

Realization of the early promise of gene therapy has been elusive. Long and eagerly anticipated, particularly for orphan diseases with few available therapeutic alternatives, only within recent years have such treatments begun to reach phase III clinical trials.1 Two articles in this issue of Molecular Therapy identify inhibition of glycolate oxidase as a potential treatment for type 1 primary hyperoxaluria (PH1), an autosomal recessive condition in which excess oxalate produced by the liver causes kidney damage and severe systemic disease. Both teams of investigators demonstrated reduction in oxalate production in a mouse knockout model of the disease through absence or inhibition of glycolate oxidase (GO). Hepatic GO converts glycolate to glyoxylate, which is the primary substrate for production of oxalate. Thus, substrate reduction was achieved. Martin-Higueras and colleagues2 demonstrated that the hyperoxaluria of the AGXT knockout mouse was substantially reduced following double knockout of AGXT and GO. Dutta and colleagues3 approached the problem differently, using Dicer-short interfering RNA (Dicer-siRNA) for inhibition targeting hydroxyacid oxidase 1 (HAO-1) messenger RNA, which encodes GO.3 Using this approach, they were able to demonstrate normalization of oxalate production in hepatocytes of AGXT knockout mice. These observations suggest an innovative approach to treatment for patients with PH1 and impart new hope for patients with PH, their families, and the physicians who care for them. This is especially true in that early experience with other applications of siRNA for treatment of human diseases, including amyloidosis, hypercholesterolemia, and α1-antitrypsin deficiency, has been encouraging.1,4
The primary hyperoxalurias (PHs) are autosomal recessive inborn errors of metabolism characterized by marked hepatic overproduction of oxalate. Three types are known: PH1 is caused by deficiency of hepatic peroxisomal alanine-glyoxylate aminotransferase (AGT) resulting from mutations in the AGXT gene, type 2 (PH2) by deficiency of cytosolic and mitochondrial glyoxylate reductase, and type 3 (PH3) by deficiency of mitochondrial 4-hydroxy-2-oxoglutarate aldolase.5 Absence or deficiency of each of these enzymes results in marked overproduction of oxalate, which cannot be broken down in humans and must be eliminated primarily by kidney excretion. As a result of the relative insolubility of oxalate in urine, calcium oxalate kidney stones form in the collecting system of the kidneys. Calcium oxalate crystals in the glomerular filtrate are injurious to cells of the proximal renal tubules and are incorporated into the kidney interstitium, where they cause further damage with loss of kidney function over time.6 Progressive kidney damage reduces the capacity of the kidneys to excrete the excess oxalate. High concentration of oxalate in the blood then leads to calcium oxalate deposition in blood vessels, myocardium, cardiac conduction system, bone, retina, and other organs.5 Severe multisystem disease (oxalosis) ensues and can be fatal. AGT deficiency (PH1) accounts for 80% of patients with PH.7 The most severe of the three types, it is associated with the highest levels of oxalate production and the greatest frequency of end-stage renal failure.7 PH1 can cause irreversible renal failure and severe systemic oxalosis at any age, including infancy. In most patients with renal failure caused by PH, dialysis is unable to keep pace with daily production.8
Some patients with PH1 experience significant improvement or even normalization of urine oxalate excretion (and thus presumably hepatic production) when receiving pharmacological doses of pyridoxine.9 However, for most PH1 patients, liver transplantation is the only effective method of AGT enzyme replacement. Combined liver and kidney transplants are the standard of care for PH1 patients who progress to end-stage renal disease. Experience in managing this challenging disease, including early initiation of pyridoxine and medications to inhibit crystalluria, attention to recurrent nephrolithiasis, and PH-specific management of advanced chronic kidney disease, has improved outcomes.10 Nonetheless, by the fourth decade of life, 50% of PH1 patients will have progressed to end-stage renal failure, and by age 60, nearly all will have done so.7 Although improvements in clinical care have brought benefits, no new treatments have emerged in over 25 years. The absence of effective alternatives, particularly treatments that reduce hepatic oxalate production, has been discouraging.
The novel siRNA inhibition of GO described in this issue is thus of considerable interest. The results in the rodent model are striking, and the technique described by Martin-Higueras et al.2 offers an avenue for development of a primate model of PH1 for further testing of both efficacy and safety of this approach. Recent work from the Rare Kidney Stone Consortium suggests that even if reduction in urine oxalate achieved by inhibition of GO is partial and some degree of hyperoxaluria persists, there is potential to improve patient outcomes.12 In a retrospective study of PH patients who presented before the onset of end-stage renal disease, urine oxalate excretion rates of <1.6 mmol/1.73 m2/day (normal range <0.45) were associated with better preservation of kidney function over several decades of follow-up than those with higher excretion rates (Figure 1). Although glycolate production increased following inhibition of GO, this compound did not appear to cause harm in the mice studied, nor in an 8-year-old boy recently reported with isolated and asymptomatic glycolic aciduria due to congenital absence of GO caused by loss-of-function mutations.11 Finally, siRNA inhibition is now in phase II and III clinical trials for treatment of several other conditions.1 Thus far, the efficacy of these agents in humans has been noteworthy, and their tolerability is encouraging.
Figure 1.
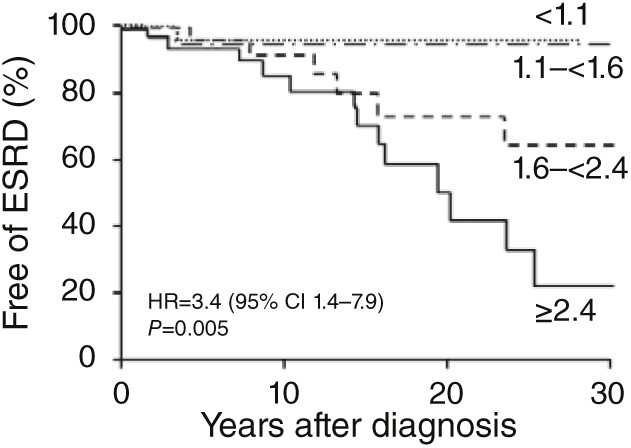
Kaplan–Meier plots of kidney survival among 168 patients with primary hyperoxaluria (PH) who did not have end-stage renal disease (ESRD) at diagnosis. Patients are shown by quartile of urine oxalate excretion (normal <0.45 mmol/1.73 m2/day). Patients in the highest quartile of urine oxalate excretion of ≥2.4 had the worst renal survival (42% at 20 years following diagnosis), compared with renal survival of 95% 20 years following diagnosis in those who had <1.6 mmol/1.73 m2/day. Reprinted from ref. 12.
There is also reason for caution. Metabolic pathways of glyoxylate generation and its disposition differ in rodents and humans, and the relative contribution of GO to oxalate production in humans is unknown. Previous proposed treatments that appeared effective in rodent models have been disappointing in clinical trials with PH1 patients, including attempted substrate reduction using (L)-2-oxothiazolidine-4-carboxylate, which is converted to cysteine and can then form an adduct with glyoxylate,13 or administration of Oxalobacter formigenes to promote enteric elimination of oxalate.14 Importantly, safety as well as efficacy of GO inhibition is unknown and may have unanticipated effects due to alteration of metabolic pathways that are incompletely understood. Nonspecific targeting of siRNA or activation of an immune response due to the agent itself could complicate use of these agents. Other important questions arise. Given that a lifetime of treatment will be needed, how well will it be tolerated over time? What will be the effects of siRNA in growing children? If parenteral administration is required, will that be sustainable over decades? Finally, comparative effectiveness with enzyme replacement by liver transplantation will need to be established. Clinical trials of siRNA inhibition of GO in PH1 patients are anticipated within a year and will provide answers to at least some of these questions soon. One is left with optimism, tempered by the requirement for thoughtful, rigorous, and long-term assessment before the role of GO inhibition in treatment of PH is understood. The valuable contributions of Martin-Higueras and Dutta and their respective teams2,3 represent a healthy beginning.
References
- Bobbin, ML and Rossi, JJ (2016). RNA interference (RNAi)-based therapeutics: delivering on the promise? Annu Rev Pharmacol Toxicol 56: 103–122. [DOI] [PubMed] [Google Scholar]
- Martin-Higueras, C, Luis-Lima, S and Salido, E (2016). Glycolate oxidase is a safe and efficient target for substrate reduction therapy in a mouse model of primary hyperoxaluria type I. Mol Ther 24: 719–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta, C, Avitahl-Curtis, N, Pursell, N, Larsson Cohen, M, Holmes, B, Diwanji, R et al. (2016). Inhibition of glycolate oxidase with Dicer-substrate siRNA reduces calcium oxalate deposition in a mouse model of primary hyperoxaluria type I. Mol Ther 24: 770–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho T, Adams, D, Silva, A, Lozeron, P, Hawkins, PN, Mant, T et al. (2013). Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med 369: 819–829. [DOI] [PubMed] [Google Scholar]
- Cochat, P and Rumsby, G (2013). Primary hyperoxaluria. N Engl J Med 369: 649–658. [DOI] [PubMed] [Google Scholar]
- Knauf, F, Asplin, JR, Granja, I, Schmidt, IM, Moeckel, GW, David, RJ et al. (2013). NALP3-mediated inflammation is a principal cause of progressive renal failure in oxalate nephropathy. Kidney Int 84: 895–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopp, K, Cogal, AG, Bergstralh, EJ, Seide, BM, Olson, JB, Meek, AM et al. (2015). Phenotype-genotype correlations and estimated carrier frequencies of primary hyperoxaluria. J Am Soc Nephrol 26: 2559–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, X, Voskoboev, NV, Wannarka, SL, Olson, JB, Milliner, DS and Lieske, JC (2014). Oxalate quantification in hemodialysate to assess dialysis adequacy for primary hyperoxaluria. Am J Nephrol 39: 376–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppe, B, Beck, BB and Milliner, DS (2009). The primary hyperoxalurias. Kidney Int 75: 1264–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergstralh, EJ, Monico, CG, Lieske, JC, Herges, RM, Langman, CB, Hoppe, B et al. (2010). Transplantation outcomes in primary hyperoxaluria. Am J Transplant 10: 2493–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frishberg, Y, Zeharia, A, Lyakhovetsky, R, Bargal, R and Belostotsky, R (2014). Mutations in HAO1 encoding glycolate oxidase cause isolated glycolic aciduria. J Med Genet 51: 526–529. [DOI] [PubMed] [Google Scholar]
- Zhao, F, Bergstralh, EJ, Mehta, RA, Vaughan, LE, Olson, JB, Seide, BM et al. (2016). Predictors of incident ESRD among patients with primary hyperoxaluria presenting prior to kidney failure. Clin J Am Soc Nephrol 11: 119–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes, RP, Assimos, DG, Wilson, DM and Milliner, DS (2001). (L)-2-oxothiazolidine-4-carboxylate in the treatment of primary hyperoxaluria type 1. BJU Int 88: 858–862. [DOI] [PubMed] [Google Scholar]
- Hoppe, B, Groothoff, JW, Hulton, S-A, Cochat, P, Niaudet, P, Kemper, MJ et al. (2011). Efficacy and safety of Oxalobacter formigenes to reduce urinary oxalate in primary hyperoxaluria. Nephrol Dial Transplant 26: 3609–3615. [DOI] [PubMed] [Google Scholar]
- Monico, CG, Olson, JB, Bergstralh, EJ, Heilman, RL and Milliner, DS (2011). Effect of betaine on urine oxalate in primary hyperoxaluria, type I [abstr.]. J Am Soc Nephrol 22 (suppl.):303A. [Google Scholar]