

Abstract
Chronic granulomatous disease (CGD) is an inherited immunodeficiency, caused by the inability of neutrophils to produce functional NADPH oxidase required for fighting microbial infections. The X-linked form of CGD (X-CGD), which is due to mutations in the CYBB (gp91phox) gene, a component of NADPH oxidase, accounts for about two-thirds of CGD cases. We derived induced pluripotent stem cells (iPSCs) from X-CGD patient keratinocytes using a Flp recombinase excisable lentiviral reprogramming vector. For restoring gp91phox function, we applied two strategies: transposon-mediated bacterial artificial chromosome (BAC) transgenesis and gene targeting using vectors with a fixed 5′ homology arm (HA) of 8 kb and 3′HA varying in size from 30 to 80 kb. High efficiency of homologous recombination (up to 22%) was observed with increased size of the 3′HA. Both, BAC transgenesis and gene targeting resulted in functional restoration of the gp91phox measured by an oxidase activity assay in X-CGD iPSCs differentiated into the myeloid lineage. In conclusion, we delivered an important milestone towards the use of genetically corrected autologous cells for the treatment of X-CGD and monogenic diseases in general.
Introduction
Chronic granulomatous disease (CGD) is a rare heritable disorder that affects 1 in 250,000 individuals.1 Patients with CGD suffer from severe infections and deregulated inflammation, both often associated with granuloma formation.2 After phagocytosis of pathogens by neutrophils, the multienzyme NADPH oxidase complex normally generates reactive oxygen species, which are indispensible for effective killing of certain opportunistic pathogens such as Aspergillus spp. and Staphylococcus aureus. Mutations in any of the five genes (CYBB, gp91phox; CYBA, p22phox; NCF1, p47phox; NCF2, p67phox; and NCF4, p40phox), all encoding components of the NADPH oxidase complex, lead to reduced or abolished function of this enzyme. The X-linked form of CGD (X-CGD) is caused by mutations in the Xp21.1-located gene CYBB coding for gp91phox (91-kDa subunit of the phagocyte oxidase) and accounts for two-thirds of CGD cases. Mutations in the CYBB gene are scattered in exons but also in other genomic regions including promoter, introns, and splice sites.3,4,5,6,7 Mutations in exonic regions include deletions or insertions causing frameshifts and nonsense and missense substitutions. Most of the mutations result in absence of gp91phox expression (X910).
While the outcome of human leukocyte antigen–matched allogeneic hematopoietic stem cell (HSC) transplantation with reduced-intensity conditioning regimens for CGD patients has improved considerably, for many patients, a suitable donor is missing.8 Because only a small fraction of circulating neutrophils (less than 10%) with normal gp91phox levels is enough to fight microbial infections, treatment of CGD by gene therapy holds great promise.9 Initial attempts to treat X-CGD using gamma-retroviral vectors did not exceed the 0.2% long-term gene marking, and later trials with preconditioning of the bone marrow did not result in significant long-term engraftment of gene-corrected cells without side effects.10 Unfortunately, the classical pitfalls associated with retroviral-mediated gene therapy, such as insertional transactivation of proto-oncogenes with clonal expansion, prevailed and led to undesired outcomes.11,12,13,14 This stimulated the development of safer alternatives based on self-inactivating retroviral or lentiviral vectors.15
The advent of induced pluripotent stem cells (iPSCs) opened new therapeutic concepts for the treatment of monogenic diseases. Patient-specific iPSCs can be derived from various somatic cell types, expanded, and genetically manipulated without losing their potential to differentiate into various lineages.16,17 By these means, it is conceivable to transplant autologous differentiated cells produced from genetically corrected iPSCs back to the patient. So far, iPSCs have been generated from different forms of CGD, including X-CGD and could be successfully differentiated in vitro into neutrophils and macrophages.18,19,20,21 Currently, there are two strategies to compensate for loss of gene function caused by a mutation in human pluripotent stem cells. The first strategy aims at the correction of the mutation by homologous recombination and has been successfully applied in different types of disease-specific iPSCs, including beta-thalassemia, laminopathy, and gyrate atrophy.16,17,22 The second strategy entangles the targeted insertion of a normal copy of the affected gene into a “safe harbor” locus, such as AAVS1, the genomic site of adeno-associated virus type 2 integration.23,24 So far, the NADPH oxidase activity in X-CGD neutrophils produced from iPSCs has been restored by zinc-finger nuclease–mediated targeting of the CYBB minigene into a “safe harbor” locus.18,21 However, this strategy does not guarantee long-term physiological levels of gp91phox expression because a constitutive heterologous promoter regulates the CYBB minigene. Myeloid-specific promoters similar to the ones used in viral vectors for restoring gp91phox expression in iPSCs generated from a mouse model of X-CGD25 or for transducing HSCs from X-CGD patients26 represent promising alternatives.
Here, we describe the restoration of the NADPH oxidase activity in X-CGD patient iPSCs by two different genetic approaches: (i) transposon-mediated integration of a bacterial artificial chromosome (BAC) vector carrying the genomic region of the CYBB gene and (ii) by correcting the X-CGD–causing mutation in the CYBB gene via homologous recombination. With both approaches, we could show that the genetically manipulated cells were functionally corrected and NADPH oxidase activity was restored after differentiation into neutrophils.
Results
Derivation of X-CGD iPSCs
We isolated keratinocytes from plucked hair of a patient with X-CGD (X910) carrying a double mutation c.958delG and c.962T > G in exon 9 of the CYBB gene. For the generation of iPSCs, we transduced keratinocytes with the LV-OKSM-Tomato vector, which contains the four reprogramming factors (OCT4, KLF4, SOX2, and c-MYC separated by 2A peptides) (Figure 1a,b). Two stable iPSC clones from this patient were chosen for further validation (S22 and J14). Southern blot analysis showed that clone S22 contained three and clone J14 two lentiviral integrations (Figure 1c). Cytokeratin-14 (K14), which marks primary keratinocytes, was not detectable in iPSCs (Supplementary Figure S1a) and the expression of the exogenous reprogramming factors was silenced (Supplementary Figure S1b). Both clones expressed endogenous pluripotency-associated genes (Figure 1d,e) and markers (SSEA-3, SSEA-4, TRA-1–60, TRA-1–81) and did not express the differentiation-associated glycoprotein SSEA-1 (Figure 1e). To test the in vivo differentiation potential of the iPSC clones, we performed teratoma assays by injecting cells intramuscularly into immunodeficient mice. Histological analysis of teratoma sections revealed the presence of cells of all three germ layers (endoderm, mesoderm, and ectoderm; Figure 1f). The above results confirm the establishment of iPSCs from keratinocytes of X-CGD patients.
Figure 1.

Derivation of X-linked chronic granulomatous disease (X-CGD) induced pluripotent stem cells (iPSCs) from plucked hair keratinocytes. (a) Diagram of the lentiviral reprogramming vector (LV-OKSM-Tomato). (b) Representative images of hair outgrowth, keratinocytes, and a derived iPSC colony (bars: left panel = 200 µm; middle and right panels = 100 µm). (c) Southern blot hybridized with the probe indicated in a showing lentiviral integrations in X-CGD iPSCs clones. Clone S22 carries three integrations and clone J14 two integrations. (d) The X-CGD iPSCs express endogenous pluripotency genes in similar levels to hESC line H7.S6. Data represent mean and SD of samples collected at day 100 and at day 125 and analyzed in triplicates. (e) X-CGD iPSCs express the pluripotency markers OCT4, NANOG, SSEA-3, SSEA-4, TRA-1–60, and TRA-1–81 and are negative for SSEA-1. Counterstaining with Hoechst 33342 and overlay is shown (bar = 100 µm). (f) X-CGD iPSCs gave rise to all three germ layers in teratoma formation assays. hESC, human embryonic stem cell.
Compensation of the gp91phox deficiency by BAC transposition
As a first approach for restoring the gp91phox deficiency, we transfected a BAC containing the CYBB gene into the X-CGD iPSCs. To guarantee a single-copy, full-length BAC integration into the genome, we used the BAC transposition method, which was pioneered previously in our lab.27 For this, we retrofitted the BAC bacterial backbone with piggyBac (PB) inverted terminal repeats (ITRs) using recombineering technology. On one side of the bacterial backbone, we inserted the PB3 ITR together with a blasticidin resistance gene driven by the Ubiquitin-C promoter. On the other side, we inserted the PB5 ITR together with the ampicillin resistance gene and the herpes simplex virus thymidine kinase (HSV-TK) gene driven by the HSV promoter for negative selection (Figure 2a). The modified BACs were transfected together with the hyperactive version of the PB transposase (hyPBase) by lipofection into human embryonic stem cells (hESCs) for testing transgenesis efficiency and then into X-CGD iPSCs. Blasticidin-resistant colonies were screened by PCR for transposition events as described previously.27 Upon transposition, the BAC integrates as a single copy, extending from PB3 ITR to PB5 ITR and excluding the bacterial backbone (Figure 2a). We detected transposition events in both iPSC clones (S22 and J14) and in one clone (S22), transposition efficiency was comparable to the H7.S6 hESC line (Figure 2b). We identified the genomic integration site of the BAC in 12 out of 15 clones analyzed by Splinkerette PCR from both sides. BAC integrations were randomly distributed across the genome (Figure 2c), being both in intragenic and intergenic regions (Supplementary Table S1). Moreover, all sites revealed the consensus motif (TTAA) of PB integration (Figure 2d). In three of the clones (#5, #6, and #21), we also confirmed the integration of the BAC in the long arms of chromosome 2, 1, and 8, respectively, by fluorescence in situ hybridization. A representative fluorescence in situ hybridization for clone #5 is shown in Figure 2e. Conventional cytogenetic analysis (G-banding) displayed an apparently normal male karyotype (46,XY) in all three clones.
Figure 2.

Transposition-mediated bacterial artificial chromosome (BAC) transgenesis. (a) Schematic representation of the 175-kb BAC containing the CYBB gene. The CYBB exons are shown as black rectangles. The bacterial backbone contains the chloramphenicol resistance gene (CmR) and the origin of replication (ori). The ubiquitin-C-BSD-pA selection cassette together with the PB3 inverted terminal repeat (ITR) was inserted on one side of the bacterial backbone. On the other side, a cassette was inserted containing the herpes simplex virus thymidine kinase negative selection marker (HSV-TK) together with an ampicillin resistance gene (ampR) and the PB5 ITR. The modified CYBB BAC was co-lipofected with the hyPBase transposase expression vector. Upon transposition, the BAC integrates as a full-length copy from the PB3 to PB5 ITR excluding the bacterial backbone. Inverted arrows indicate the PCRs for detecting transposition signature. Splinkerette PCR was used to identify the genomic integration site. (b) Table showing transposition events in H7.S6 human embryonic stem cells (hESCs) and X-linked chronic granulomatous disease (X-CGD) induced pluripotent stem cell (iPSC) clones S22 and J14. (c) Mapping of the BAC integration sites in the human genome. Red triangles mark the integration site into the human chromosomes. (d) BACs integrated into TTAA sites, a characteristic of piggyBac transposition. (e) Mapping of the BAC integration sites using fluorescence in situ hybridization for clone 5. DNA of the CYBB BAC was directly labeled in red and hybridized together with a control probe for the centromere of chromosome X (where the endogenous CYBB gene is located) labeled in green. A representative metaphase is shown with a red and a green signal on the X chromosome (endogenous CYBB gene and centromere region of chromosome X) and an additional red signal on one chromosome 2 that corresponds to the integration of the CYBB BAC (arrow).
Correction of the X-CGD–causing mutation by gene targeting
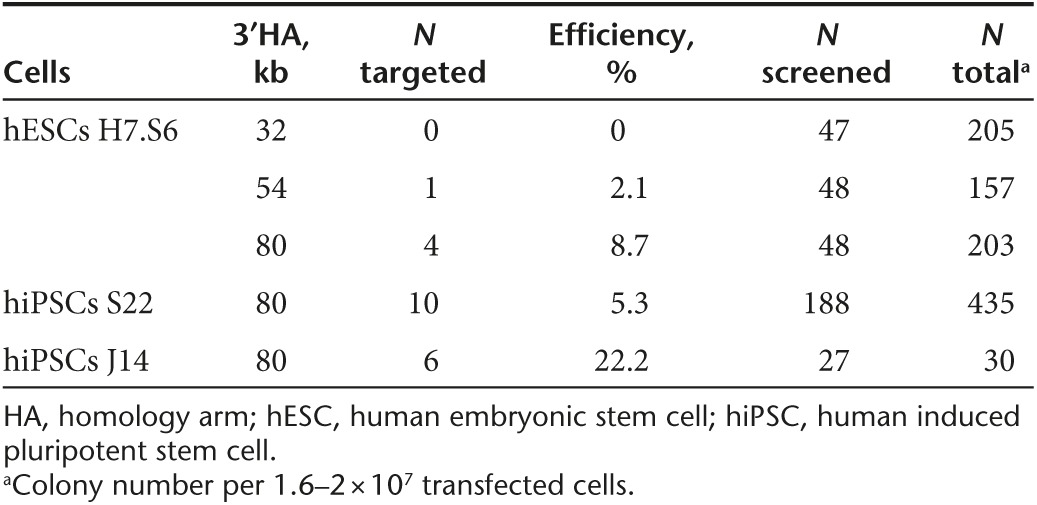
As a second approach, we aimed to correct the X-CGD–causing mutation by gene targeting in iPSCs. The CYBB gene is not expressed in human pluripotent stem cells (assayed by reverse transcription–PCR; data not shown), therefore we used a promoter-driven selection strategy for targeting. First, we inserted in the CYBB-containing BAC a PGK-neo-pA selection cassette flanked by FRT sites in intron 8, in a position that does not interfere with splicing and which is close to the mutations found in exon 9 of the gene in this patient. Then we subcloned from the BAC a targeting vector with short homology arms (HAs) (2.7-kb 5′HA and 1.7-kb 3′HA) using recombineering technology. We tested the targeting efficiency in H7.S6 hESCs but could not obtain any correct targeted events out of 168 screened colonies (data not shown). Consequently, we changed our targeting strategy and trimmed the modified BAC containing the PGK-neo-pA cassette from the 5′ end and obtained a targeting vector that has a short 5′HA (8 kb). The maximal length of the 3′HA from the downstream FRT site until the end of the genomic fragment in the BAC is 80 kb. Using single restriction sites in the 3′ region, we could shorten the length of the 3′HA to 54 kb (SbfI) and to 32 kb (SalI), respectively. This strategy allowed the use of a 5′ external probe for detecting correct targeting events by Southern blot (Figure 3a). We tested the BAC-based targeting vector in H7.S6 hESCs and experienced an increase in targeting frequency with longer 3′HAs (Table 1). Consequently, we decided to use the version with the longer 3′HA (80 kb) for targeting the iPSC clones. We confirmed correct targeting by Southern blot (Figure 3b) and detected relatively high targeting frequencies (5 and 22% in two clones) for a gene that is not expressed in human pluripotent stem cells (Table 1).
Figure 3.

Correction of the X-linked chronic granulomatous disease (X-CGD)–causing mutation by gene targeting. (a) Schematic representation of the targeting strategy and Flp recombination. In the targeting construct, a PGK-neo-pA selection cassette flanked by FRT sites was inserted in intron 8 of the CYBB gene close to the mutation in exon 9 (asterisks) in a bacterial artificial chromosome (BAC) vector. The BAC was trimmed upstream of the selection cassette to get a 5′HA of 8 kb. To obtain variable sizes of the 3′HA, the BAC was digested with SalI (32-kb 3′ homology arm (HA)), SbfI (54-kb 3′HA), or SfiI (80-kb 3′HA). (b) Southern blot of genomic DNA from G418 resistant clones digested with ApaLI and hybridized with an external 5′ probe shown in a. Homologous recombination result in a 11.6-kb band (TG), whereas the endogenous unrecombined is 10 kb. Asterisks indicate the targeted clones. (c) Southern blot analysis of X-CGD–targeted clones after Flp recombination. Upper panel: excision of the PGK-neo cassette results in a band (ΔneoFlp) that runs at the same size as the endogenous unmodified allele. Middle panel: Flp recombination resulted in the simultaneous excision of the reprogramming lentiviral vector in all clones that lost the PGK-neo cassette. Arrows point at the three lentiviral integrations. Lower panel: as a loading control, the same blot in middle panel was hybridized with the 5′ external probe. (d) Sequencing results confirming the correction of the mutation in six targeted clones. (e) Conventional cytogenetics (G-banding) of one correctly targeted and Flp-recombined clone revealed a normal male karyotype (46,XY).
Table 1. Gene targeting efficiency.
Removal of the selection cassette and of the reprogramming vectors by Flp-mediated recombination
The lentiviral vector used for reprogramming of the patient keratinocytes into iPSCs and the selection cassette (PGK-neo-pA) of our BAC-based targeting vector are both flanked by FRT sites. Hence, we aimed at excising the reprogramming vector and the selection cassette in the targeted clones simultaneously by lipofection of a codon-optimized Flp recombinase (Flpo) expression plasmid.28 To confirm excision, we screened the transfected clone (S22 #77) by Southern blot and for sensitivity to G418. As expected upon Flp-mediated excision of the selection cassette, the targeted band (11.6 kb—ApaLI digestion with 5′ external probe) shifted to the same size as the wild-type band (Figure 3c, upper blot). Surprisingly, in 56 out of 94 screened colonies (59.6%), we detected simultaneous excision of the selection cassette and all three copies of the reprogramming vector (Figure 3b, middle blot; BsrGI digestion with PRE lentiviral probe). In 2 colonies out of 94, we detected either excision of the selection cassette or of the reprogramming vector only (Supplementary Figure S2). All colonies that excised the selection cassette also lost resistance to G418. To show that the absence of signal is not due to absence of DNA in the respective lanes, we hybridized the membrane in which the genomic DNA was digested by BsrGI with the 5′ external probe (Figure 3c, lower blot). Finally, to confirm that our gene targeting approach corrected the mutation, we PCR-amplified a region surrounding the mutation and sequenced it. In all targeted and Flp-recombined clones, we confirmed the correction of the X-CGD–causing mutation (Figure 3d) (and also the presence of the single FRT site; Supplementary Figure S3a,b). Conventional cytogenetics (G-banding) of one correctly targeted and Flp-recombined clone revealed a normal male karyotype (46,XY), which was further confirmed by spectral karyotyping analysis.29 No recurrent structural (in particular, no translocations) or numerical aberrations were observed (Figure 3e and Supplementary Figure S3c).
Restoration of the NADPH oxidase activity in iPSC-differentiated neutrophils
To confirm functional restoration of the oxidase activity in the corrected X-CGD iPSCs, we performed differentiation into the myeloid lineage (Figure 4a). To monitor the differentiation efficiency into the hematopoietic lineage, we analyzed the cells for expression of CD34 and CD45 between days 18 and 20. As a positive control, we used human hematopoietic stem and progenitor cells from healthy donors. We detected high levels of CD34/CD45 expression (40–50% double-positive cells) in both patient X-CGD iPSCs and those carrying the BAC transgene (Figure 4b). For terminal differentiation, the cells were further cultured on OP9 feeder cells in differentiation medium III for 8–10 days until suspension cells appeared, followed by culture in medium supplemented with G-CSF for additional 18–24 days. At this stage, the differentiated cells were assayed for the expression of CD45 and CD13, a marker for the myeloid lineage. We detected high levels of CD13 (>90%) expression in the differentiated iPSCs, confirming the efficiency of our differentiation protocol (Figure 4c). We also monitored the expression of CD10, CD31, and CD123 (IL-3 receptor) at days 20 and 49 of iPSC differentiation. The expression of CD31 and CD123 was maintained at day 49 of differentiation, whereas expression of the lymphoid precursor–specific marker CD10 was lost (Supplementary Figure S4a). To see whether the X-CGD patient's iPSCs carrying the transgenic BAC have restored gp91phox expression, we analyzed the cells by flow cytometry. As expected, we did not detect gp91phox expression in granulocytes isolated from the X-CGD patient, confirming that the c.958delG and c.962T > G mutations lead to complete loss of the protein (X910). In contrast, the patient's differentiated iPSCs carrying the BAC transgene and the ones corrected by targeting expressed high levels of gp91phox (Figure 4d and Supplementary Figure S4b). As expected, the corrected iPSCs expressed gp91phox after differentiation into myeloid cells (day 43) but not before (day 16) (Supplementary Figure S4b). To further test whether the restored gp91phox expression is also functional, we performed an oxidase activity assay based on chemiluminescence. As expected, the X-CGD patient granulocytes did not show any oxidase activity as compared to healthy donor granulocytes, which produced superoxide in a cell-number–dependent manner (Figure 4e). Importantly, the oxidase activity in X-CGD iPSCs was also restored by the BAC transgene to levels comparable to the in vitro differentiated normal hematopoietic stem and progenitor cells (Figure 4f). Physiological levels of NADPH oxidase activity were also detected in the differentiated X-CGD iPSCs in which the mutation was corrected by gene targeting and the selection cassette together with the reprogramming vectors had been removed by Flp recombination (Figure 4g). We have also analyzed the colony-forming unit potential of the differentiated patient's iPSCs and of one BAC transgenic and one targeted iPSC clone. We only obtained granulocyte macrophage megakaryocyte (GMM) colonies, which in the case of the corrected cells showed reactive oxygen species activity as determined by nitroblue tetrazolium staining after phorbol myristate acetate stimulation (Supplementary Figure S5a). We next asked the question how many of the BAC transgenic iPSC clones were functional. We chose six BAC transgenic clones with intergenic integrations and performed nitroblue tetrazolium assay after differentiation into granulocytes. All clones that showed efficient differentiation to granulocytes (four out of six BAC transgenic and also five out of six targeted) were positive in nitroblue tetrazolium assay, whereas the patient cells were not (Supplementary Figure S5b,c).
Figure 4.

Functional restoration of oxidase activity in X-linked chronic granulomatous disease (X-CGD) induced pluripotent stem cells (iPSCs) differentiated into the hematopoietic lineage. Representative analyses are shown. (a) Diagram of the differentiation protocol. (b) Flow cytometry analysis of the hematopoietic markers CD34 and CD45 at differentiation days 18–20. (c) Flow cytometry analysis of the myeloid marker CD13 at differentiation days 43–44. (d) Expression of gp91phox at differentiation days 43–44. Note that the X-CGD granulocytes do not express gp91phox, whereas the differentiated iPSCs containing the CYBB bacterial artificial chromosome (BAC) express high levels. Differentiated human hematopoietic stem and progenitor cells (HSPCs) and granulocytes from healthy individuals serve as positive control. (e) X-CGD granulocytes do not show any oxidase activity measured by a chemiluminescence assay. Granulocytes from unaffected individuals produced superoxide in a cell-number–dependent manner. (f) Oxidase activity was undetectable in differentiated X-CGD iPSCs but was restored in the ones carrying the BAC transgene. In vitro differentiated HSPCs serve as positive control. (g) Restoration of the oxidase activity was also observed with the correctly targeted and Flp-recombined differentiated X-CGD iPSCs.
Discussion
In this study, we describe the derivation of iPSCs from a male patient with X-CGD and the subsequent restoration of the NADPH oxidase deficiency by BAC transgenesis or by homologous recombination–mediated correction of the mutation. The efficiency of iPSCs generation depends on the differentiation state of the somatic cell type and on the reprogramming protocol.30 Here, we applied a noninvasive method by isolating keratinocytes from plucked hair of the patient to avoid any disease-related complications such as disturbed wound healing that could arise after skin biopsies in the CGD patient. An important prerequisite for using iPSCs in therapeutic applications is reprogramming by integration-free methods. This can be achieved by episomal vectors, by nonintegrating viral vectors and by mRNA transfection or protein transduction of the reprogramming factors, albeit with low efficiency.31 An alternative to the above-mentioned methods is to transfect the cells with excisable vectors, which are either flanked by site-specific recombination target sites or transposon ITRs.32 Our initial attempts to reprogram keratinocytes using the PB transposon vector containing the four reprogramming factors resulted in unstable iPSC colonies, which did not completely silence the exogenous factors (data not shown). Therefore, we decided to use a Flp recombinase excisable lentiviral vector and derived iPSCs in which the exogenous reprogramming factors were silenced and successfully removed.
For restoration of the gp91phox activity in X-CGD iPSCs, we applied two strategies: BAC transgenesis and gene targeting by homologous recombination. In contrast to viral vectors in which either constitutively active heterologous promoters or myeloid-specific promoters express cDNAs, BACs depending on their size contain most of the regulatory sequences of the gene of interest and thus most likely provide physiological expression levels. Furthermore, transposon-mediated BAC transgenesis results in most of the cases in full-length, single-copy integrations.27 The genomic integration site can be easily determined by Splinkerette PCR or other similar methods and thus allowing selection of clones in which BAC integration is most likely not mutagenic. However, to exclude that the BAC insertion is not mutagenic, transplantation of the differentiated cells into surrogate models in vivo is necessary. Moreover, BAC transgenesis can be universally applied to correct gp91phox deficiency irrespective of the underlying CYBB mutation. Nonetheless, depending on the site of genomic integration, BACs may also be prone to silencing and therefore long-term monitoring of the expression level of the gene of interest in the transfected cells is required.
An alternative to BAC transgenesis is the correction of the genetic mutation by homologous recombination. However, the efficiency of gene targeting via homologous recombination in human pluripotent stem cells using classical nonviral vectors is low as compared to their mouse counterparts.33 Targeting frequencies are improved by using helper-dependent adenoviral (HdAVs) or adeno-associated viral (AAV) vectors,16,34 BAC-based vectors,17,35 and engineered nucleases.24,36 HdAVs and BAC vectors carry long HAs, a parameter that influences positively the targeting efficiency. Targeting of the CYBB locus, which is not expressed in pluripotent cells with vectors containing short HAs, was not feasible. Also, a 3′HA of 32 kb was not sufficient for homologous recombination in the CYBB locus. By extending the 3′HA to 54 kb, we were able to detect homologous recombination events with an efficiency of 2.1% in hESCs, which was further increased to 8.7% with a 3′HA of 80 kb. This increase in homologous recombination efficiency can be due to the existence of open chromatin in the downstream genomic region. Indeed, 23 kb downstream of the last CYBB exon (33 kb from the end of the selection cassette) lies the last exon of the dynein light chain 3 (DYNLT3) gene, which is transcribed from the reverse strand and is expressed in hESCs (ENCODE database37). The promoter region of DYNLT3 is located more than 34 kb downstream of the last CYBB exon (43 kb from the selection cassette), implying that the 3′HA of 32 kb is not reaching this region. Our targeting strategy is not relying on the use of engineered nucleases which can increase targeting frequencies but can also generate new mutations due to off-target cleavage.38 Another advantage of targeting vectors with long HAs is that they can be universally applied to correct other patient-specific mutations dispersed along the gene.
The single FRT site left in the intronic sequence after Flp recombination did not interfere with CYBB gene expression, because the corrected cells showed functional gp91phox activity. Nevertheless, residual site-specific recombination target sites can disrupt regulatory elements, as was the case where insertion of a loxP/FRT site in the third intron of Tnr gene altered the expression of the neighboring gene Gas5.39 Therefore, a safer alternative is the use of selection cassettes flanked by ITRs of the PB transposon, which can be seamlessly excised after transfection of the transposase.40
Although we show that correction of the X-CGD–causing mutation by homologous recombination in iPSCs and their subsequent differentiation to functional neutrophils is feasible, there is still a long way to go before employing this strategy for cell-based therapy. The reprogramming process and also the prolonged expansion required for the genetic manipulation of the iPSCs in culture may lead to the accumulation of new mutations,41 some of which could lead to malignancies. Interestingly, Howden et al.17 identified several mutations in iPSCs reprogrammed by episomal vectors as compared to the parental somatic cells, but the accumulation of new mutations by the subsequent expansion of the cells for gene targeting and excision of the selection cassette was minimal. Although the X-CGD–corrected iPSC clones were karyotypically normal in our study, a more careful in-depth analysis of their genomic integrity is required before using these cells for future therapeutic applications. In vivo testing in animal models will allow long-term assessment of the risk imposed by potential novel mutations acquired during iPSC generation and the risk of tumorigenesis by pluripotent cell contamination in the differentiated graft.
A further prerequisite for the utility of genetically corrected disease-specific iPSCs in future therapeutic applications is their capacity to efficiently differentiate into the required cell type. Various differentiation protocols into the hematopoietic lineage have been established using hESCs.42,43,44,45,46,47 However, iPSCs retain an epigenetic memory of their cell type of origin, which can compromise the differentiation efficiency into certain lineages.48 We derived iPSCs from keratinocytes, which are of ectodermal origin and could show that they can be differentiated into the hematopoietic lineage. Using our modified differentiation media, we were able to achieve a yet unprecedented high percentage of CD34+CD45+ hematopoietic stem and progenitor cells from iPSCs. These cells demonstrated colony-forming unit potential and gave rise to myeloid cells in suspension cultures. However, we also experienced that not all iPSC clones could be efficiently differentiated into granulocytes. It has previously been reported that hematopoietic cells derived from X-GCD patients mimic the CGD.18,19,20 Indeed, our iPSC-derived X-CGD neutrophils displayed complete oxidase deficiency as suggested from the initial analyses of the native X-CGD donor neutrophils that did not show any gp91phox expression nor oxidase function. Restoration of the gp91phox deficiency with both the BAC and homologous recombination approach resulted in a strong oxidase activity, comparable to that of peripheral blood granulocytes from a healthy donor. This high level of functional correction is due to the native promoter and enhancer elements leading to physiological levels of gp91phox expression. For future X-CGD gene therapy studies, the level of gp91phox expression per cell is crucial for therapeutic success, since the efficiency on eliminating pathogens depends on both, the ratio of engrafted corrected HSCs compared to the total number of resident HSCs and to the efficacy of each neutrophil to produce reactive oxygen species.
Methods to generate readily transplantable HSCs have to be established to enable routine testing of the efficacy and safety of differentiated iPSCs in vivo. There are conflicting results about engraftment of iPSCs-derived HSCs. While Wang et al.22 successfully report intraosseous injection and Hanna et al.49 used intravenous transplantation of corrected hematopoietic cells into mice, most of other groups have not been able to achieve bone marrow reconstitution from iPSCs-derived hematopoietic cells, unless they used alternative techniques such as derivation of HSCs through teratoma formation.44 In vivo testing in animal models will allow long-term assessment of the risk imposed by potential novel mutations acquired during iPSC generation and the risk of tumorigenesis by pluripotent cell contamination in the differentiated graft.
In conclusion, we established a universal method for efficient correction of any mutation in the CYBB gene in X-CGD iPSCs by homologous recombination and BAC transgenesis. After differentiation into neutrophils, the repaired cells restored the capacity to produce physiological levels of oxidase activity.
Materials and Methods
Keratinocyte culture. Outgrowths of keratinocytes from the X-CGD patient's hair carrying the deletion c.958delG and the substitution c.962T > G in exon 9, classified as X910, were done as previously described.50 The use of human material in this study is approved by the ethics committee of the Technische Universität Dresden (EK 99032010) and is in compliance with the guidelines of the Federal Government of Germany and the Declaration of Helsinki concerning Ethical Principles for Medical Research Involving Human Subjects.
Virus vector particle production. The pRRL.PPT.SF.hOct34.hKlf4.hSox2.hcmyc.i2dTomato.pre (LV-OKSM-Tomato) vector containing human reprogramming factors—OCT4, KLF4, SOX2, and c-MYC—was kindly donated by Axel Schambach (MHH, Universität Hannover).51 Virus vector particle production was performed as previously described.52
Generation and culture of iPSCs. Human keratinocytes from X-CGD patient were cultured on Matrigel (BD Biosciences, Brussels, Belgium) for 2–3 days before and 3–4 days after lentiviral vector transduction in Keratinocyte Growth Medium 2 (PromoCell, Heidelberg, Germany). Thereafter, the cells were cultured in mTeSR1 (Stem Cell Technologies, Cologne, Germany) with daily medium change until colonies appeared. The iPSCs were validated by immunostaining and quantitative reverse transcription–PCR for the expression of pluripotency-associated genes and by teratoma formation assays.
Generation of modified BACs and targeting vectors. All BAC modifications and the subsequent generation of targeting vectors were done using recombineering technology.53 Detailed information is presented in Supplementary Materials and Methods. Oligonucleotides were purchased from Biomers (Ulm, Germany) and are listed in Supplementary Table S2.
Transfection and screening of clones. PB transposase expression vector (hyPBase), a kind gift from Alan Bradley, Sanger Institute, UK54 and BAC co-lipofection was performed as previously described.27 Positive selection with 2 µg/ml blasticidin (Life Technologies, Darmstadt, Germany) and negative selection with 200 nmol/l FIAU (Fialuridine; Moravek Biochemicals, Brea, CA) started 2 and 6 days after transfection, respectively. Colonies were picked after 2 weeks, expanded, and screened by PCR for transposition events. The BAC integration sites were determined using Splinkerette PCR and sequencing as already described.27,55 For electroporation, plasmid DNA or trimmed BAC were linearized, purified by phenol–chloroform extraction, and dissolved in phosphate-buffered saline. About 3 × 106 cells were electroporated in 750 µl culture medium with 25 µg DNA at 320 V and 250 µF using a Biorad Gene Pulser. Selection with 100 µg/ml G418 started 2 days after transfection and after 2 weeks, resistant colonies were picked, expanded, and screened by Southern blot.
Conventional cytogenetic (G-banding), spectral karyotyping, and fluorescence in situ hybridization. G-banding and spectral karyotyping analysis was performed as described previously.29 Metaphase chromosomes were hybridized with a self-made spectral karyotyping hybridization probe cocktail for human chromosomes as described.56 For fluorescence in situ hybridization analysis of the integration site, the CYBB BAC was labeled via nick translation using tetramethyl-rhodamine-5-dUTP (Roche, Mannheim, Germany) and hybridized on chromosome spreads together with a commercial probe targeting the centromere of chromosome X labeled in green (DXZ1; Kreatech, Amsterdam, The Netherlands).
In vitro differentiation. For the in vitro differentiation of hiPSCs into the hematopoietic lineage, we adapted the protocol provided by Stem Cell Technologies based on Ng et al.46 and Chadwick et al.45 with small modifications. Detailed composition of the differentiation media are found in Supplementary Materials and Methods. In brief, cells were incubated with differentiation medium I and after 4–5 days with differentiation medium II. Between days 18 and 20, cells were analyzed for expression of hematopoietic markers (CD34/CD45). For further differentiation, we followed the protocol published by Yokoyama et al.47 with slight modifications. Cells were dissociated and replated on Mitomycin-C (Sigma-Aldrich, Schnelldorf, Germany) inactivated OP9 cells in differentiation medium III. From day 28 to 30 onwards, the cells were cultured with 50 ng/ml G-CSF (Miltenyi Biotec, Bergisch Gladbach, Germany) in RPMI medium (PAA, Cölbe, Germany). Between days 43 and 44, cells were analyzed by flow cytometry for expression of CD13 and gp91phox as well as for oxidase activity.
Oxidase activity assay. Freshly isolated normal or X-CGD neutrophils, differentiated normal hematopoietic stem and progenitor cells as well as in vitro differentiated patient-specific and genetically corrected iPSCs were stimulated with phorbol myristate acetate and analyzed for superoxide production by chemiluminescence using the Diogenes kit (National Diagnostics, Atlanta, GA) according to the manufacturers instructions. About 1 × 104 to 1 × 105 cells were placed into a 96-well plate and the response was measured sequentially on a Mithras LB940 (Berthold, Bad Wildbad, Germany).
SUPPLEMENTARY MATERIAL Figure S1. Expression of K14 and exogenous reprogramming factors in iPSCs. Figure S2. Flp-mediated excision of the selection cassette and lentiviral integrations. Figure S3. Confirmation of the genetic correction by sequencing. Figure S4. Expression of surface markers and gp91phox during differentiation. Figure S5. ROS activity in CFUs and differentiated granulocytes assayed by NBT staining. Table S1. CYBB-BAC genomic integration sites. Table S2. List of recombineering oligos and primers. Materials and Methods
Acknowledgments
We would like to thank Mandy Obst, Isabell Kolbe, Katrin Navratiel, and Jenny Marzahn for excellent technical assistance. We would like to thank Martin Wermke and Xenia Lojewski for sharing methods and protocols. We are thankful to Dirk Hoffmann and Axel Schambach, Medizinische Hochschule Hannover, for advice and for kindly donating the LV-OKSM-Tomato reprogramming vector. Further, we would like to thank A. Francis Stewart for advice and support. This project was financially supported by the Bundesministerium für Bildung und Forschung (BMBF) grant number 01GN1007 to K.A. and S.B. The authors have no competing financial interests. M.L., M.R., K.N., A.Z., and C.R. derived and validated X-CGD iPSCs from patient keratinocytes; M.R., S.V., and K.A. designed and generated constructs and performed BAC transgenesis and targeting; M.H., M.L., C.R., A.Z., and S.T. performed teratoma assays; B.K. and E.S. performed karyotyping and FISH; J.R. provided patient samples and helped with functional analysis; M.L., M.R., and C.R. established the hematopoietic differentiation; M.L., M.R., S.B., and K.A. designed experiments and wrote the manuscript.
Supplementary Material
References
- Winkelstein, JA, Marino, MC, Johnston, RB Jr., Boyle, J, Curnutte, J, Gallin, JI et al. (2000). Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine (Baltimore) 79: 155–169. [DOI] [PubMed] [Google Scholar]
- Holland, SM (2013). Chronic granulomatous disease. Hematol Oncol Clin North Am 27: 89–99, viii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noack, D, Heyworth, PG, Newburger, PE and Cross, AR (2001). An unusual intronic mutation in the CYBB gene giving rise to chronic granulomatous disease. Biochim Biophys Acta 1537: 125–131. [DOI] [PubMed] [Google Scholar]
- Heyworth, PG, Curnutte, JT, Rae, J, Noack, D, Roos, D, van Koppen, E et al. (2001). Hematologically important mutations: X-linked chronic granulomatous disease (second update). Blood Cells Mol Dis 27: 16–26. [DOI] [PubMed] [Google Scholar]
- Noack, D, Heyworth, PG, Curnutte, JT, Rae, J and Cross, AR (1999). A novel mutation in the CYBB gene resulting in an unexpected pattern of exon skipping and chronic granulomatous disease. Biochim Biophys Acta 1454: 270–274. [DOI] [PubMed] [Google Scholar]
- Rae, J, Newburger, PE, Dinauer, MC, Noack, D, Hopkins, PJ, Kuruto, R et al. (1998). X-Linked chronic granulomatous disease: mutations in the CYBB gene encoding the gp91-phox component of respiratory-burst oxidase. Am J Hum Genet 62: 1320–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos, D, Kuhns, DB, Maddalena, A, Roesler, J, Lopez, JA, Ariga, T et al. (2010). Hematologically important mutations: X-linked chronic granulomatous disease (third update). Blood Cells Mol Dis 45: 246–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Güngör, T, Teira, P, Slatter, M, Stussi, G, Stepensky, P, Moshous, D et al.; Inborn Errors Working Party of the European Society for Blood and Marrow Transplantation (2014). Reduced-intensity conditioning and HLA-matched haemopoietic stem-cell transplantation in patients with chronic granulomatous disease: a prospective multicentre study. Lancet 383: 436–448. [DOI] [PubMed] [Google Scholar]
- Ryser, MF, Roesler, J, Gentsch, M and Brenner, S (2007). Gene therapy for chronic granulomatous disease. Expert Opin Biol Ther 7: 1799–1809. [DOI] [PubMed] [Google Scholar]
- Grez, M, Reichenbach, J, Schwäble, J, Seger, R, Dinauer, MC and Thrasher, AJ (2011). Gene therapy of chronic granulomatous disease: the engraftment dilemma. Mol Ther 19: 28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein, S, Ott, MG, Schultze-Strasser, S, Jauch, A, Burwinkel, B, Kinner, A et al. (2010). Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med 16: 198–204. [DOI] [PubMed] [Google Scholar]
- Nienhuis, AW (2013). Development of gene therapy for blood disorders: an update. Blood 122: 1556–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee, S and Thrasher, AJ (2013). Gene therapy for PIDs: progress, pitfalls and prospects. Gene 525: 174–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann, KB, Büning, H, Galy, A, Schambach, A and Grez, M (2013). Gene therapy on the move. EMBO Mol Med 5: 1642–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicalese, MP and Aiuti, A (2015). Clinical applications of gene therapy for primary immunodeficiencies. Hum Gene Ther 26: 210–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, GH, Suzuki, K, Qu, J, Sancho-Martinez, I, Yi, F, Li, M et al. (2011). Targeted gene correction of laminopathy-associated LMNA mutations in patient-specific iPSCs. Cell Stem Cell 8: 688–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howden, SE, Gore, A, Li, Z, Fung, HL, Nisler, BS, Nie, J et al. (2011). Genetic correction and analysis of induced pluripotent stem cells from a patient with gyrate atrophy. Proc Natl Acad Sci USA 108: 6537–6542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou, J, Sweeney, CL, Chou, BK, Choi, U, Pan, J, Wang, H et al. (2011). Oxidase-deficient neutrophils from X-linked chronic granulomatous disease iPS cells: functional correction by zinc finger nuclease-mediated safe harbor targeting. Blood 117: 5561–5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, Y, Cowley, SA, Siler, U, Melguizo, D, Tilgner, K, Browne, C et al. (2012). Derivation and functional analysis of patient-specific induced pluripotent stem cells as an in vitro model of chronic granulomatous disease. Stem Cells 30: 599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brault, J, Goutagny, E, Telugu, N, Shao, K, Baquié, M, Satre, V et al. (2014). Optimized generation of functional neutrophils and macrophages from patient-specific induced pluripotent stem cells: ex vivo models of X(0)-linked, AR22(0)- and AR47(0)- chronic granulomatous diseases. Biores Open Access 3: 311–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merling, RK, Sweeney, CL, Chu, J, Bodansky, A, Choi, U, Priel, DL et al. (2015). An AAVS1-targeted minigene platform for correction of iPSCs from all five types of chronic granulomatous disease. Mol Ther 23: 147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y, Zheng, CG, Jiang, Y, Zhang, J, Chen, J, Yao, C et al. (2012). Genetic correction of β-thalassemia patient-specific iPS cells and its use in improving hemoglobin production in irradiated SCID mice. Cell Res 22: 637–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, JR, Maguire, S, Davis, LA, Alexander, M, Yang, F, Chandran, S et al. (2008). Robust, persistent transgene expression in human embryonic stem cells is achieved with AAVS1-targeted integration. Stem Cells 26: 496–504. [DOI] [PubMed] [Google Scholar]
- Hockemeyer, D, Soldner, F, Beard, C, Gao, Q, Mitalipova, M, DeKelver, RC et al. (2009). Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat Biotechnol 27: 851–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee, S, Santilli, G, Blundell, MP, Navarro, S, Bueren, JA and Thrasher, AJ (2011). Generation of functional neutrophils from a mouse model of X-linked chronic granulomatous disorder using induced pluripotent stem cells. PLoS One 6: e17565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiriaco, M, Farinelli, G, Capo, V, Zonari, E, Scaramuzza, S, Di Matteo, G et al. (2014). Dual-regulated lentiviral vector for gene therapy of X-linked chronic granulomatosis. Mol Ther 22: 1472–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostovskaya, M, Fu, J, Obst, M, Baer, I, Weidlich, S, Wang, H et al. (2012). Transposon-mediated BAC transgenesis in human ES cells. Nucleic Acids Res 40: e150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranz, A, Fu, J, Duerschke, K, Weidlich, S, Naumann, R, Stewart, AF et al. (2010). An improved Flp deleter mouse in C57Bl/6 based on Flpo recombinase. Genesis 48: 512–520. [DOI] [PubMed] [Google Scholar]
- Schröck, E, du Manoir, S, Veldman, T, Schoell, B, Wienberg, J, Ferguson-Smith, MA et al. (1996). Multicolor spectral karyotyping of human chromosomes. Science 273: 494–497. [DOI] [PubMed] [Google Scholar]
- Eminli, S, Foudi, A, Stadtfeld, M, Maherali, N, Ahfeldt, T, Mostoslavsky, G et al. (2009). Differentiation stage determines potential of hematopoietic cells for reprogramming into induced pluripotent stem cells. Nat Genet 41: 968–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao, MS and Malik, N (2012). Assessing iPSC reprogramming methods for their suitability in translational medicine. J Cell Biochem 113: 3061–3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaji, K, Norrby, K, Paca, A, Mileikovsky, M, Mohseni, P and Woltjen, K (2009). Virus-free induction of pluripotency and subsequent excision of reprogramming factors. Nature 458: 771–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockemeyer, D and Jaenisch, R (2010). Gene targeting in human pluripotent cells. Cold Spring Harb Symp Quant Biol 75: 201–209. [DOI] [PubMed] [Google Scholar]
- Mitsui, K, Suzuki, K, Aizawa, E, Kawase, E, Suemori, H, Nakatsuji, N et al. (2009). Gene targeting in human pluripotent stem cells with adeno-associated virus vectors. Biochem Biophys Res Commun 388: 711–717. [DOI] [PubMed] [Google Scholar]
- Song, H, Chung, SK and Xu, Y (2010). Modeling disease in human ESCs using an efficient BAC-based homologous recombination system. Cell Stem Cell 6: 80–89. [DOI] [PubMed] [Google Scholar]
- Hockemeyer, D, Wang, H, Kiani, S, Lai, CS, Gao, Q, Cassady, JP et al. (2011). Genetic engineering of human pluripotent cells using TALE nucleases. Nat Biotechnol 29: 731–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein, BE, Birney, E, Dunham, I, Green, ED, Gunter, C and Snyder, M (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y, Foden, JA, Khayter, C, Maeder, ML, Reyon, D, Joung, JK et al. (2013). High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol 31: 822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier, ID, Bernreuther, C, Tilling, T, Neidhardt, J, Wong, YW, Schulze, C et al. (2010). Short DNA sequences inserted for gene targeting can accidentally interfere with off-target gene expression. FASEB J 24: 1714–1724. [DOI] [PubMed] [Google Scholar]
- Yusa, K (2013). Seamless genome editing in human pluripotent stem cells using custom endonuclease-based gene targeting and the piggyBac transposon. Nat Protoc 8: 2061–2078. [DOI] [PubMed] [Google Scholar]
- Hussein, SM, Batada, NN, Vuoristo, S, Ching, RW, Autio, R, Närvä, E et al. (2011). Copy number variation and selection during reprogramming to pluripotency. Nature 471: 58–62. [DOI] [PubMed] [Google Scholar]
- Vodyanik, MA, Bork, JA, Thomson, JA and Slukvin, II (2005). Human embryonic stem cell-derived CD34+ cells: efficient production in the coculture with OP9 stromal cells and analysis of lymphohematopoietic potential. Blood 105: 617–626. [DOI] [PubMed] [Google Scholar]
- Matsumoto, K, Isagawa, T, Nishimura, T, Ogaeri, T, Eto, K, Miyazaki, S et al. (2009). Stepwise development of hematopoietic stem cells from embryonic stem cells. PLoS One 4: e4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amabile, G, Welner, RS, Nombela-Arrieta, C, D'Alise, AM, Di Ruscio, A, Ebralidze, AK et al. (2013). In vivo generation of transplantable human hematopoietic cells from induced pluripotent stem cells. Blood 121: 1255–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadwick, K, Wang, L, Li, L, Menendez, P, Murdoch, B, Rouleau, A et al. (2003). Cytokines and BMP-4 promote hematopoietic differentiation of human embryonic stem cells. Blood 102: 906–915. [DOI] [PubMed] [Google Scholar]
- Ng, ES, Davis, R, Stanley, EG and Elefanty, AG (2008). A protocol describing the use of a recombinant protein-based, animal product-free medium (APEL) for human embryonic stem cell differentiation as spin embryoid bodies. Nat Protoc 3: 768–776. [DOI] [PubMed] [Google Scholar]
- Yokoyama, Y, Suzuki, T, Sakata-Yanagimoto, M, Kumano, K, Higashi, K, Takato, T et al. (2009). Derivation of functional mature neutrophils from human embryonic stem cells. Blood 113: 6584–6592. [DOI] [PubMed] [Google Scholar]
- Kim, K, Zhao, R, Doi, A, Ng, K, Unternaehrer, J, Cahan, P et al. (2011). Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells. Nat Biotechnol 29: 1117–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna, J, Wernig, M, Markoulaki, S, Sun, CW, Meissner, A, Cassady, JP et al. (2007). Treatment of sickle cell anemia mouse model with iPS cells generated from autologous skin. Science 318: 1920–1923. [DOI] [PubMed] [Google Scholar]
- Aasen, T and Izpisúa Belmonte, JC (2010). Isolation and cultivation of human keratinocytes from skin or plucked hair for the generation of induced pluripotent stem cells. Nat Protoc 5: 371–382. [DOI] [PubMed] [Google Scholar]
- Voelkel, C, Galla, M, Maetzig, T, Warlich, E, Kuehle, J, Zychlinski, D et al. (2010). Protein transduction from retroviral Gag precursors. Proc Natl Acad Sci USA 107: 7805–7810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugarte, F, Ryser, M, Thieme, S, Fierro, FA, Navratiel, K, Bornhäuser, M et al. (2009). Notch signaling enhances osteogenic differentiation while inhibiting adipogenesis in primary human bone marrow stromal cells. Exp Hematol 37: 867–875.e1. [DOI] [PubMed] [Google Scholar]
- Hofemeister, H, Ciotta, G, Fu, J, Seibert, PM, Schulz, A, Maresca, M et al. (2011). Recombineering, transfection, Western, IP and ChIP methods for protein tagging via gene targeting or BAC transgenesis. Methods 53: 437–452. [DOI] [PubMed] [Google Scholar]
- Yusa, K, Zhou, L, Li, MA, Bradley, A and Craig, NL (2011). A hyperactive piggyBac transposase for mammalian applications. Proc Natl Acad Sci USA 108: 1531–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn, C, Hansen, J, Schnütgen, F, Seisenberger, C, Floss, T, Irgang, M et al. (2007). Splinkerette PCR for more efficient characterization of gene trap events. Nat Genet 39: 933–934. [DOI] [PubMed] [Google Scholar]
- Schröck, E, Veldman, T, Padilla-Nash, H, Ning, Y, Spurbeck, J, Jalal, S et al. (1997). Spectral karyotyping refines cytogenetic diagnostics of constitutional chromosomal abnormalities. Hum Genet 101: 255–262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.