4, free thyroxine; THBR, thyroid hormone binding ratio; TPO Ab, thyroid peroxidase antibody.
Fig. 1.
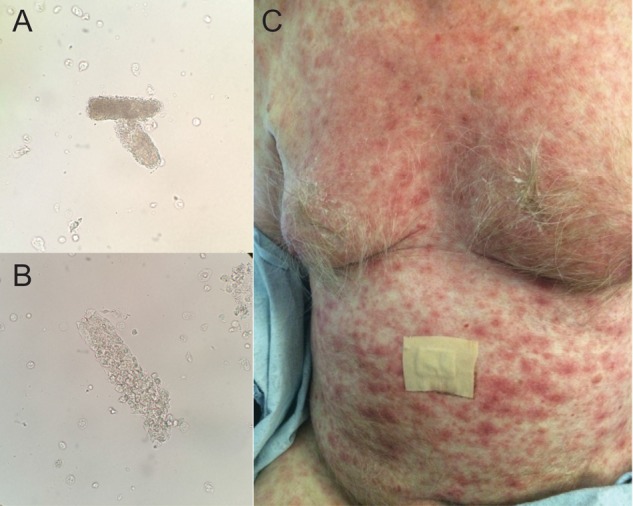
(A and B) Urine sediment analysis revealed granular cast (A) and WBC cast (B). (C) Non-pruritic morbilliform erythematous rash on patient's chest and abdomen.
On physical examination, vitals revealed a temperature of 36.4°C, a pulse of 93/min, a blood pressure at 178/80 mmHg, a respiratory rate of 20/min and body weight of 73.1 kg (an increase of 5 kg from his pre-treatment weight). A non-tender submandibular mass was palpable. His lungs were clear to auscultation and 1+ bilateral lower extremity edema were noted. A morbilliform eruption rash was noted on his chest, abdomen and back (Figure 1C). There was no change in the oral mucosa. A renal ultrasound showed no evidence of hydronephrosis, although the kidneys were slightly enlarged bilaterally (right 12.4 cm and left 13.0 cm). The decision was then made to pursue a kidney biopsy to further define the etiology of the patient's severe AKI.
Kidney biopsy
The kidney biopsy contained 34 glomeruli, of which 4 were globally sclerosed. The remaining glomeruli showed wrinkled glomerular capillary loops and narrowing of Bowman's space, suggestive of hypoperfusion. Cellularity and architecture was normal and no active inflammation of the tuft, necrotizing lesions or cellular crescents was noted (not shown). There was diffuse extensive interstitial inflammation associated with moderate interstitial edema (Figure 2A). The infiltrates were primarily composed of lymphocytes and plasma cells with admixed eosinophils and neutrophils. Granuloma or granulomatous lesions were not noted. There were frequent tubulitis and various degenerative changes, with focal flattening of the epithelium and occasional mitoses. Several tubules contained necrotic cellular debris, granular casts and periodic acid–Schiff-positive hyaline casts.
Fig. 2.

Light microscopy of the kidney biopsy: (A) Periodic acid–Schiff staining showed severe interstitial infiltration of inflammatory cells, mainly lymphocytes and neutrophils. (B–F) Immunohistochemistry revealed extensive T-cell infiltration of CD4+ (B) and CD8+ T cells (C). Ten to 20% of the infiltrates were positive for granzyme B (D) and perforin (E). Foxp3-positive regulatory T cells were also present (F). Scale bars, 100 μm.
Immunofluorescence staining revealed no significant immune deposits in the glomeruli. Tubular basement membranes showed focal fine granular deposition of C3 (1+). The interstitium revealed intense fibrin deposits (not shown). Immunohistochemistry showed extensive T-cell-dominant infiltrates (Figure 2B and C), of which ∼10–20% were strongly positive for cytotoxic enzymes including granzyme B (Figure 2D) and perforin (Figure 2E). Foxp3+ regulatory T cells were also present in the interstitium (Figure 2F).
Clinical follow-up
The patient was treated with a steroid pulse [solumedrol 500 mg intravenous (IV) × 3 days] followed by a taper (prednisone 60 mg daily). Although his creatinine initially improved to 2.41 mg/dL over the course of a week, it had increased to 4.64 mg/dL when he was readmitted the following week with symptoms of a fever and a rash (Table 1 and Figure 3). There was no laboratory evidence of hypophysitis or hepatitis at the time (Table 1). Skin biopsy revealed interface dermatitis involving adnexal structures with superficial perivascular lymphohistiocytic infiltrates. Peripheral immune characterization revealed highly proliferative circulating CD4+ and CD8+ T cells (measured by the percentage of Ki67+ cells, Figure 4A–C) in combination with an expansion of effector memory T cells (TEM, CD45RA-negative CCR7-negative, Figure 4D and E). Furthermore, serum cytokine analysis revealed a strikingly high level of pro-inflammatory cytokines (IL-1Ra, CXCL10 and TNF-α) (Figure 4F–H) and soluble IL-2 receptor (32 400 pg/mL; reference range <1033 pg/mL). Due to these findings, the patient's steroid dose was increased (solumedrol 140 mg IV twice a day) and mycophenolate mofetil (MMF; 1000 mg twice a day) was added to intensify treatment. Creatinine peaked at 7.31 mg/dL and then improved to 3.37 mg/dL over 10 days (Figure 3). The patient's rash dramatically improved and eventually resolved. Two weeks later, the patient was again admitted with a fever of 38.4°C and 3 days of bloody diarrhea. While his creatinine had remained stable, he was found to be pancytopenic (WBC 1.25 × 103/µL with 92% neutrophils, hemoglobin 9.2 g/dL, platelets 37 × 103/µL). The patient subsequently developed septic shock and deceased in spite of aggressive treatment with broad-spectrum antibiotics and vasopressor support. Blood culture turned out to be positive for pan-sensitive Pseudomonas aeruginosa. Autopsy demonstrated hemorrhagic colitis with no histological evidence of viable tumor in his liver.
Fig. 3.

Clinical course of the patient. Serum creatinine, timing of nivolumab and ipilimumab administration, kidney biopsy procedure and treatment course are shown. Time course was described as weeks after the first dose of nivolumab/ipilimumab.
Fig. 4.

Immunological characterization of T cells upon anti-PD-1/anti-CTLA-4 therapy. (A) Gating strategy of peripheral blood mononuclear cells (PBMCs) in flow cytometry analysis. Patient's CD8+ or CD4+ T cells carried an extremely proliferative phenotype recognized as Ki67+ cells (B and C), and more effector-memory (TEM) T cells identified as CD45RA-negative CCR7-negative population (D and E) compared with healthy controls. Serum cytokine profile was analyzed using Luminex multiplex panel. Pro-inflammatory cytokines (IL-1Ra, CXCL10 and TNF-α) were strikingly higher in the patient (F–H). HC; healthy controls.
Discussion
Immune-checkpoint molecules play important roles in tolerance and cancer immunity. One of the mechanisms by which cancer cells evade immune surveillance and destruction is cell surface expression of co-inhibitory molecules such as PD-L1. Immune-checkpoint inhibitors exhibit immense tumor immunity via enhancing effector lymphocyte functions to target cancer cells by uncovering these molecular ‘shields’ [6]. Nevertheless, this immune enhancement comes at the expense of an increased risk of irAE (Table 2) [7]. A recent study of nivolumab and ipilimumab combination therapy for melanoma reported higher treatment-related severe adverse events rates [Common Terminology Criteria for Adverse Events (CTCAE) grade 3 or 4] for dual therapy (55%) compared with nivolumab or ipilimumab alone (16.3 and 27.3%, respectively) [8]. However, severe renal adverse events (grade 3–4) were still rare (6 cases out of 313) in the combination therapy group.
Table 2.
Immune-related adverse events associated with immune-checkpoint inhibitors in melanoma patients
| Nivolumab |
Ipilimumab |
Nivolumab + ipilimumab |
||||
|---|---|---|---|---|---|---|
| All grade | Grade 3/4 | All grade | Grade 3/4 | All grade | Grade 3/4 | |
| Skin | ||||||
| Pruritus (%) | 9–23 | <1 | 24–36 | 0–1 | 33–47 | 2 |
| Rash (%) | 9–26 | 0–1 | 15–33 | 0–2 | 28–55 | 3–5 |
| Vitiligo (%) | 3–11 | 0 | 2–4 | 0 | 7 | 0 |
| Gastro-intestinal | ||||||
| Colitis (%) | 1–3 | <1–2 | 7–12 | 5–9 | 9–23 | 4–17 |
| Diarrhea (%) | 8–20 | 0–2 | 23–33 | 3–10 | 34–45 | 6–11 |
| Endocrinopathy | ||||||
| Hypothyroidism (%) | 2–9 | <1 | 1.5–4 | 0 | 15–16 | <1 |
| Hyperthyroidism (%) | 1–5 | <1 | 1–2 | <1 | 10 | 1 |
| Hypophysitis (%) | <1 | <1 | 2–4 | 2 | 8–12 | 2–7 |
| Pneumonitis (%) | 1–9 | 0–1 | 0–2 | <1 | 0–6 | 1–2 |
| Renal injury (%) | 0–1 | 0–1 | 0–1 | <1 | <1 | <1 |
To our knowledge, this is the first report of biopsy-proven acute interstitial nephritis (AIN) caused by combination immune-checkpoint inhibitor therapy. Seven cases of ipilimumab-associated AIN were reported in the literature [9–14], which were recently reviewed by Izzedine et al. [13]. Most of the cases presented with minimally symptomatic elevation of creatinine with mild proteinuria 6–12 weeks after initiation of ipilimumab. Of the four cases where biopsy was available, three had granulomatous tubulointerstitial nephritis while one had membranous lupus nephritis. Unlike these patients, who responded well to steroid therapy, our patient relapsed 2 weeks later with a more severe presentation accompanied by systemic rash and fever. Further immune suppression with combination of high dose steroid and MMF was required to control the disease activity. What made our case different from previous cases of AIN?
One possibility is related to the dual immune-checkpoint inhibition. CTLA-4 regulates peripheral tolerance by modulating the interaction between antigen-presenting cells and T cells in secondary lymphoid organs, and CTLA-4 deficiency was reported to trigger early-onset severe lymphoproliferative autoimmune syndromes both in human and mouse [15–18] via tissue self-antigen-specific T-cell activation [19]. On the other hand, PD-1 contributes to tolerance primarily at the level of target organs. PD-L1 or PD-1 deficient animals do develop autoimmune phenotype, but in a milder form and later in life [20, 21], which corroborates data in humans showing that anti-PD-1 therapy was associated with distinct and less severe irAE profile compared with anti-CTLA-4 therapy [22]. Blocking both pathways could synergistically potentiate antigen recognition and T-cell proliferation at lymph nodes [23], and provoke untethered cytotoxic T-cell effects in the periphery, not only against tumor but also against normal tissues (Figure 5). This mechanism is particularly relevant given mounting evidence that the clonal deletion of auto-reactive T-cell clones in the thymus is incomplete, and a few auto-reactive T cells remain dormant in all individuals [24, 25].
Fig. 5.

Dual PD-1/CTLA-4 blockade synergistically breaks the tolerance by unleashing quiescent tissue-specific self-reactive T cells, which express high levels of PD-1. In the draining lymph nodes (right), regulatory T cells (Tregs) suppress T cells/APCs in a CTLA-4-dependent manner. With CTLA-4 blockade, Tregs lose their suppressive capacity and an uncontrolled activation of auto-reactive T cells occurs. Those cells then migrate and infiltrate the target tissue (e.g. kidney). In the renal tissue (left, top), IFN-γ triggers the upregulation of PD-L1 by renal cells, which will bind and signal through PD-1 expressed by T cells, trying to prevent those cells from proliferating and damaging the tissue. However, when the self-reactive T cells have their PD-1 receptor blocked by the antibody, the PD-1/PD-L1 signaling will be interrupted and T cells will further proliferate and mature. These T cells will then produce perforin and granzyme B (GrzB), feeding the pathogenic cycle further.
In our patient, a highly proliferative effector-memory T-cell phenotype along with aggressive cytotoxic T-cell infiltration in the kidney (Figures 2 and 4) raises the intriguing possibility that immune-checkpoint inhibitors unleashed these dormant auto-reactive T cells, leading to selective tissue injury (Figure 5).
There are unanswered questions on clonality and antigen-specificity of tissue-infiltrating cells. The observation that certain tissues seem more prone to irAE while others are spared [5] suggests tissue-antigen specificity as shown in CTLA-4-deficient animals [19]. Moreover, some patients develop irAE while others do not. T-cell receptor repertoire analyses and detailed immune profile characterization may enable us to risk-stratify patients and better manage these complications in the future.
The mainstay of irAE management is immune suppression, including high-dose steroids, MMF and potentially TNF-α inhibitors [5], interventions which do not seem to worsen cancer-specific outcomes [26]. Nevertheless, as our patient tragically demonstrated, one must be vigilant about the possibility of severe systemic infections in these patients.
In summary, our case highlights the aggressive T-cell-mediated pathophysiology of irAE, in the setting of immune-checkpoint inhibitor combination therapy. Early intervention and fine adjustment of immune modulatory agents is the key to the management of these complications.
Conflict of interest statement
None declared.
Acknowledgements
N.M. is supported by a grant from the National Institutes of Health (T32DK007527) and L.V.R is supported by a Research Grant (12FTF120070328) from the American Heart Association.
References
- 1.Hodi FS, O'Day SJ, McDermott DF et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363: 711–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brahmer J, Reckamp KL, Baas P et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med 2015; 373: 123–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garon EB, Rizvi NA, Hui R et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med 2015; 372: 2018–2028 [DOI] [PubMed] [Google Scholar]
- 4.Motzer RJ, Escudier B, McDermott DF et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med 2015; 373: 1803–1813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weber JS, Kahler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol 2012; 30: 2691–2697 [DOI] [PubMed] [Google Scholar]
- 6.Murakami N, Riella LV. Co-inhibitory pathways and their importance in immune regulation. Transplantation 2014; 98: 3–14 [DOI] [PubMed] [Google Scholar]
- 7.Spain L, Diem S, Larkin J. Management of toxicities of immune checkpoint inhibitors. Cancer Treat Rev 2016; 44: 51–60 [DOI] [PubMed] [Google Scholar]
- 8.Larkin J, Chiarion-Sileni V, Gonzalez R et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015; 373: 23–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beck KE, Blansfield JA, Tran KQ et al. Enterocolitis in patients with cancer after antibody blockade of cytotoxic T-lymphocyte-associated antigen 4. J Clin Oncol 2006; 24: 2283–2289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fadel F, El Karoui K, Knebelmann B. Anti-CTLA4 antibody-induced lupus nephritis. N Engl J Med 2009; 361: 211–212 [DOI] [PubMed] [Google Scholar]
- 11.Forde PM, Rock K, Wilson G et al. Ipilimumab-induced immune-related renal failure—a case report. Anticancer Res 2012; 32: 4607–4608 [PubMed] [Google Scholar]
- 12.Voskens CJ, Goldinger SM, Loquai C et al. The price of tumor control: an analysis of rare side effects of anti-CTLA-4 therapy in metastatic melanoma from the ipilimumab network. PLoS One 2013; 8: e53745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Izzedine H, Gueutin V, Gharbi C et al. Kidney injuries related to ipilimumab. Invest New Drugs 2014; 32: 769–773 [DOI] [PubMed] [Google Scholar]
- 14.Thajudeen B, Madhrira M, Bracamonte E et al. Ipilimumab granulomatous interstitial nephritis. Am J Ther 2015; 22: e84–e87 [DOI] [PubMed] [Google Scholar]
- 15.Waterhouse P, Penninger JM, Timms E et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 1995; 270: 985–988 [DOI] [PubMed] [Google Scholar]
- 16.Tivol EA, Borriello F, Schweitzer AN et al. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 1995; 3: 541–547 [DOI] [PubMed] [Google Scholar]
- 17.Kuehn HS, Ouyang W, Lo B et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science 2014; 345: 1623–1627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schubert D, Bode C, Kenefeck R et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med 2014; 20: 1410–1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ise W, Kohyama M, Nutsch KM et al. CTLA-4 suppresses the pathogenicity of self antigen-specific T cells by cell-intrinsic and cell-extrinsic mechanisms. Nat Immunol 2010; 11: 129–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nishimura H, Nose M, Hiai H et al. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 1999; 11: 141–151 [DOI] [PubMed] [Google Scholar]
- 21.Nishimura H, Okazaki T, Tanaka Y et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 2001; 291: 319–322 [DOI] [PubMed] [Google Scholar]
- 22.Robert C, Schachter J, Long GV et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med 2015; 372: 2521–2532 [DOI] [PubMed] [Google Scholar]
- 23.Das R, Verma R, Sznol M et al. Combination therapy with anti-CTLA-4 and anti-PD-1 leads to distinct immunologic changes in vivo. J Immunol 2015; 194: 950–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malhotra D, Linehan JL, Dileepan T et al. Tolerance is established in polyclonal CD4T cells by distinct mechanisms, according to self-peptide expression patterns. Nat Immunol 2016; 17: 187–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuchroo VK, Ohashi PS, Sartor RB et al. Dysregulation of immune homeostasis in autoimmune diseases. Nat Med 2012; 18: 42–47 [DOI] [PubMed] [Google Scholar]
- 26.Horvat TZ, Adel NG, Dang TO et al. Immune-related adverse events, need for systemic immunosuppression, and effects on survival and time to treatment failure in patients with melanoma treated with ipilimumab at Memorial Sloan Kettering Cancer Center. J Clin Oncol 2015; 33: 3193–3198 [DOI] [PMC free article] [PubMed] [Google Scholar]