

Abstract
We describe a supramolecular surface competition assay for quantifying glutamine uptake from single cells. Cy3-labeled cyclodextrins were immobilized on a glass surface as a supramolecular host/FRET donor, and adamantane-BHQ2 conjugates were employed as the guest/quencher. An adamantane-labeled glutamine analog was selected through screening a library of compounds and validated by cell uptake experiments. When integrated onto a single cell barcode chip (SCBC) with a multiplex panel of 15 other metabolites, associated metabolic enzymes, and phosphoproteins, the resultant data provided input for a steady state model that describes energy potential in single cells, and correlates that potential with receptor tyrosine kinase signaling. We utilize this integrated assay to interrogate a dose-dependent response of model brain cancer cells to EGFR inhibition. We find that low dose (1 μM erlotinib) drugging actually increases cellular energy potential even as glucose uptake and phosphoprotein signaling is repressed. We also identify new interactions between phosphoprotein signaling and cellular energy processes that may help explain the facile resistance exhibited by certain cancer patients to EGFR inhibitors.
1. Introduction
Cancer may be considered a metabolic disease, where multiple deregulated metabolic pathways contribute to disease progression and development of drug resistance[1-4]. The prototypical metabolic alternation in cancer is the Warburg effect, where cells exhibit an elevated glycolysis in aerobic or anaerobic environments[1,4]. Upregulated glutamine metabolism has recently been recognized as another unique feature of many tumors [5]. In those cases, glutamine can participate in the TCA cycle through conversion to α-ketoglutarate, and provide an alternative energy source to glucose [6-10]. Certain cases of drug resistance in cancer are accompanied by heightened glutamine metabolism [10-14].
Methods for the analysis of cellular glutamine uptake have provided powerful biological insights, but they are largely limited to isotopic labeling followed by radioactivity or mass spectrometric analysis of bulk samples [15, 16]. Here we extend glutamine uptake assays to the single cell level to help resolve the heterogeneous nature of bulk tissues or tissue cultures that can mask deeper meanings. Additionally, we combine those measurements with assays for a panel of 15 other metabolites and proteins to help resolve relationships that are hard to establish when the analytes are measured independently [17]. The glutamine uptake assay is based upon a novel supramolecular chemistry approach. For the first part of this paper we describe the development and validation of that assay. We then describe how the addition of the glutamine uptake assay to a combined metabolite/protein panel that includes glucose uptake and assays for the levels of glutathione (GSH), cAMP, and cGMP, plus a number of associated enzymes, permits the construction of a semi-quantitative metabolic model for understanding the dose-dependent response of model glioblastoma multiforme cancer cells to receptor tyrosine kinase inhibition.
2. Results
2.1 Surface patterned supramolecular FRET pair
A major objective of this work was to develop a glutamine uptake assay that was compatible with existing surface assays for glucose uptake [17] and for assessing the levels of various other metabolites, proteins, and phosphoproteins [18]. The glutamine uptake measurement is a competition surface assay based upon a supramolecular interactions and Fluorescence Resonant Energy Transfer (FRET). The chemical structures of the supramolecular FRET pair used in this work and the detection method are shown in Fig. 1. Cy3-labeled β-cyclodextrin molecules (Supporting information Fig S1) were conjugated to a single-strain DNA through a hydrazine/aldehyde linker to serve as the supramolecular host/FRET donor. Another single strain DNA with a complementary sequence was covalently patterned onto a glass slide. The cyclodextrin-Cy3 conjugate was then immobilized to the surface through DNA hybridization (Fig. 1a). Different numbers of cyclodextrin-Cy3 moieties were conjugated to the single strain DNA, and the fluorescence intensities after hybridization were evaluated. A 2:1 ratio of cyclodextrin/DNA was determined to be the most optimal (Supporting information Fig. S2).
Fig. 1.

Illustration of the supramolecular glutamine assay. (a) The chemical structures and the functioning mechanism of the supramolecular FRET pair. The cyclodextrin-Cy3 was conjugated to a single strain DNA and then immobilized onto the glass slide through DNA hybridization. The Cy3 groups serve as FRET donor and the cyclodextrins as supramolecular host. A dark quencher group, BHQ2, was conjugated to an adamantane to form the FRET acceptor / supramolecular guest. The binding between adamantane and cyclodextrin brings BHQ2 to the vicinity of Cy3 and quenches the fluorescence. (b) Fluorescence intensities of the surface Cy3 with and without 100 nM of adamantane-BHQ2. (c) An adamantane-labeled glutamine analog competes with the adamantane-BHQ2 for the binding site of surface cyclodextrin, thus inhibiting the quenching process and retaining Cy3 fluorescence. (d) Fluorescence intensities of the surface Cy3 with 100 nM of adamantane-BHQ2 and different concentrations of the glutamine analog.
A dark quencher molecule (BHQ2) was conjugated to an adamantane group to form the supramolecular guest/FRET acceptor. PEG moieties were attached to the molecule to aid the aqueous solubility and also to minimize the non-specific binding between the FRET pair (Supporting information Fig.S3). When the adamantane-BHQ2 solution is introduced to the surface cyclodextrin-Cy3, the adamantane group forms a supramolecular host-guest complex with the cyclodextrin [19], which brings the BHQ2 group to the vicinity of the Cy3 fluorophore and quenches the fluorescence. As shown in Fig. 1b, adamantane-BHQ2 can effectively quench the Cy3 fluorescence at a low concentration.
On the basis of the supramolecular FRET pair, an adamantane-labeled glutamine analog will compete with the adamantane-BHQ2 molecule for the binding site of surface cyclodextrin. This causes an attenuated quenching efficiency and a retained fluorescence signal (Fig. 1c). Because of the nature of competition, the intensity of the residue fluorescence is proportional to the amount of glutamine analog in the solution, and the response can be tuned through varying the adamantane-BHQ2 concentration (Supporting information Fig S4). Fig. 1d shows the fluorescence intensity increases upon adding more glutamine analog (in this case, compound 3).
2.2 Screening of glutamine analogs
A library of eight different adamantane-labeled amino acids were generated to screen for the best glutamine analog candidate (Fig. 2A). All candidates have an adamantane label at the terminus of a side chain for anchoring the amino acid to surface-attached cyclodextrin. These candidates were tested using cell-uptake experiments conducted on a human glioblastoma cancer cell line with amplified epidermal growth factor receptor (EGFR) expression and an EGFR variant 3 (vIII) gene mutation – U87/EGFRvIII. These cells were first incubated with the glutamine analog candidates for 20 minutes to allow uptake. The cells were washed to remove excess analog in the solution, and then lysed to release the intracellular glutamine analog back into the solution. That analog then competes with the adamantane-BHQ2 for the surface cyclodextrin binding and results in retained Cy3 fluorescence (Fig. 2b).
Fig. 2.

The screening of the glutamine analogs. (a) The structures of glutamine and the library of glutamine analogs. (b) The illustration and the results of the cell uptake experiments. Compound 3 and 8 exhibit expected temperature-dependency and glutamine-dependency. Compound 3 was selected as the glutamine analog due to the better response. (c) The uptake of compound 3 in control, sodium-free media, and SLC1A5-knock down U87 cells.
Side-by-side uptake experiments were performed in complete culture medium at 37 °C and 4 °C respectively to validate the active transport process of the compound. Because the cellular uptake of glutamine is an active process, lowering the temperature will inhibit the uptake due to the slower metabolic rate. Simultaneously, cells were incubated with the compound in a glutamine-free medium at 37 °C to validate the competitive glutamine substrate feature of the analog. A suitable glutamine analog will compete with glutamine for the transporter binding, therefore a glutamine-depleted environment should result in an elevated uptake of the glutamine analog. The results of the cell uptake experiments are shown in Fig. 2b. Compounds 1, 2, 4, 5 and 7 exhibited minimal changes between the different culturing environments, and are thus poor candidates as glutamine analogs. Compound 6 had a significantly inhibited uptake at decreased temperature, but glutamine-depletion did not strongly influence uptake. Thus, Compound 6 either strongly out-competes glutamine, or it enters the cell through a glutamine transporter-independent pathway, and so is not a good glutamine surrogate. Compound 3 and 8 both yielded satisfactory temperature-dependence and glutamine-dependence. Due to the relatively lower background signal at 4 °C, compound 3 was selected as the glutamine analog for additional validation studies (Fig. 2b).
Compound 3 was selected for further testing as a glutamine surrogate through additional experiments (Fig 2c). SLC1A5 is a sodium-dependent neutral amino acid transporter that provides the major transportation for glutamine. siRNA knockdown experiments of SLC1A5 in U87 cells, or U87 cells incubated in sodium free media, both showed decreased uptake of compound 3. This proves that compound 3 is transported into the cell through primarily the SLC1A5 transporter. The existence of compound 3 also yielded less glutamate in the cells (Supporting information Fig S5), further showing that compound 3 can inhibit glutamine uptake. Finally, the results of compound 3 uptake closely resemble the glutamate assay results under various drug conditions (Supporting information Fig S6). The glutamate assay provides insight into the amount of glutamine that is being processed by the cell, rather than uptaken and then pumped back out. Cell proliferation studies showed that compound 3 has no observable toxicity (Supporting information Fig S7).
2.3 Glutamine uptake measurements on an integrated metabolic/proteomic single cell barcode chip
The glutamine uptake assay was incorporated onto an integrated metabolic/proteomic single cell barcode chip (SCBC) platform along with assays for 15 other analytes (Fig. 3a, Supporting information Fig S8). The SCBC consists of a two-layer PDMS chip and a DNA barcoded glass slide. The PDMS chip allows the spatial separation of single cells into ~1 nanoliter volume microchambers, with each microchamber designed for on-chip single cell lysis. The DNA barcode slide provides, within each microchamber, a scaffold for a multiplexed immunofluorescence assay for different analytes through a DNA-encoded antibody library (DEAL) method [20]. U87EGFRvIII cells were treated for 24 hrs with 1 μM or 10 μM of erlotinib (EGFR inhibitor) or DMSO (control) for analysis. 10 μM erlotinib treatment is effective in inhibiting cell proliferation, while 1 μM erlotinib has little to no effect (Supporting information Fig S9). In clinical studies, 1 μM of erlotinib in patient plasma is well tolerated, while 10 μM is beyond the normal tolerance level. In general, single agent erlotinib treatments at low concentrations are ineffective [21-23].
Fig. 3.
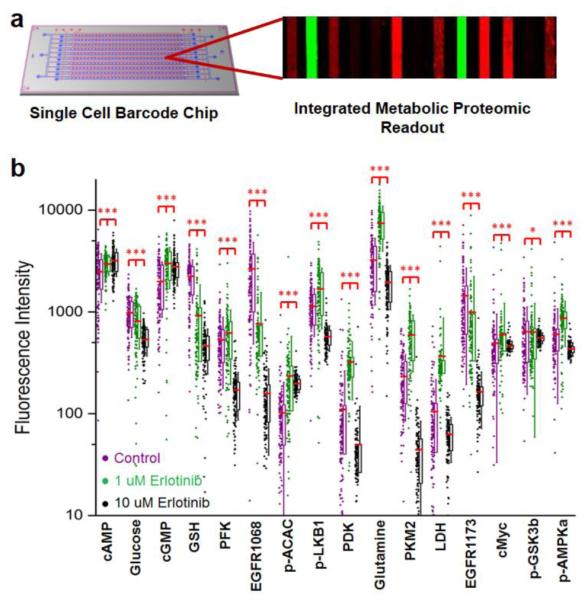
SCBC experiments on the U87EGFRvIII cells. (a) Illustration of the SCBC platform. (b) SCBC results of U87EGFRvIII cells treated with 1 μM and 10 μM of erlotinib for 24 hr. Compound 3 was used as the glutamine analog. The red horizontal lines denote the mean fluorescence value of the measured analyte, the boxes cover the second and the third sample quartiles and the whiskers label the standard deviation. (*, p < 0.1; ***, p < 0.001)
As shown in Fig. 3b, both high dose and low dose erlotinib treatments suppressed the phospho(p)-EGFR level at two phosphorylation sites (Y1068 & Y1173), indicating that the drug has engaged its target. The glucose uptake decreased upon drug treatment and higher dose further inhibited glucose uptake levels. This result is consistent with previous studies, including 18F-fluorodeoxyglucose (FDG) in vivo molecular imaging studies of EGFRvIII mutant GBM tumors using Positron Emission Tomography (PET), with images collected 24 hr following dosing[17]. The glutamine uptake levels increased upon 1 μM drug treatment and decreased in the 10 μM samples. The levels of several intracellular protein levels showed similar trends to that of glutamine. By comparison, the cAMP and cGMP levels increased while the GSH level decreased upon drug treatment, collectively indicating that the cells were under stress [24, 25]. Additionally, the distributions of many analyte levels in the 1 μM drug treated cells broadened relative to those from the control, implying an increase in cellular heterogeneity following low dose treatment. Similarly, high dose treatment lead to much narrower distribution, corresponding to a decreased heterogeneity.
2.4 A simplified model for analyzing integrated metabolic/proteomic data
The data of Fig. 3b indicate that although glucose uptake is repressed in the cells following low-dose erlotinib treatment, the activity of enzymes associated with glucose-based energy production increases. Further, glutamine uptake increases, although the cellular function that is served by this increase is not clear. One possibility is that the cells respond to erlotinib by increasing energy production, but that is not directly measured in these SCBC assays. In order to better understand the SCBC data, we carried out a few dose-dependent functional and metabolic assays on bulk cell cultures to assess relative changes in signatures of energy production (ATP levels and O2 consumption), as well as bulk glucose uptake, and cellular proliferation (Figure 4). First, cell proliferation is not significantly repressed for erlotinib dosing of 2 μM or less (Fig 4a). Second, glucose uptake is repressed in proportion to erlotinib dosing, for dosing from 1 μM to 20 μM (Fig 4b). Third, two key metrics of energy production – O2 consumption (reflective of TCA cycle activity) and intracellular ATP concentration (a more global energy metric), both increase for low (≤ 2μM) dosing (Fig. 4c,d). The implication is that cellular energy production actually increases upon low dose erlotinib treatment, and glucose uptake is a poor metric of that production. This prompted us to seek a semi-quantitative metabolic model, based upon the SCBC assays, to help guide the interpretation of the response of the GBM cells to erlotinib treatment, in terms of energy production potential (Fig. 5a).
Fig. 4.
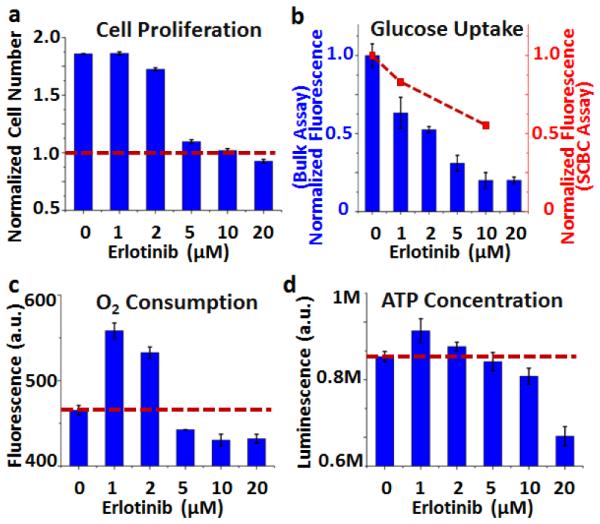
Functional and metabolic bulk assays on U87EGFRvIII cells treated with various concentrations of erlotinib for 24 hrs. (a) Normalized cell numbers after 24 hours. The red line shows the plating number. (b) Glucose uptake results from bulk (blue bars) and SCBC assays (red dots). (c) Oxygen consumption and (d) ATP concentration assay results. The red lines show the levels from the control samples.
Fig. 5.

Data analysis based on the metabolic model. (a) The simplified metabolic model. The production number of various high energy molecules (ATP, NADH, FADH2, etc) are converted to the production of ATP equivalents. (b) The definition of the energy indices. (c) Scatter plots of the indices. (d) The correlations networks for different samples. (e) The clustering result of the combined SCBC dataset.
The model of Fig 5a is focused on the glycolysis pathway and the TCA cycle, which cover the major contributors of cellular energy generation. Glucose is used by cells for many tasks, but by combining the measurement of glucose uptake with assays of those enzymes that process glucose for energy and lactate production, we can estimate the contributions of glucose to glycolysis and to the TCA cycle (Fig 5a,b). We assume that each glucose molecule consumed yields 8 ATP equivalents through glycolysis, while 6 ATP equivalents are further consumed through lactate production, and an additional 22 ATP equivalents through the TCA cycle. The numbers here are not the actual ATP production number, but an energy-currency conversion, which describes the energy potential from the uptaken fuel molecules. The first part of glycolysis generates 2 ATP and 2 NADH molecules per glucose, and each NADH is ultimately worth 3 ATP, thus making 8 ATP total. Similar treatments are applied for lactate generation and the TCA cycle to obtain ATP numbers. For glutamine that is taken up by the cell, there are multiple possible outcomes. It can enter the TCA cycle to generate energy, it can be exported in exchange for other amino acids, or it can participate in biosynthesis of glutathione and proteins, as examples. On this basis, we consider glutamine as a special currency, which can either be used directly by the cell to produce energy, or in exchange of other necessary nutrients or to perform some crucial metabolic function. Even though our method only probes the glutamine uptake, and does not provide information on the fate of the uptaken glutamine; from a global viewpoint, the active uptake of glutamine reflects a metabolic demand from the cell, regardless of how it is used. In order to have an integrated picture of the potential energy production from glutamine and glucose, we simply assume that upon the regulation of cMyc,[26] all glutamine participates in the TCA cycle, so that each glutamine molecule affords 8.5 ATP equivalents at most. To calculate the energy production for each pathway, glucose and glutamine uptake levels as well as related metabolic enzyme levels are normalized into the range between 0 and 1 across all samples. We then used the normalized analyte levels (α) and a steady-state approximation (Supporting information Fig. S10) to calculate the energy production rate for each step (Fig. 5b). We extracted several energy indices from the model. The energy potential index (EPI) assesses the potential contribution to the energy pool from glucose and glutamine metabolism, while the energy source index (ESI) measures the energy contribution ratio between glucose and glutamine. The glycolysis index (GI) weighs the contribution of glycolysis in total glucose-based energy production, and depends upon the levels of lactate dehydrogenase (LDH) as well as pyruvate dehydrogenase kinase (PDK). In addition to the energy indices, we use the cAMP and cGMP levels to define an energy frustration index (EFI), and use the GSH level to define a redox frustration index (RFI).
The calculated indices provided a functional assessment of how the cellular heterogeneity is altered by drugging. Fig. 5c shows the scatter plot of the indices. Both the GI and the EPI increased upon 1 μM erlotinib treatment. Glutamine uptake is increased at 1 μM dosing. Both glucose and glutamine uptake are decreased at 10 μM erlotinib treatment. The implication is that low dose erlotinib induced a higher energy production rate in cells, as those cells adapt to the repressed p-EGFR levels. A consequence is that the cells become more glycolytic, in spite of repressed glucose uptake. On the other hand, higher drug concentration inhibits cellular energy production and the cells become less glycolytic. The ESI dropped as drug concentration was increased, possibly suggesting that higher drug concentration increases the cellular dependency on glutamine metabolism. Both the EFI and RFI values increase upon drug treatment, providing validation of these indices as measures of cellular stress.
Multiplex single cell assays also permit direct extraction of analyte-analyte correlations. Fig. 5d displays the correlation networks for each sample. Both low and high dose drug treatments clearly induced significant changes within the correlation networks. In the control sample, there is a strong correlation between pEGFR1068 and pEGFR1173, which is expected. There are also correlations between the ESI and the pEGFR1068 levels that point to a relationship between EGFR signaling and cellular glutamine dependency – cells with higher pEGFR1068 levels are less dependent on glutamine. The possible implication is that the upstream signaling of EGFR plays a strong role in determining cellular glutamine dependency.
When cells were treated with 1 μM erlotinib, correlations between signaling proteins are significantly reduced relative to the control, while relationships among energy indices are strengthened. The correlation between pEGFR1068 and the ESI is strengthened. We can also deduce that cells with unsuppressed pEGFR1068 are more glycolytic (GI-pEGFR1068), produce more energy (EPI-pEGFR1068), and have less glutamine dependence (ESI-pEGFR1068). The strong correlation between RFI and EPI and the strong anti-correlation between EFI and EPI indicates that metabolically active cells have higher redox stress and lower energy stress.
Following 10 μM erlotinib treatment, pEGFR1173 is further decoupled from the correlation network. Similarly, only one analyte exhibits correlation with the EPI, suggesting that these cells exhibit disrupted regulation of energy production. The correlation between the ESI and pEGFR1068 is retained, yet weakened, indicating a potential regulatory effect of EGFR signaling on the cellular glutamine dependence that is not fully repressed by high dose erlotinib. Correlations between the signaling proteins are significantly activated relative to those in the low dose sample, even as the levels of those same proteins are repressed.
Agglomerative hierarchical clustering analysis of the three treatment conditions is represented as a heat map in Fig. 5e. This plot indicates that the differences between the control and 1 μM erlotinib treated cells are much smaller than those between 10 μM and 1 μM samples, implying that 1 μM erlotinib treatment is a relatively weak perturbation. Individual clustering of each sample group reveals that the sample heterogeneity increased upon 1 μM erlotinib treatment, but significantly decreased with the higher dose treatment (Supporting information, y-axes of Fig S11). The clustering analysis also indicates that the GI and EPI provide excellent markers for differentiating control and drug treated populations. There is also a strong correlation between ESI and p-EGFR levels across all three samples, again pointing to the influence of EGFR signaling on cellular glutamine dependence.
3. Discussion
Glutamine plays an important role in cellular metabolism, and has special significance in cancer cell maintenance and survival. Although the ability to quantify glutamine uptake at a single cell level is meaningful, the value of that assay is significantly amplified when it is integrated into a multiplex panel of related metabolites, metabolic enzymes, and signaling proteins. Such an integrated analysis was enabled by the compatibility of the supramolecular-based glutamine uptake assay with the core DNA barcoding method that is used in SCBCs.
The design of the DNA-cyclodextrin-Cy3 donor was engineered for competitive binding, and required a balance between high surface coverage of cyclodextrin and steric access to enable the DNA hybridization-driven surface-assembly of the donor. Thus, there is a tradeoff on the amount of cyclodextrins per DNA oligomer, and this tradeoff was experimentally optimized (Supporting information Fig S2). Because of the high density of cyclodextrin groups on the surface, and the confined space in the microfluidic device, the effective cyclodextrin/adamantane binding is much stronger than that in solution. For the quencher molecule, both BHQ2 and adamantane are hydrophobic and so PEG moieties are appended to improve the aqueous solubility. Additionally, the p-nitroazobenzene group on BHQ2 can also bind with cyclodextrin [27], further increasing the binding affinity between cyclodextrin and adamantane-BHQ2. This enhances the quenching efficiency and improves assay sensitivity.
The design of the small library of glutamine analogs (Fig 2a) was based on the substrate-specificity of the human glutamine transporter (SLC1A family), that is, a neutral amino acid side chain with hydrogen bonding capabilities [28, 29]. Because the binding mechanism and the exact binding pocket structure of SLC1A is unknown, it is difficult to interpret the differing performances for each analog. In addition, because there are other potential carriers for glutamine transport in different types of cells, these analogs may not serve well as universal glutamine uptake surrogates.
Multiplex single cell measurements of functional analytes (metabolites and enzymes) yield several classes of information, in addition to the average value of the analyte (which is also obtained from bulk assays). First, the distribution of each analyte level provides insight into the sample heterogeneity. Second, analyte-analyte correlations can provide information regarding signaling interactions that are activated or repressed between different experimental conditions. Finally, clustering analysis can provide a more global view for the statistical analysis of sets of analytes, or permit comparisons across different experimental conditions. In our case, the inclusion of metabolites with intrinsically correlated metabolic proteins within the panel permits the use of a semi-quantitative and simplified physical chemistry model (Fig 5a) for guiding the interpretation of the single cell data. Although the model excludes many other metabolic pathways that may be active, it should have some meaning within the context of global cellular energy demand.
The metabolic model, combined with insights from bulk metabolic assays, yields energy indices that help guide the interpretation of the dose-dependent response of the GBM cells to erlotinib (Fig. 5c). For example, the energy potential index (EPI) increases upon low dose drug treatment. This is in seeming contrast to the decreased glucose uptake, but the EPI provides a more global view of energy flux. The increased level of metabolic enzymes and the heightened glutamine uptake point to elevated metabolic activities of the cells under low (but clinically relevant) dosage drug stress. At the higher dose, the metabolic activity of the cells is repressed, by all measures reported here. The low dosage results are further validated through intracellular ATP concentration measurements and extracellular oxygen consumption assays (Fig 4). This result may have clinical relevance. At low dose, erlotinib successfully engages its target (decreased p-EGFR level) and inhibits glucose uptake (corresponding to a decreased 18F-FDG PET signal in clinical molecular imaging). However, the cells are still very metabolically active, and the cell growth is not inhibited in vitro, thus anticipating a poor patient response. The value of the EPI index as a gauge of drug effectiveness, relative to protein assays and/or glucose uptake assays alone, is seen in the clustering analysis of Fig. 5e. Here, the EPI index readily differentiates control/ low dose drug/ high dose drug populations, and correlates well with cell survival.
The single-cell resolved metabolic model also provides deeper biological information. For instance, a linkage between receptor tyrosine kinase signaling and GBM cell metabolism is resolved. The ESI is highly correlated with the p-EGFR level of the cells (Fig. 5e), while no single metabolite or metabolic protein exhibited such a strong correlation. This suggests that EGFR signaling may regulate the cellular glutamine dependence. Further, the pEGFR1068 and the pEGFR1173 sites are decoupled via drugging, and we can infer different functional consequences of these two phosphorylation sites, with the pEGFR1068 possibly playing a role in regulating cellular glutamine dependency, and thus cell survival upon erlotinib treatment. This information may be useful in terms of understanding the rapid development of resistance to erlotinib that is seen in GBM patients.
The metabolic protein panel and the metabolic model were established with the motivation for establishing a picture of the metabolic demand that can be resolved at the single cell level, using metabolites that are especially important for cellular energy consumption. It is straightforward to assay more proteins on the SCBC platform [30]. It should also be possible to develop additional chemical methods to expand the panel of metabolites that are simultaneously assayed. For instance, quantifying ATP/ADP ratio would complement the assays reported here. Further, a more comprehensive panel with metabolic intermediates and products, such as lactate, pyruvate, alpha-ketoglutarate, etc., would significantly help resolve a more sophisticated and quantitative picture of cellular energy flux, thus removing some of the assumptions that went into the semi quantitative model provided here. .
4. Experimental Section
Chemicals and reagents. Fmoc-Rink Amide resin (0.6 mmol/g loading), 2-Chlorotrityl Chloride resin (0.5 mmol/g loading), Fmoc-L-Lys(Alloc)-OH, Fmoc-L-Gly(Propargyl)-OH, Fmoc-NH-PEG5-CH2CH2COOH (PEG5), Fmoc-Lys(Boc)-OH were purchased from ChemPep Inc. (Wellington, FL, U.S.A.). 1-[Bis(dimethyl amino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU, 97%), N,N-diisopropylethylamine (DIEA, 99%), trifluoroacetic acid (TFA, 99%), triethylsilane (99%), piperidine (99.5%), 1-adamantaneacetic acid (Adaa, 98%), 1-adamantylamine (97%), 1-adamantanecarboxylic acid (99%), copper(I) iodide (CuI, 98%), sodium L-ascorbate (98%), tetrakis(triphenylphosphine)palladium(0) (Pd0, 99%), phenylsilane (97%), Fmoc-L-Dap(Alloc)-OH (98.5), Fmoc-L-Dab(Alloc)-OH (99%), Fmoc-L-Orn(Alloc)-OH (99%), Fmoc-L-Glu(OAll)-OH (96%), Fmoc-L-Asp(OAll)-OH (98%) were purchased from Sigma Aldrich (St. Louis, MO, U.S.A.). Cyanine3 carboxylic acid (Cy3) was purchased from Lumiprobe (Hallandale Beach, FL, U.S.A.). BHQ2 carboxylic acid (BHQ2) was obtained from Biosearch Technologies (Petaluma, CA, U.S.A.). Succinimidyl 4-formylbenzoate (S-4FB) and 6-Boc-hydrazinonicotinic acid (Boc-HNA) were purchased from Solulink (San Diego, CA, U.S.A.). Glutamate assay kit (ab83389), oxygen consumption assay kit (ab197243) and ATP assay kit (ab65313) were purchased from Abcam (Cambridge, MA, U.S.A.). siRNA against SLC1A5 and the siRNA transfection kit were purchased from ThermoFisher (Grand Island, NY, U.S.A.).
Cyclodextrin-Cy3
A sequence of Lys(Alloc)-PEG5-Gly(Propargyl)-PEG5-Cy3 was synthesized on 100 mg of Rink Amide resin following standard solid phase peptide synthesis protocol using dimethylformamide as solvent [31]. The deprotection of the Fmoc group was achieved by piperidine (20% v/v in DMF) treatment (5 mL, 3 × 10 min) and the amide bond coupling was carried out through HATU/DIEA process (2 mL of 200 mM HATU/DMF, 2 mL of 200 mM aminoacid/DMF, 0.5 mL of DIEA, 1 hr incubation at room temperature). The Alloc protecting group was removed through treating the resin with 100 mg of Pd0 and 0.5 mL of phenylsilane in 5 mL of dichloromethane for 2 hr. Subsequently, Boc-HNA was coupled to the peptide. The peptide was cleaved from the resin using 5 mL of TFA with 2.5% water, 2.5% acetone and 2.5% triethylsilane and dried under vacuum. Then, the peptide was dissolved in 10 mL of DMF and mixed with 200 mg of CuI, 300 mg of sodium ascorbate, 2 mL of piperidine and 500 mg of mono-6-azido-6-deoxy-β-cyclodextrin [32]. The reaction was incubated at room temperature for 48 hrs and the product was purified through reverse phase HPLC. MOLTI-TOF, C118H182O50N14, [MH+] calc. 2596.22, found 2596.26.
Cyclodextrin-Cy3-DNA
0.1 mg of S-4FB was dissolved in 5 μL of anhydrous DMF and added into 20 μL of ssDNA oligo solution (200 μM in PBS). This solution was incubated at room temperature for 2 hours and then buffer exchanged to a pH 5.0 citrate buffer using Zeba spin columns (7K MWCO, Thermo Fisher Scientific, Pittsburgh, PA). The resulting solution was mixed with 50 μL of Cyclodextrin-Cy3 solution (50 μM in pH 5.0 citrate buffer). After 12 hr of incubation at room temperature, the product was purified through FPLC.
Adamantane-BHQ2
A sequence of Lys(Boc)-Lys(Boc)-Lys(Alloc)-Adaa was synthesized on 100 mg of Rink Amide resin using aforementioned protocols. After Alloc deprotection, BHQ2 was conjugated to the peptide through HATU coupling. The peptide was then cleaved from the resin using 95% TFA with 2.5% water and 2.5% triethylsilane. After removing solvent under vacuum, the peptide was dissolved in 2 mL of acetonitrile and coupled with PEG5 through HATU coupling (2 mL of 200 mM HATU/acetonitrile, 2 mL of 200 mM PEG5/acetonitrile, 0.5 mL of DIEA, 1 hr incubation at room temperature). Subsequently, 5 mL of piperidine was added to perform Fmoc deprotection (room temperature, 20 min). After removing the solvent under vacuum, the remaining solid was extracted with 5 mL of 50% water/49.9% acetonitrile/0.1% TFA. The product was then purified through reverse phase HPLC. MOLTI-TOF, C81H129N15O21, [MH+] calc. 1648.96, found 1648.96.
3-(adamantane-1-carboxamido)-2-aminopropanoic acid (compound 1)
1.5 mmol of Fmoc-Dap(Alloc)-OH was dissolved in 20 mL of dry dichloromethane and 1 mL of DIEA was added into the solution. 2 g of 2-Chlorotrityl Chloride resin was added into the solution and the mixture was stirred at room temperature for 1 hr before 2 mL of methanol was added into the solution. After 15 min, the solution was filtered and the resin was washed with dichloromethane and dried under vacuum. The Alloc group was removed through aforementioned protocol, and 1-adamantanecarboxylic acid was coupled to the side chain amine through HATU/DIEA reaction. The Fmoc protecting group was subsequently removed by piperidine treatment. The modified amino acid was cleaved from the resin using 1% TFA in dichloromethane (1 hr). The product was purified through reverse phase HPLC. 1H-NMR 400 MHz, D2O, δ = 3.98 (dd, 1H, J= 5.8, 4.4Hz), 3.65-3.54 (m, 2H), 1.89-1.93 (m, 3H), 1.70 (d, 6H, J=2.9 Hz), 1.66-1.50 (m, 6H). HR-FAB, C14H22O3N2, [MH+] calc. 267.1709, found 267.1700.
4-(adamantane-1-carboxamido)-2-aminobutanoic acid (compound 2)
Compound 2 was synthesized with protocols similar to compound 1, with Fmoc-Dab(Alloc)-OH as the starting material. 1H-NMR 400 MHz, D2O, δ = 3.76 (t, 1H, J=6.5 Hz), 3.37-3.17 (m, 2H), 2.02 (qd, 2H, J=6.7, 4.8 Hz), 1.95-1.83 (m, 3H), 1.70 (d, 6H, J=2.9Hz), 1.68-1.52 (m, 6H). HR-FAB, C15H24O3N2, [MH+] calc. 281.1865, found 281.1860.
5-(adamantane-1-carboxamido)-2-aminopentanoic acid (compound 3)
Compound 3 was synthesized with protocols similar to compound 1, with Fmoc-Orn(Alloc)-OH as the starting material. 1H-NMR 400 MHz, D2O, δ = 3.90 (t, 1H, J=6.1 Hz), 3.13 (td, 2 H, J=6.7, 2.0 Hz), 1.90 (m, 3H), 1.86-1.73 (m, 2H), 1.73-1.66 (m, 6H), 1.69 (d, 6H, J=2.9Hz), 1.50-1.40 (m, 2H). HR-FAB, C16H26O3N2, [MH+] calc. 295.2022, found 295.2026.
N5-(adamantan-1-yl)glutamine (compound 4)
Compound 4 was synthesized with protocols similar to compound 1, with Fmoc-Glu(OAll)-OH as the starting material. The OAll deprotection procedure is the same as that of Alloc deprotection. After removing OAll group, 1-adamantylamine was coupled to the side chain carboxylic acid through HATU/DIEA procedure. After cleaving, the modified amino acid was purified by reverse phase HPLC. 1H-NMR 400 MHz, D2O, δ = 3.77 (t, 1H, J=6.3Hz), 2.24 (td, 2H, J=7.5, 3.5 Hz), 2.02 (qd, 2H, J= 7.2, 6.7, 2.0 Hz), 1.94 (s, 3H), 1.88-1.78 (m, 6H), 1.63-1.39 (m, 6H). HR-FAB, C15H24O3N2, [MH+] calc. 281.1865, found 281.1862.
3-(2-(adamantan-1-yl)acetamido)-2-aminopropanoic acid (compound 5)
Compound 5 was synthesized with protocols similar to compound 1, with Adaa coupled to the side chain amine. 1H-NMR 400 MHz, D2O, δ = 4.04 (dd, 1H, J=5.7, 4.0 Hz), 3.74-3.48 (m, 2H), 2.00-1.88 (m, 2H), 1.87-1.75 (m, 3H), 1.64-1.34 (m, 12H). HR-FAB, C15H24O3N2, [MH+] calc. 281.1865, found 281.1858.
4-(2-(adamantan-1-yl)acetamido)-2-aminobutanoic acid (compound 6)
Compound 6 was synthesized with protocols similar to compound 2, with Adaa coupled to the side chain amine. 1H-NMR 400 MHz, D2O, δ = 3.84 (dd, 1H, J=7.4, 5.9Hz), 3.38-3.11 (m, 2H), 2.16-1.93 (m, 2H), 1.83 (s, 3H), 1.67-1.32 (m, 12H). HR-FAB, C16H26O3N2, [MH+] calc. 295.2022, found 295.2035.
5-(2-(adamantan-1-yl)acetamido)-2-aminopentanoic acid (compound 7)
Compound 7 was synthesized with protocols similar to compound 3, with Adaa coupled to the side chain amine. 1H-NMR 400 MHz, D2O, δ = 3.95 (t, 1H, J=6.2 Hz), 3.12 (td, 2H, J=7.0, 1.2 Hz), 1.98-1.75 (m, 7H), 1.67-1.35 (m, 14H). HR-FAB, C17H28O3N2, [MH+] calc. 309.2178, found 309.2167.
N4-(adamantan-1-yl)asparagine (compound 8)
Compound 8 was synthesized with protocols similar to compound 4, with Fmoc-Asp(OAll)-OH as the starting material. 1H-NMR 400 MHz, D2O, δ = 3.96 (dd, 1H, J=6.9, 4.7Hz), 2.78-2.56 (m, 2H), 1.98-1.90 (m, 3H), 1.80-1.88 (m, 6H), 1.50-1.62 (m, 6H). HR-FAB, C14H22O3N2, [MH+] calc. 267.1709, found 267.1709.
DNA-encoded antibody library (DEAL) and surface barcoding on glass slides
The DNA-encoded antibody library was constructed through well-established procedure [18]. The antibodies used in this paper are listed in Supporting information (Table S1, S2). The complementary ssDNA were patterned on to polylysine-coated glass slides through microfluidic-guided covalent crosslinking procedure [17]. The conversion of surface ssDNA barcodes to capture antibody microarrays were achieved through incubating a mixture of DEAL conjugates with the glass slide at 37 °C for 1 hr. The Cyclodextrin-Cy3-DNA can also be incorporated onto the surface barcode through the same procedure.
Cell culture and drug treatment
U87/EGFRV3 cells were provided by Prof. Paul S. Mischel (UCSD, San Diego, U.S.A.) and cultured in Dulbecco’s modified eagle media (DMEM, Gibco, ThermoFisher Scientific, Grand Island, NY, U.S.A.) supplemented with 10% of fetal bovine serum (Thermo Fisher Scientific) and 100 U/mL of penicillin and streptomycin in a humidified 5% CO2 (v/v) incubator, at 37 °C. For the drug treatment, 1 million cells were first cultured for 1 day and then the medium was changed with 10 mL of new media containing varies concentrations of erlotinib (ChemieTek, Indianapolis, Indiana, U.S.A.) and cultured for 24 hr before the bulk assays or SCBC measurements. All bulk assays were carried out following the kit protocols. For the oxygen consumption assay, the cells were suspended in 96 wells and the oxygen consumption was monitored over a 3 hr period.
Single cell suspension preparation
The media in a U87/EGFRV3 culture was removed and the cells were treated with 0.25% Tripsin/EDTA (Thermo Fisher Scientific) for 10 min at 37 °C. The original media was added back to the disassociated cells and the suspension was centrifuged at 500 g for 5 min. After removing the supernatant, the cells were resuspended in warm media at a concentration of 1 million/mL.
Glutamine analog uptake
U87/EGFRV3 single cells were resuspended in DMEM media or glutamine-free DMEM media (Thermo Fisher Scientific) with 1 mM of glutamine analog and incubated at 37 °C or 4 °C for 20 min. Subsequently, the cells were collected via centrifugation (500 g, 5 min) and washed with cold PBS for three times. Then, the cells were lysed using cell lysis buffer (Cell Signaling, Boston, MA, U.S.A.). To quantify the analog uptake, the lysate was mixed with an Adamantane-BHQ2 solution (100 nM in water) incubated on the glass slide with surface hybridized Cyclodextrin-Cy3-DNA at room temperature for 1 hr. the glass slide was then washed and spin dried. The fluorescence signal on the glass slide was read out using an Axon GenePix 4400A scanner.
Single cell metabolic/proteomic measurements
The integrated single cell metabolic/proteomic measurements were performed on single cell barcode chips (SCBC, Supporting information Fig S4). The chip was fabricated and operated following previously-established protocols [17] with two minor modifications: 1) cells were incubated with 10 μg/mL of Gluc-Bio and 1 mM of compound 3 in glutamine-free DMEM media at 37 °C for 20 min; 2) the lysis buffer formulation was changed to contain 200 nM of Adamantane-BHQ2.
Data processing and statistical analysis
All the error bars in the figures show the standard deviation of the dataset. The SCBC dataset is an m × n matrix table where each row (m) represents a specific microchamber address and each column (n) represents the abundance of a specific analyte. Group comparison was carried out through Kruskal-Wallis ANOVA method (*, p<0.1; ***, p<0.001). The correlation coefficients were calculated using Spearman’s rank method. Bonferroni corrected p-value was used to filter the correlation network through statistical significance (Fig. 5d). The correlation networks were drawn using Circos software [33]. Agglomerative hierarchical clustering analysis was carried out by Matlab software (Mathworks) on the combined single cell dataset using Euclidian distance and complete linkage. The result was presented as a heatmap (Fig. 5e). Additional clustering analysis was performed by XLSTAT software (Addisoft) on SCBC datasets from each sample, where the proximity among single cell readouts were measured by the dissimilarity coefficients of Euclidian distance with Ward’s minimum variance method (Fig S11). The calculated dissimilarity coefficients were employed to quantify the heterogeneity of the cells from each sample.
Supplementary Material
ACKNOWLEDGMENT
We thank Dr. David Nathanson for the suggestions on glioblastoma cell line culturing and drug treatments. We acknowledge the following agencies and foundations for support: The National Cancer Institute (1U54 CA199090-01 JRH PI, WW and R01-CA170689 JRH PI), The Ben and Catherine Ivy Foundation (JRH), the Jean Perkins Foundation (JRH PI), and the Phelps Family Foundation (JRH and WW).
Footnotes
Supporting Information. Synthetic schemes, additional validation experiments, illustration of SCBC device and detection methods, clustering results for each of the single cell dataset, list of antibodies used in this work. This material is available free of charge via the Internet at http://pubs.acs.org.
Conflict of Interest Disclosure
JRH is a board member of IsoPlexis, which is a company seeking to commercialize the SCBC technology.
REFERENCES
- 1.Cairns RA, Harris IS, Mak TW. Nat. Rev. Cancer. 2011;11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Svensson RU, Shaw RJ. Nature. 2012;485:509–591. doi: 10.1038/485590a. [DOI] [PubMed] [Google Scholar]
- 4.Vander Heiden MG, Cantley LC, Thompson CB. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wise DR, Thompson CB. Trends. Biochem. Sci. 2010;35:427–433. doi: 10.1016/j.tibs.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hensley CT, Wasti AT, DeBerardinis RJ. J. Clin. Invest. 2013;123:3678–3684. doi: 10.1172/JCI69600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alberghina L, Gaglio D. Cell Death Disease. 2014;5:e1561. doi: 10.1038/cddis.2014.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB, Thompson CB. Proc. Natl. Acad. Sci. U.S.A. 2008;105:18782–18787. doi: 10.1073/pnas.0810199105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, Kang Y, Fleming JB, Bardeesy N, Asara JM, Haigis MC, A. DePinho R, Cantley LC, Kimmelman AC. Nature. 2013;496:101–105. doi: 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeBerardinis RJ, Cheng TQ. Oncogene. 2010;29:313–324. doi: 10.1038/onc.2009.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao Y, Butler EB, Tan M. Cell Death Disease. 2013;4:e532. doi: 10.1038/cddis.2013.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seltzer MJ, Bennett BD, Joshi AD, Gao P, Thomas AG, Ferraris DV, Tsukamoto T, Rojas CJ, Slusher BS, Rabinowitz JD, Dang CV, Riggins GJ. Cancer Res. 2010;70:8981. doi: 10.1158/0008-5472.CAN-10-1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tanaka K, Sasayama T, Irino Y, Takata K, Nagashima H, Satoh N, Kyotani K, Mizawaki T, Imahori T, Ejima Y, Masui K, Gini B, Yang H, Hosoda K, Sasaki R, Mischel PS, Kohmura E. J. Clin. Invest. 2015;125:1591–1602. doi: 10.1172/JCI78239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang C, Sudderth J, Dang T, Bachoo RG, McDonald JG, DeBerardinis RJ. Cancer Res. 2009;69:7986–7993. doi: 10.1158/0008-5472.CAN-09-2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benntt BD, Yuan J, Kimball EH, Rabinowitz JD. Nat. Protoc. 2008;3:1299–1311. doi: 10.1038/nprot.2008.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, Turay AM, Frauwirth KA. J. Immunol. 2010;185:1037–1044. doi: 10.4049/jimmunol.0903586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xue M, Wei W, Su Y, Kim J, Shin YS, Mai WX, Nathanson DA, Heath JR. J. Am. Chem. Soc. 2015;129:1959–1967. doi: 10.1021/jacs.5b00944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shin YS, Ahmad H, Shi Q, Kim H, Pascal TA, Fan R, Goddard WA, Heath JR. Chem. Phys. Chem. 2010;11:3063–3069. doi: 10.1002/cphc.201000528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma X, Zhao Y. Chem. Rev. 2015;115:7794–7839. doi: 10.1021/cr500392w. [DOI] [PubMed] [Google Scholar]
- 20.Bailey RC, Kwong GA, Radu CG, Witte ON, Heath JR. J. Am. Chem. Soc. 2007;129:1959–1967. doi: 10.1021/ja065930i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mellinghoff IK, Wang MY, Vivanco I, Haas-Kogan DA, Zhu S, Qia EQ, Lu KV, Yoshimoto K, Huang JHY, Chute DJ, Riggs BL, Horvath S, Liau LM, Cavenee WK, Rao N, Beroukhim R, Peck TC, Lee JC, Sellers WR, Stokoe D, Prados M, Cloughesy TF, Sawyers CL, Mischel PS. N. Engl. J. Med. 2005;353:2012–2024. doi: 10.1056/NEJMoa051918. [DOI] [PubMed] [Google Scholar]
- 22.Raizer JJ, Abrey LE, Lassman AB, Chang SM, Lamborn KR, Kuhn JG, Alfred Yung WK, Gilbert MR, Aldape KD, Wen PY, Fine HA, Mehta M, DeAngelis LM, Lieberman F, Cloughesy TF, Robins HI, Dancey J, Prados MD. Neuro Oncol. 2010;12:87–94. doi: 10.1093/neuonc/nop017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raizer JJ, Abrey LE, Lassman AB, Chang SM, Lamborn KR, Kuhn JG, Alfred Yung WK, Gilbert MR, Aldape KD, Wen PY, Fine HA, Mehta M, DeAngelis LM, Lieberman F, Cloughesy TF, Robins HI, Dancey J, Prados MD. Neuro Oncol. 2010;12:95–103. doi: 10.1093/neuonc/nop015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kato K, Ito H, Hasegawa K, Inaguma Y, Kozawa O, Asano T. J. Neurochem. 1996;66:946–950. doi: 10.1046/j.1471-4159.1996.66030946.x. [DOI] [PubMed] [Google Scholar]
- 25.Swartling FJ, Ferletta M, Kastermar M, Weiss WA, Westermark B. Oncogene. 2009;28:3121–3131. doi: 10.1038/onc.2009.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dang CV. Cancer Res. 2010;70:859–862. doi: 10.1158/0008-5472.CAN-09-3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamaguchi H, Kobayashi Y, Kobayashi R, Takashima Y, Hashidzume A, Harada A. Nat. Commun. 2012;3:603. doi: 10.1038/ncomms1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Esslinger CS, Cybulski KA, Rhoderick JF. Bioorg. Med. Chem. 2005;13:1111–1118. doi: 10.1016/j.bmc.2004.11.028. [DOI] [PubMed] [Google Scholar]
- 29.Scopelliti AJ, Heinzelmann G, Kuyucak S, Ryan RM, Vandenberg RJ. J. Biol. Chem. 2014;289:17468–17479. doi: 10.1074/jbc.M114.565242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu Y, Xue Q, Eisele MR, Sulistijo ES, Brower K, Han L, Amir el-A.D., Pe’er D, Miller-Jensen K, Fan R. Proc. Natl. Acad. Sci. U.S.A. 2015;112:E607–615. doi: 10.1073/pnas.1416756112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deyle KM, Farrow B, Hee YQ, Work J, Wong M, Lai B, Umeda A, Millward SW, Nag A, Das S, Heath JR. Nat. Chem. 2015;7:455–462. doi: 10.1038/nchem.2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tang W, Ng S-C. Nat. Protoc. 2008;3:691–697. doi: 10.1038/nprot.2008.37. [DOI] [PubMed] [Google Scholar]
- 33.Krzywinski MI, Schein JE, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.