

Abstract
The aim of this study is to investigate the influence of cisplatin on the efficacy of natural killer (NK) cells immunotherapy to suppress HCC progression, and provide valuable information on better application of cisplatin in clinical settings. By using in vitro cell cytotoxicity test and in vivo liver orthotopic xenograft mice model, we identified the role of cisplatin in modulating NK cells cytotoxicity. Luciferase report assay and chromatin immunoprecipitation assay were applied for mechanism dissection. Immunohistochemistry was performed for sample staining. We found cisplatin could enhance the efficacy of NK cell immunotherapy to better suppress HCC progression via altering the androgen receptor (AR)-UL16-binding protein 2 (ULBP2) signals both in vitro and in vivo. Mechanism dissection revealed that cisplatin could suppress AR expression via two distinct ways: increasing miR-34a-5p to suppress AR expression and altering the ubiquitination to accelerate the AR protein degradation. The suppressed AR might then function through up-regulating ULBP2, a natural-killer group 2 member D ligand, to enhance the cytotoxicity of NK cells. Together, these results indicated an unrecognized favoring effect of cisplatin in HCC treatment. By suppressing AR in HCC, cisplatin could up-regulate cytotoxicity of NK cells to better target HCC. This finding may provide a potential new approach to control HCC by combining traditional chemotherapy with immunotherapy.
Keywords: Androgen receptor, Liver cancer, Natural killer cells
Introduction
Hepatocellular carcinoma (HCC) is difficult to control by current clinical treatment, especially among later-stage patients, and most liver diseases, including viral hepatitis, cirrhosis and alcoholic liver disease, may eventually develop into HCC [1]. Unfortunately, very few drugs, except sorafenib, a multiple-target kinase inhibitor, are currently used with some effect to treat the advanced HCC [2,3]. Results from immunotherapy using natural killer (NK) cells yield some positive response [4] and some chemotherapy agents like doxorubicin were reported to be able to induce expression of the NKG2D ligand, on multiple myeloma (MM) cells to activate NK cells and to enhance its efficacy to better kill the tumor cells [5]. Meanwhile, cisplatin is a common drug used for transcatheter arterial chemoembolization (TACE) in advanced HCC patients [6–8]. Its potential effects on NK cells remained unaddressed though cytotoxic T lymphocytes immunotherapy for liver cancer was reported to be enhanced by it in a tumor-specific, antigen-independent manner [9].
Gender disparity of HCC suggests androgen and AR may play important roles to influence the HCC initiation and progression [10–13]. The linkage of AR signals to the currently used chemotherapy or immunotherapy for HCC, however, remains unclear. Here we found chemotherapy agent cisplatin could decrease AR that normally suppresses the expression of NKG2D ligand, UL16 binding protein-2 (ULBP2) in HCC cells. A derepression of ULBP2 in turn may enhance the efficacy of NK cells immunotherapy to better suppress HCC progression.
Materials and methods
Cell culture and transfection
The human HCC cells were maintained in DMEM (Invitrogen, Grand Island, NY) with 10% fetal bovine serum (FBS), 1% Glutamine, and 1% penicillin/streptomycin. NK-92MI cells (ATCC, Manassas, VA) were maintained in α-MEM (Invitrogen) with 0.2 mM inositol, 0.1 mM 2-mercaptoethanol, 0.02 mM folic acid, horse serum to a final concentration of 12.5% and FBS to a final concentration of 12.5% based on ATCC guidelines. All cell lines were cultured ina5%(v/v) CO2 humidified incubator at 37 °C The SK-Hep1 (ATCC, Manassas, VA) and SNU423 (ATCC, Manassas, VA) AR stable transfectants were established based on a previous procedure [14]. Cisplatin (479306), MG132 (M8699) and Cycloheximide (CHX, 227048) were purchased from Sigma.
To generate AR knock-down stable clones of SK-Hep1 (ATCC, Manassas, VA) and SNU423 cells (ATCC, Manassas, VA), HEK-293T cells were transfected with lentiviral vectors, pLKO1-sh-AR/pLKO1-scr, with the psAX2 packaging plasmid, and pMD2G envelope plasmid for 48 hrs to obtain the lentivirus supernatant, which was frozen at −80 °C for later use.
For the luciferase reporter assay, cells were transfected using Lipofectamine 3000 (Invitrogen) reverse transfection protocol, according to the manufacturer's instructions. See Supplemental data for detailed sequence information.
Cellular cytotoxicity assay
NK cells cytotoxicity against HCC cells was analyzed using a standard lactate dehydrogenase release assay. An aliquot of 50 μL media after NK cells performed their function, cytotoxicity was used for detection of lactate dehydrogenase activity using the lactate dehydrogenase cytotoxic assay kit (Thermo Scientific). Spontaneous release of target cells alone was <15% of the maximum release as determined with target cells lysed in lysis buffer. The experimental release was corrected by subtraction of the spontaneous release of effector cells at corresponding dilutions. %Cytotoxicity = (Experimental value - Effector Cells Spontaneous Control - Target Cells Spontaneous Control)/(Target Cell Maximum Control-Target Cells Spontaneous Control) × 100.
ELISA
Conditioned media (CM) collected from interaction between HCC cells and NK-92MI cells were tested with IFN-γ ELISA kit (Thermo Scientific). The standard curve was made to determine the IFN-γ concentration. All the procedures were performed according to the manufacturer's instructions.
Quantitative real-time PCR analysis
Total RNAs were isolated using Trizol reagent (Invitrogen). One μg of total RNA was subjected to reverse transcription using Superscript III transcriptase (Invitrogen). Quantitative real-time PCR (qRT-PCR) was conducted using a Bio-Rad CFX96 system (Bio-Rad, Hercules, CA) with SYBR green to determine the mRNA expression level of a gene of interest. Expression levels were normalized to the expression of GAPDH RNA (see Supplemental data for primer sequence information).
Western blot analysis
Cells were lysed in RIPA buffer and proteins (60 μg) were separated on 8–10% SDS/PAGE gel and then transferred onto PVDF membranes (Millipore, Billerica, MA). After blocking membranes, they were incubated with appropriate dilutions of specific primary antibodies, the blots were incubated with HRP-conjugated secondary antibodies and visualized using ECL system (Thermo Fisher Scientific, Rochester, NY). Anti-α-tubulin (1:1000, TU-02), Anti-GFP (1:1000, B-2) antibody and anti-AR (1:1000, N20) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-ULBP2 (1:500, bs-2729R) antibody was purchased from Bioss Antibodies (Woburn, MA).
Plasmid construction and luciferase assay
A 903bp promoter of ULBP2 was obtained from genomic DNA of 293T cells by Phusion® High-Fidelity DNA Polymerase (NEB, Beverly, NY) and cloned into pGL3-basic vector (Promega, Madison, WI) by Gibson assembly method. For generating androgen response elements (ARE) mutation, quickchange™ was used according to manufacturer's instruction. For luciferase assay, cells were plated in 24-well plates and the cDNA transfected using Lipofectamine3000 (Invitrogen) according to the manufacturer's instruction. pRL-TK was used as internal control. Luciferase activity was measured by Dual-Luciferase Assay (Promega) according to the manufacturer's manual. GFP-Ub was a gift from Nico Dantuma (Addgene plasmid #11928).
Chromatin immunoprecipitation assay (ChIP)
Cell lysates were precleared sequentially with normal rabbit IgG (sc-2027, Santa Cruz Biotechnology) and protein A-agarose. Anti-AR antibody from Santa Cruz (2.0 μg) was added to the cell lysates and incubated at 4 °C overnight. IgG was used as the negative control. Specific primer sets designed to amplify a target sequence within the human ULBP2 promoter were listed in the Supplemental Table. PCR products were analyzed by agarose gel electrophoresis.
Co-immunoprecipitation for ubiquitination
293T cells were transfected with AR and GFP-ubiquitin. 48 h post-transfection, whole cell lysates were prepared in a small volume RIPA buffer supplemented with 1% SDS and protease and phosphatase inhibitors, and then heated samples to 95 °C for 5 min to denature. Next diluting the lysate to adjust SDS concentration to 0.1% followed by centrifugation, the supernatant was used for immunoprecipitation with anti-AR antibody. The immunoprecipitates were analyzed by SDS-PAGE and immunoblotted with anti-GFP and anti-AR antibodies.
Patient selection and immunohistochemistry (IHC) staining
We randomly selected 81 male patients’ HCC samples, which have been collected at the Sir Run-Run Shaw Hospital starting in February 2008 and this project was approved by the Institutional Review Board/Privacy Board of Sir Run-Run Shaw Hospital. We reviewed pathology records to identify samples with confirmed HCC. The IHC slides of all 81 patients used for AR and ULBP2 scoring were reviewed by two pathologists in a double blind manner. The staining results were measured semiquantitatively on a scale of (−), (+), (++) and (+++). A stain was scored as follows: (−), there is less than 10% staining of nuclear AR in any of the tumor cells/field or no cytoplasm and cytomembrane staining of ULBP2 in tumor cells; (+), there is nuclear AR staining in 10%-30% of the tumor cells with any intensity, or faint, barely discernable cytoplasmic and cytomembrane staining for ULBP2; (++), there is staining in 30%-50% of the tumor cells with moderate to strong intensity of nuclear AR, or moderate, smooth cytoplasmic and cytomembrane staining of the tumor cells with moderate intensity for ULBP2; and (+++), there is staining in more than 50% of the tumor cells nuclei with strong intensity of AR, or apparent granularity, dark brown staining seen in cytoplasm and cytomembrane for ULBP2. Representative examples of (-), (+), (++) and (+++) IHC staining for AR and ULBP2 are demonstrated in Fig. 5.
Fig. 5.
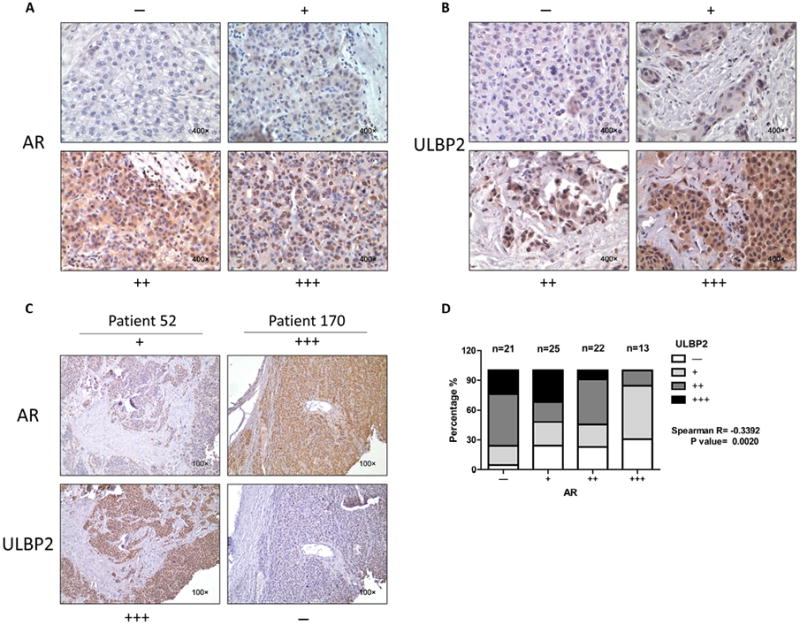
AR and ULBP2 expressions were negatively correlated in human HCC samples. (A) Representative images for scoring the AR IHC staining. (B) Representative images for scoring the ULBP2 IHC staining. (C) Representative images to show the comparison of AR and ULBP2 staining in a same patient. (D) Spearman correlation analysis for AR and ULBP2 based on our stained clinical samples (P value = 0.0020).
IHC stains for AR and ULBP2 were performed using the standard streptavidin-biotin-peroxidase immunostaining procedure. The antibodies used for anti-AR and anti-ULBP2 were the same as used with Western blot with the concentration raised to 1:100.
In vivo orthotopic tumor model
A total of 32 male 6–8 week old nude mice were used. SK-Hep1 cells were engineered to express luciferase reporter gene (PCDNA3.0-luciferase) by stable transfection and the positive stable clones were selected with G418 and expanded in culture.
Two groups of 6 mice were injected with SK-Hep1 cells (w/wo 1 μg/mL cisplatin treated for 48 hrs) only (2 × 106 of luciferase expressing cells per mouse, as a mixture with Matrigel, 1:1), 2 groups of 6 mice were co-injected with SK-Hep1 cells (w/wo 1 μg/mL cisplatin treated for 48 hrs) and NK-92MI cells (E:T ratio = 1:5), into the left lobe of liver. Tumor formation and metastasis were monitored by a Luminescence Imager (IVIS Spectrum, Caliper Life Sciences, Hopkinton, MA) starting 2 weeks after tumor injection and using i.p. injection of 150 mg/kg Luciferin. These four groups of 24 mice were sacrificed after 8 weeks and liver tumors were isolated for further analysis.
An additional 8 mice were injected with SK-Hep1 cells (no other treatment before injection), after tumors formed as detected by IVIS, we divided them into 2 groups randomly. These mice were given an intraperitoneal injection of either saline or cisplatin (4 mg/kg body weight, dissolved in saline) every 4 days for total of 4 doses, at days 6,11,16 and 21 after tumors formed. After treatment finished, the mice were sacrificed and liver tumors were collected.
All animal studies were performed under the supervision and guidelines of the University of Rochester Medical Center Animal Care and Use Committee.
Statistical analysis
Data are expressed as mean ± SEM from at least 3 independent experiments. Statistical analyses involved unpaired t-test, one-way ANOVA and Spearman correlation with SPSS 17.0 (SPSS Inc., Chicago, IL). P < 0.05 was considered statistically significant.
Results
Cisplatin enhanced NK cells immunotherapy efficacy to better suppress HCC progression
An early study indicated doxorubicin and melphalan might enhance NK cells therapy efficacy via the induction of NKG2D ligands in multiple myeloma (MM) cells to activate NK cells [5]. We were interested to see the potential impact of cisplatin, a chemotherapy agent used to treat advanced HCC [15], on the NK cell immunotherapy efficacy to suppress HCC. We first used different dosages of cisplatin (0.5 μg/mL–2.0 μg/mL) to treat HCC cells for 48 hrs and found little effect on HCC cell viability (data not shown). This is expected since these dosages are relatively low as compared to the IC50 of cisplatin in these two HCC cell lines (11.34 μg/mL for SK-Hep1 and 7.95 μg/mL for SNU423). Interestingly, results from the lactate dehydrogenase (LDH) cytotoxic assay (see Fig. 1A for detailed procedure) revealed that adding NK-92MI cells to these cisplatin-treated HCC cells resulted in better efficacy with more HCC cells lysed (Fig. 1B). Moreover, conditioned media collected from interaction between tumor cells and NK-92MI cells indicated higher IFN-γ release after tumor cells were treated with cisplatin (Fig. 1C). These results suggested that cisplatin could enhance the cytotoxicity of NK cells that further improves its immunotherapy efficacy to better suppress HCC cells.
Fig. 1.
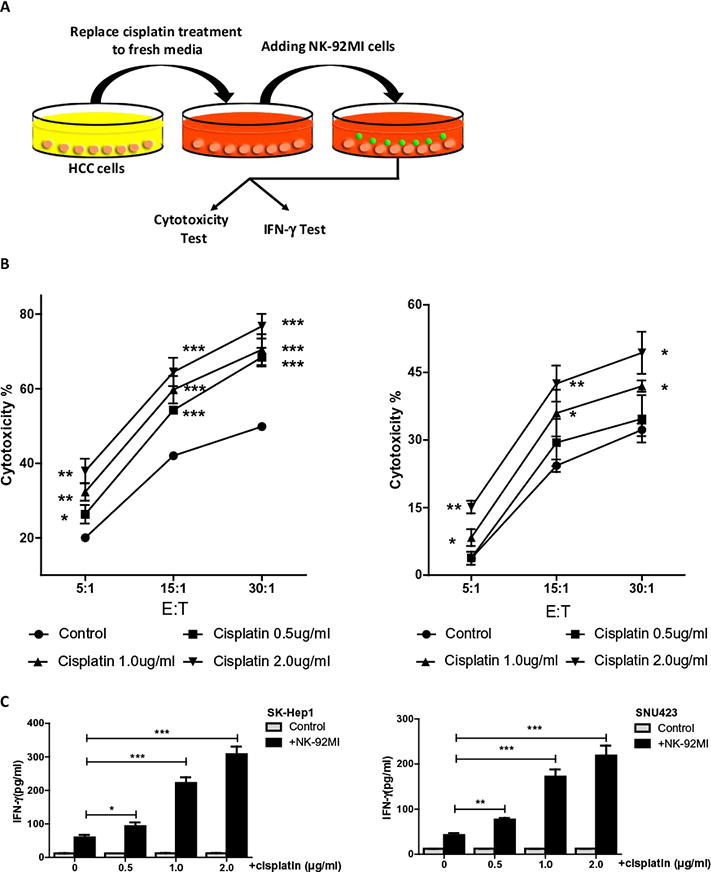
Cisplatin enhances NK cells susceptibility of HCC cell. (A) Schematic for cytotoxicity test procedure. (B) We used two HCC cell lines, SK-Hep1 (left panel) and SNU-423 (right panel), and treated with cisplatin at different dosages (0.5 μg/mL, 1.0 μg/mL, and 2.0 μg/mL) for 48 hrs, then added NK-92MI cells with multiple E:T ratios (5:1, 15:1, and 30:1) for 4 hrs before performing LDH cytotoxic assay, compared to control group. (C) Conditioned media were also collected to test IFN-γ release from NK-92MI cells. Control groups had no NK-92MI cells. Data shown are mean ± SEM. ***P < 0.001, **P < 0.01, *P < 0.05.
Cisplatin enhanced NK cells immunotherapy efficacy to suppress HCC cells via up-regulation of ULBP2 expression in HCC cells
To dissect the potential molecular mechanism why cisplatin could enhance NK cell immunotherapy, we focused on NKG2D related signals since early studies suggested they might be altered during the chemotherapy [5]. We found that among the NKG2D ligands on HCC cells after cisplatin treatment, the ULBP2 mRNA had a significant increase after 48 hrs of cisplatin treatment in HCC SK-Hep1 and SNU423 cells (Fig. 2A). Similar results were also obtained with protein expression showing increased ULBP2 protein in a dose-dependent manner after 48 hrs of cisplatin treatment (Fig. 2B, left panel), and quantification was performed since the internal control was not very consistent (Fig. 2B, right panel).
Fig. 2.
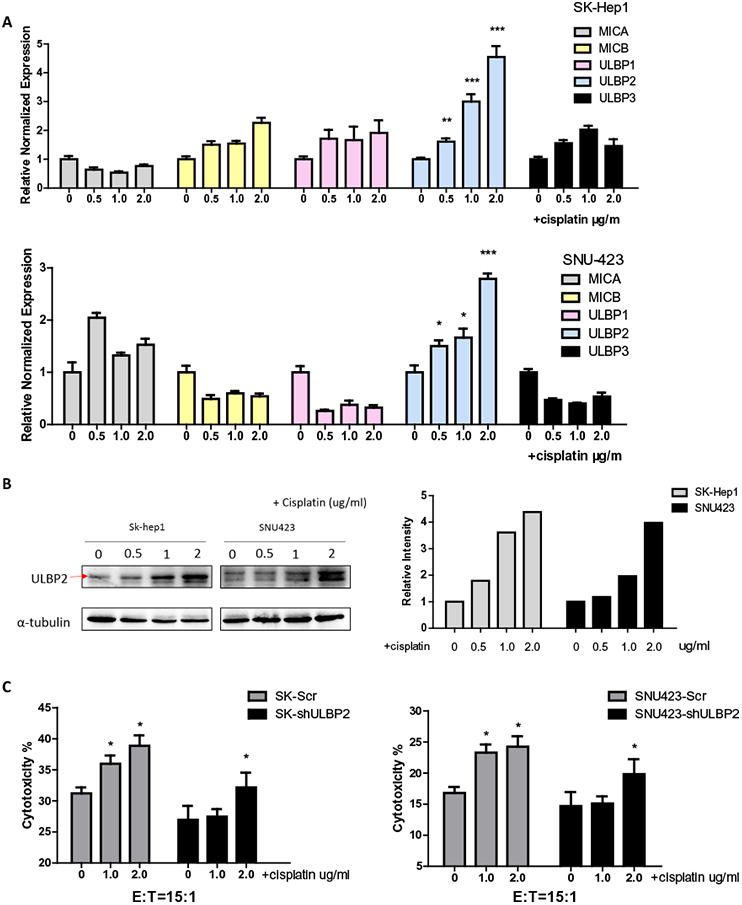
Cisplatin up-regulates ULBP2 expression in HCC cells which results in better targeting of HCC by NK cells. (A) We treated SK-Hep1 and SNU423 cells with cisplatin at different dosages (0.5 μg/mL, 1.0 μg/mL, and 2.0 μg/mL) for 48 hrs, then extracted mRNA to run RT-Q-PCR to test NKG2D ligands expression panel. Cisplatin treatment groups were compared to non-treatment group. (B) After 48 hrs of cisplatin treatment (0.5 μg/mL, 1.0 μg/mL, 2.0 μg/mL), protein was extracted from SK-Hep1 and SNU423 cells for western blots with ULBP2 specific antibody (left panel). Right panel is quantification data. (C) Before treating SK-Hep1 (left panel) and SNU423 (right panel) cells with multiple dosages of cisplatin, we knocked down AR or scrambled with sh-RNA. After treatment, we performed LDH cytotoxic assay using NK92-MI cells to target HCC cells. Cisplatin treatment groups were compared to non-treatment group. Data shown are mean ± SEM. ***P < 0.001, *P < 0.05.
We then applied the interruption approach using lentivirus ULBP2-shRNA to knock down ULBP2 in HCC cells, and results revealed that blocking ULBP-2 could interrupt the increase of NK cytotoxicity induced by cisplatin in SK-Hep1 cells (Fig. 2C, left panel). Similar results were also obtained when we replaced SK-Hep1 cells with SNU423 cells (Fig. 2C, right panel).
Together, results from Figs. 1 and 2 suggest that cisplatin can make HCC cells more vulnerable during NK cells immunotherapy via up-regulating ULBP2 expression in the HCC cells.
Cisplatin decreases AR, which is negatively correlated with ULBP2 expression in HCC cells
To dissect the mechanism how cisplatin can increase ULBP2 expression in HCC cells, we focused on AR since we initially found the ULBP2 mRNA expression was negatively correlated with AR expression in the HCC cell lines that we examined (Fig. 3A). We then examined cisplatin's effects on AR expression and western blot results showed a down-regulation of AR within a very short period of time (Fig. 3B). And such AR decrease was also observed in a dose-dependent manner after 24 hrs of treatment (Fig. 3C).
Fig. 3.
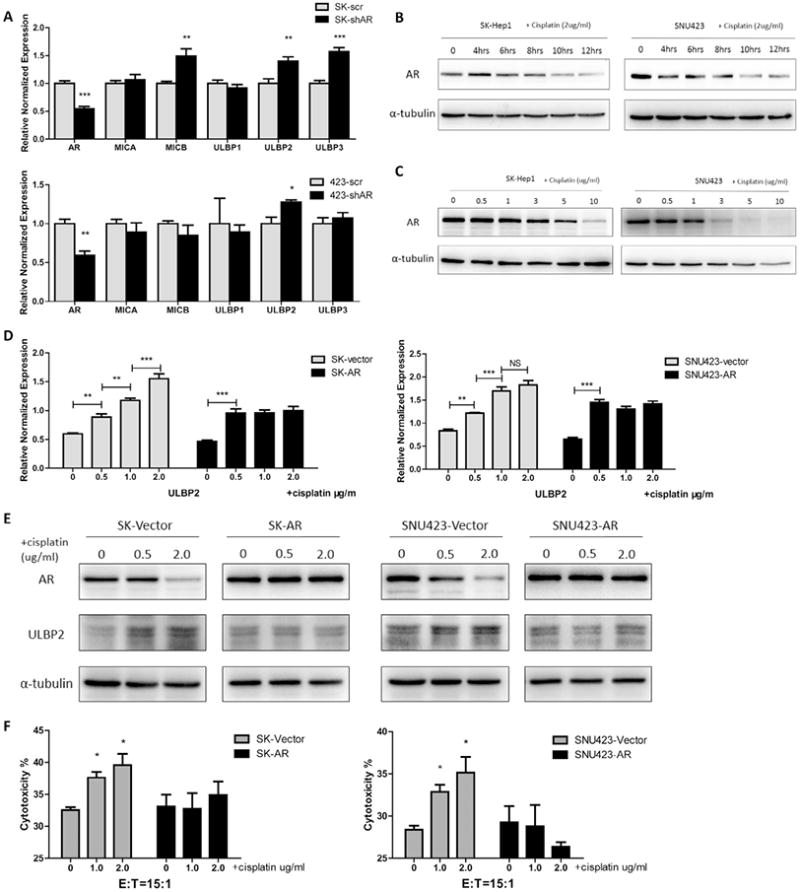
Cisplatin increased ULBP2 in tumor cells through suppressing AR in HCC cells. (A) We knocked down AR in SK-Hep1 and SNU423 cells then extracted mRNA to perform RT-Q-PCR screening of NK cell activation related molecules, compared to scramble group. (B) We used 2.0 μg/mL cisplatin to treat SK-Hep1 (left panel) and SNU423 (right panel) cells and extracted protein at different time points (4 hrs, 6 hrs, 8 hrs, 10 hrs, 12 hrs) to test AR changes by western blots. (C) After multiple dosage (0.5 μg/mL, 1.0 μg/ mL, 3.0 μg/mL, 5.0 μg/mL, and 10 μg/mL) of cisplatin treatment for 24 hrs, SK-Hep1 (left panel) and SNU423 (right panel) cells were lysed to extract protein. Western blots were performed to test AR changes. (D, E) We overexpressed full length AR in SK-Hep1 cells and SNU423 cells and then treated cells with cisplatin (0.5 μg/mL, 1.0 μg/mL and 2.0 μg/mL) for 48 hrs, then performed Q-PCR and western blot to test ULBP2 changes compared with vector group. (F) After overexpressing AR and cisplatin treatment, HCC cells were collected to perform cytotoxic assay with an E:T ratio at 15:1. Cisplatin treatment groups were compared to non-treatment group. Data shown are mean ± SEM. ***P < 0.001, **P < 0.01, *P < 0.05.
Cisplatin functions through suppressing AR to increase ULBP2 expression in HCC
To prove cisplatin-increased ULBP2 expression may function through suppressing AR expression, we overexpressed AR in SK-Hep1 and SNU423 cell lines, and treated these cells with multiple doses of cisplatin for 48 hrs. The results showed that adding AR partially suppressed cisplatin-increased ULBP2 expression at both mRNA (Fig. 3D) and protein levels (Fig. 3E). As expected, exogenous AR also interrupted the cisplatin-enhanced cytotoxicity of NK cells against HCC (Fig. 3F).
Together, results from Fig. 3 suggest cisplatin may function through decreasing AR expression to increase ULBP2 expression to exert its function to enhance NK cells cytotoxicity to better suppress HCC.
Mechanism dissection how AR suppresses ULBP2 expression
We first confirmed knocking down AR with AR-shRNA not only increased ULBP2 mRNA (Fig. 3A) but also its protein levels in HCC cells (Fig. 4A). Then we further examined AR's influence on transcriptional activity by analyzing the ULBP2 promoter region (http://www.genecards.org/cgi-bin/carddisp.pl?gene=ULBP2) with ALGGEN-PROMO software (http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3) and found four potential androgen-response-element (ARE) binding sites on the 903bp ULBP2 promoter region (Fig. 4B, upper panel). Interestingly, these predicted AREs are all only half of the classical ARE binding pattern which usually consisted of two 6bp single binding sequence connected with a 3bp hinge sequence for AR to form a homo-dimer to regulate its downstream genes [16]. Such scenarios have been illustrated in recent studies which indicated that under selective conditions, AR could also regulate different AR target genes via binding to these half-site-like ARE sequences [17].
Fig. 4.
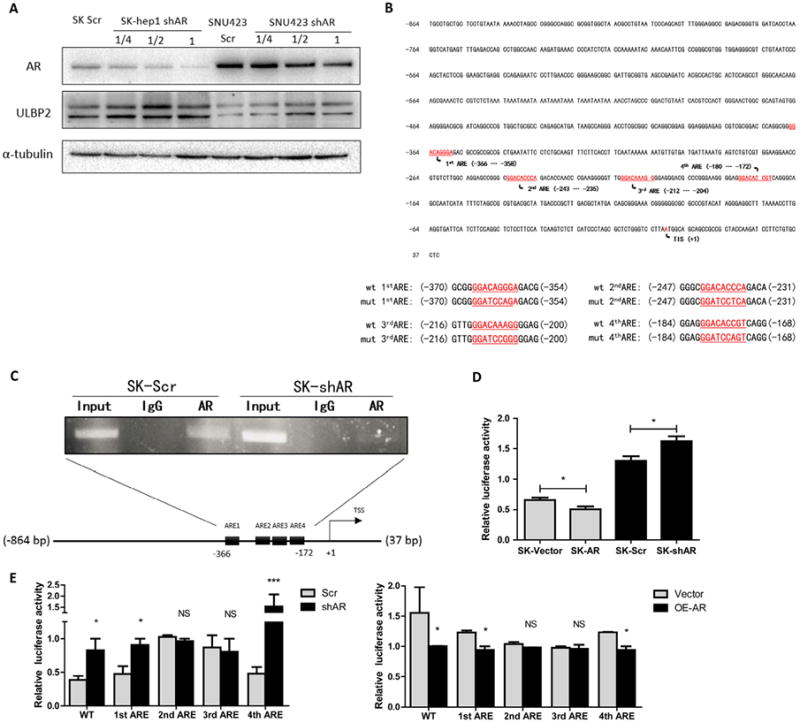
AR suppressed ULBP2 expression transcriptionally through binding to its promoter region. (A) We knocked down AR with sh-RNA at different volumes packaged into lenti-virus. After 48 hrs, we extracted protein and ran western blots to test AR knockdown efficiency and if ULBP2 was decreased. (B) We used ALGGEN-PROMO website to predict possible ARE sites in the ULBP2 903bp promoter region. The lower panel shows four ARE mutation sequences. (C) We designed a pair of primers that could detect the region including the four AR binding sites, then we performed ChIP assay in SK-Hep1 cells with sh-ARorsh-scramble. (D) We constructed ULBP2 903bp promoter region into PGL-3-Basic backbone. After overexpressing or knocking down AR in SK-Hep1 cells, we performed luciferase reporter assay with co-transfected PGL-3-ULBP2-promoter and TK Renilla reporter vector. (E) After four AREs were mutated, we performed luciferase reporter assay either in AR knocked down or overexpressed SK-Hep1 cells. Knocked down groups were compared to scramble groups and over expressed groups were compared to vector groups. Data shown are mean ± SEM. ***P < 0.001, *P < 0.05.
Using ChIP assay to examine if AR could bind to these four predicted AREs distributed within 200bp, we found that AR could bind to this region and knocking down AR led to a decrease of such binding (Fig. 4C), suggesting AR could physically bind to these potential AREs on the ULBP2 promoter region. Next we constructed this 903bp ULBP2 promoter region into PGL-3 basic backbone and performed luciferase reporter assay with overexpressing or knocking-down AR in SK-Hep1 cells. The results revealed that AR could suppress this promoter region since overexpression of AR decreased luciferase activity while knocking down AR increased it (Fig. 4D).
Finally, we mutated these 4 AREs respectively (Fig. 4B, lower panel) and tested luciferase signals after modulating AR level. The results revealed that both ARE2 and ARE3 mediated AR's suppression of ULBP2 transcription (Fig. 4E).
Together, results from Fig. 4 suggest that AR could suppress ULBP2 expression at the transcription level.
ULBP2 is negatively correlated with AR in human clinical HCC samples
To correlate the above in vitro cell lines data in human clinical samples, we studied human HCC samples from Sir Run Run Shaw Hospital and stained AR and ULBP2 for correlation analysis. We classified the results into four grades based on the reviews performed by pathologists and representative images are shown in Fig. 5A and B. The results indicated a negatively correlated expression (Fig. 5C and D) and Spearman correlation analysis also identified a moderate negative correlation between AR and ULBP2 in human HCC tumor tissues (R =−0.3392, P = 0.0020).
Mechanism dissection showing cisplatin decreased AR expression in HCC cells via altering miR-34a-5p
Early studies suggested that cisplatin could induce miR-34a-5p [18] and miR-34a-5p was reported to be able to decrease AR expression in prostate cancer [19]. We therefore examined if cisplatin could also decrease AR via inducing miR-34a-5p in HCC cells. As shown in Fig. 6A, we found cisplatin treatment could increase the miR-34a-5p in a dosage-dependent manner. We then overexpressed miR-34a-5p in HCC SK-Hep1 and SNU423 cells (Fig. 6B, left panel) and found addition of miR-34a-5p suppressed AR expression in HCC cells (Fig. 6B, right panel). Next we performed the interruption assay via adding miR34a-5p inhibitor to examine inhibitor's effect on cisplatin induced AR down-regulation for 24 hrs, and results revealed that cisplatin-decreased AR was partially reversed after adding miR34a-5p inhibitor, suggesting the essential role of miR34a-5p for mediating the cisplatin-decreased AR expression in HCC cells (Fig. 6C).
Fig. 6.
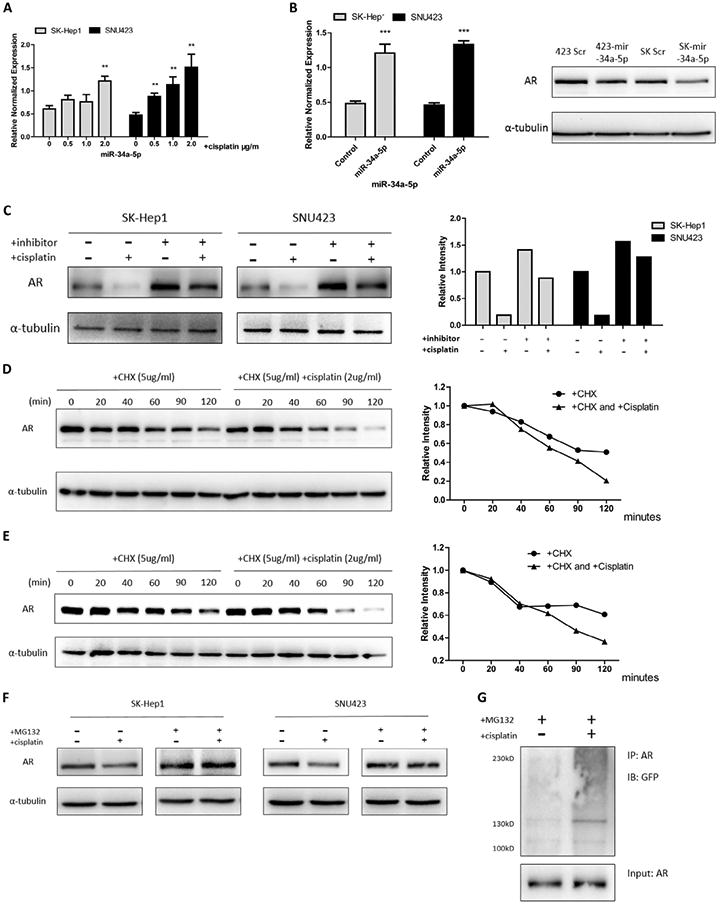
Cisplatin could induce miR-34a-5p and AR degradation. (A) We treated SK-Hep1 and SNU423 cells with different dosages of cisplatin (0.5 μg/mL, 1.0 μg/mL and 2.0 μg/ mL) for 24 hrs, then cells were collected to extract mRNA and perform microRNA RT-Q-PCR. Cisplatin treatment groups were compared to non-treatment group. (B) We constructed overexpressed miR-34a-5p by inserting it into pLV-THM and packaged lenti-virus, then infected SK-Hep1 and SNU423 cells and tested overexpression efficiency (left panel). After 48 hrs infection, protein was extracted for western blots to test AR changes (right panel). (C) 50 nM miR34-5p inhibitor was transfected into HCC cells for 48 hrs, then cells treated w/wo 2.0 μg/mL cisplatin for 12 hrs. Left panel is western blot image while right is quantification data. (D and E) After 1 hr 2.0 μg/mL cisplatin treatment, 5.0 μg/mL Cycloheximide (CHX) was added and collected cell lysates at different time points up to 2 hrs. Western blot was performed (left panel) and quantified (right panel). (F) We treated cells w/wo MG132 (10 μM) and w/wo cisplatin (2.0 μg/mL) together for 3 hrs and performed western blot. (G) AR and GFP-ubiquitin were trans-fected into 293T cells and then treated with MG132 (10 μM) and cisplatin (2.0 μg/mL) for 3 hrs, then Co-IP and immunoblot were performed. Data shown are mean ± SEM. ***P < 0.001, **P < 0.01.
Together, results from Fig. 6A–C suggest cisplatin may function through increasing miR-34a-5p14 to suppress AR expression in HCC cells.
Mechanism dissection showing cisplatin decreased AR expression in HCC cells via altering AR ubiquitination
Recently cisplatin was reported to function through altering ubiquitination to influence the protein degradation [20]. To determine the metabolic stability of AR in the presence of cisplatin, we added cycloheximide (CHX) (for 2 hrs) in HCC cells pre-treated w/wo cisplatin for 1 hr and then collected cell lysates to determine AR protein level. The results from SK-Hep1 cells (Fig. 6D) and SNU423 cells (Fig. 6E) all revealed that cisplatin could alter AR protein stability. We then applied MG132, a proteasome inhibitor, to determine its impact on cisplatin effects on AR protein stability, and results revealed that MG132 reversed cisplatin-induced AR protein degradation (Fig. 6F). To test whether this AR degradation was mediated by ubiquitin, we co-transfected AR and GFP-ubiquitin protein [21] into HEK293T cells and performed immunoprecipitation assay to examine if cisplatin could enhance ubiquitination of AR protein, and results showed cisplatin was able to increase the covalent linkage of AR and ubiquitin (Fig. 6G).
Together, results from Fig. 6D-G led to the conclusion that in addition to increasing miR-34a-5p to suppress AR expression in HCC cells, cisplatin could also decrease AR protein level through enhanced ubiquitin-mediated protein degradation.
Cisplatin enhanced cytotoxicity of NK cells to better suppress HCC in in vivo mouse models
To confirm cisplatin induced up-regulation of NK cells cytotoxicity in vivo, we developed an orthotopic xenograft nude mouse model using the following 4 groups: group 1 mice were co-injected with HCC tumor cells (SK-Hep1 treated with 1 μg/mL cisplatin for 48 hrs before injection) and NK-92MI cells; group 2 mice were co-injected with SK-Hep1 cells (no cisplatin treatment) and NK-92MI cells; group 3 mice were only injected with SK-Hep1 cells (treated with 1 μg/mL cisplatin for 48 hrs before injection) and group 4 mice were only injected with SK-Hep1 cells (no cisplatin treatment). E:T ratios were 1:5 and we injected cells into the left lobes of nude mice livers.
Using the IVIS deception system, we found that groups (#3 and #4) only injected with tumor cells had tumor formation after 2 weeks while groups (#1 and #2) with co-injected NK-92MI cells could develop tumors only after 4 weeks.
Eight weeks after injection, the IVIS detection demonstrated SK-Hep1-cisplatin tumors (in #1) were significantly smaller than SK-Hep1 control tumors (in #2) when they were co-injected into liver with NK cells (Fig. 7A), while no obvious difference of tumor size was observed on IVIS image in #3 vs #4 (Fig. 7A, left panel).
Fig. 7.
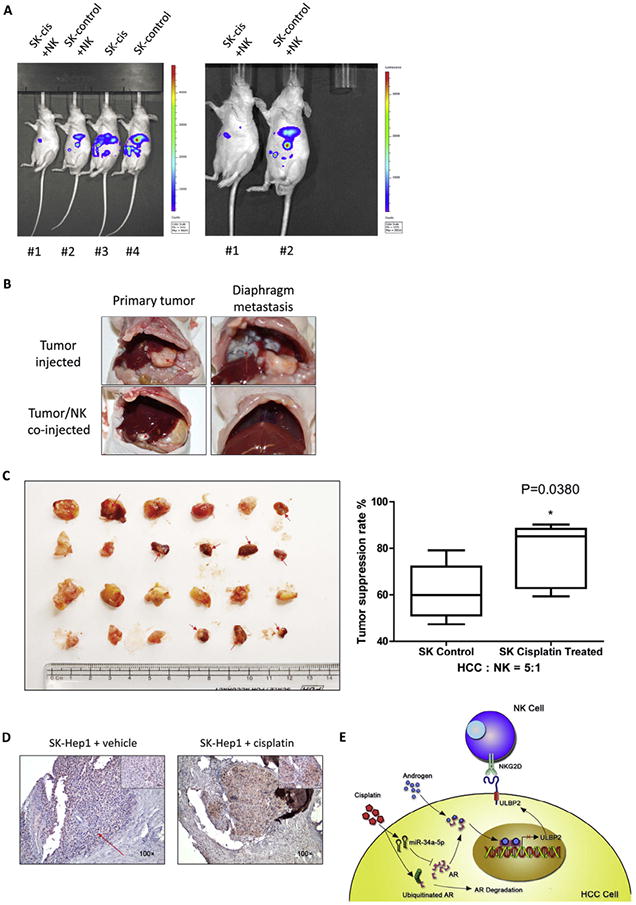
Cisplatin could up-regulate NK cells cytotoxicity in orthotopic xenograft mouse model. (A) Orthotopic HCC tumor mice model was monitored by IVIS image system before sacrifice at 8 weeks after injection. Representative images were shown with four groups together (left panel) or only NK-92MI cells co-injected group comparisons (right panel). (B, C) Isolated orthotopic tumors from mice livers (left panel). Tumors were kept in PBS before weighing. Suppression rate of NK cells was calculated based on tumor weights. Suppression rate = (weight of tumor co-injected with NK cells/average weight of same type tumor without NK cells) × 100%. (D) Representative images of IHC staining in tumors treated with cisplatin or vehicle in vivo, which showed cisplatin could induce ULBP2 expression. (E) Schematic diagram of cisplatin induced activation of NK cells. Data shown are mean ± SEM. *P < 0.05.
We then sacrificed mice and identified co-injected NK-92MI cells in #1 and #2 have significantly inhibited tumor formation and progression compared with #3 and #4 (Fig. 7B). Results from the tumor weight measurements (Fig. 7C, left panel) and suppression rate calculation (Fig. 7C, right panel) further confirmed our conclusion, showing NK cells immunotherapy has better effects against HCC tumor after low dose cisplatin pre-treatment. IHC staining results also confirmed our in vitro cell line data showing the ULBP2 expression was increased in the cisplatin treatment group (Fig. 7D).
Discussion
Tumor heterogeneity and tumor micro-environment make it difficult to control HCC only with single treatment modality for patients [22,23]. Especially for advanced HCC patients current therapy with sorafenib could not be satisfying and resistance to therapy steadily arises [24]. In Japan, it was found that a combination of low-dose 5-fluorouracil (5-FU) and cisplatin by hepatic arterial infusion could yield favorable outcomes for advanced HCC patients [25,26]. Such treatment regiment has not been widely adopted in the western countries due to a lack of randomized clinical trials [15]. Here we offered some valuable information to better apply cisplatin for HCC treatment by identifying its indirect effect on NK cells (Fig. 7E). After cisplatin treatment, ULBP2, one of the major NKG2D ligands, could be up-regulated and assisted NK cells to better target tumor cells in liver cancer. Furthermore, such up-regulation is AR dependent since we found AR is a ULBP2 suppressor by direct binding to ULBP2 promoter, meanwhile cisplatin has a potent function to decrease AR via either increasing miR-34a-5p or inducing AR protein degradation. It is likely that cisplatin represses AR expression in a temporal manner with a rapid reduction of AR through protein degradation while having a lasting effect through induction of miRNA. The exact contribution from these two mechanisms to the final outcome of enhancement of efficacy remains to be clarified. Nevertheless the implication of cisplatin-induced AR decrease is wide particularly for prostate cancer as AR has been a critical factor for cancer progression [27]. Interestingly, an early report indicated that miR-34a could serve as a ULBP2 repressor in tumors [28], which might imply miR-34a plays an opposite role in such a circumstance. But the cells they used were melanoma cells and the expression level of AR was unknown. It is possible that an inhibition of AR by cisplatin, thus an increase of ULBP2 expression, would outweigh the inhibition by miR-34a, and the net effect was up-regulation of ULBP2 in the AR-positive tumor cells.
Gender disparity has been regarded as a major issue for HCC development and treatment. In the last 10 years, androgen receptor, not androgen, has been proven to have huge impact on HCC [11,12,29]. In the current study, we identified and characterized a novel function of AR in HCC, i.e. AR can regulate HCC progression through its influence on innate immunity. Such understanding likely will help the development of new therapies by combining traditional chemotherapy and immunotherapy.
Accumulating evidence indicated that AR could suppress genes expression by directly binding DNA through AR binding site [30,31]. However, the exact mechanism remains unclear. We found that AR could directly bind to the two half-AREs in ULBP2 promoter region to suppress ULBP2 transcription, and such suppression could be diminished by cisplatin treatment. However these two half-AREs do not conform to the canonical ARE structure as they are spaced by 22 nucleotides instead of 3 nucleotides. It is not clear whether a homodimer of AR bind to these AREs with a sequence bulge in between, or AR alone, or with some unknown factor, binding to these AREs separately. The latter mode of AR binding was supported by the recent analysis of genome-wide AR binding in prostate cancer cells [17] and detailed mechanism remains to be clarified.
In summary, our findings indicated that AR expression level might serve as a biomarker for application of cisplatin during TACE treatment for HCC, and the initial AR expression level may influence the final treatment outcome. Furthermore, possible immune therapy combined with chemotherapy may generate a better efficacy against advanced liver cancer.
Supplementary Material
Acknowledgments
We thank Karen Wolf for help with manuscript preparation. We appreciate Dr. Zhinong Jiang and Dr. Tao Zhu from Sir Run Run Shaw Hospital for kindly reviewing and scoring IHC staining of patient specimens. This work was supported by NIH grants (CA155477 and CA156700), George Whipple Professorship Endowment and Taiwan Department of Health Clinical Trial, Research Center of Excellence (DOH99-TD-B-111-004 to China Medical University, Taichung, Taiwan), International scientific and technological cooperation projects (2012DFA30410, China), Zhejiang Provincial Natural Science Foundation of China (LZ14H160002), National Natural Science Foundation of China (81201942), and the National Science-technology Support Plan Projects (2012BAI14B06, China).
Abbreviations
- AR
androgen receptor
- NK cells
natural killer cells
- ULBP2
UL16-binding protein 2
- NKG2D
natural-killer group 2 member D
- HCC
hepatocellular carcinoma
- ET
effector cells:target cells
- IFN-γ
interferon-γ
- CHX
cycloheximide
- ARE
androgen receptor element
Appendix: Supplementary material
Supplementary data to this article can be found online at doi:10.1016/j.canlet.2016.01.017.
Footnotes
Conflict of interest: There is no conflict of interest with any financial organization regarding the material discussed in the manuscript.
Contributor Information
Xiujun Cai, Email: cxjzu@hotmail.com.
Chawnshang Chang, Email: chang@urmc.rochester.edu.
References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics. CA Cancer J Clin. 2012;65(2015):87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Llovet JM, Bruix J. Novel advancements in the management of hepatocellular carcinoma in 2008. J Hepatol. 2008;48(Suppl. 1):S20–S37. doi: 10.1016/j.jhep.2008.01.022. [DOI] [PubMed] [Google Scholar]
- 3.Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S, Kim JS, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10:25–34. doi: 10.1016/S1470-2045(08)70285-7. [DOI] [PubMed] [Google Scholar]
- 4.Barkholt L, Alici E, Conrad R, Sutlu T, Gilljam M, Stellan B, et al. Safety analysis of ex vivo-expanded NK and NK-like T cells administered to cancer patients: a phase I clinical study. Immunotherapy. 2009;1:753–764. doi: 10.2217/imt.09.47. [DOI] [PubMed] [Google Scholar]
- 5.Soriani A, Zingoni A, Cerboni C, Iannitto ML, Ricciardi MR, Di Gialleonardo V, et al. ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood. 2009;113:3503–3511. doi: 10.1182/blood-2008-08-173914. [DOI] [PubMed] [Google Scholar]
- 6.Yamashita T, Arai K, Sunagozaka H, Ueda T, Terashima T, Yamashita T, et al. Randomized, phase II study comparing interferon combined with hepatic arterial infusion of fluorouracil plus cisplatin and fluorouracil alone in patients with advanced hepatocellular carcinoma. Oncology. 2011;81:281–290. doi: 10.1159/000334439. [DOI] [PubMed] [Google Scholar]
- 7.Ueshima K, Kudo M, Takita M, Nagai T, Tatsumi C, Ueda T, et al. Hepatic arterial infusion chemotherapy using low-dose 5-fluorouracil and cisplatin for advanced hepatocellular carcinoma. Oncology. 2010;78(Suppl. 1):148–153. doi: 10.1159/000315244. [DOI] [PubMed] [Google Scholar]
- 8.Yeo W, Mok TS, Zee B, Leung TW, Lai PB, Lau WY, et al. A randomized phase III study of doxorubicin versus cisplatin/interferon alpha-2b/doxorubicin/fluorouracil (PIAF) combination chemotherapy for unresectable hepatocellular carcinoma. J Natl Cancer Inst. 2005;97:1532–1538. doi: 10.1093/jnci/dji315. [DOI] [PubMed] [Google Scholar]
- 9.Ramakrishnan R, Assudani D, Nagaraj S, Hunter T, Cho HI, Antonia S, et al. Chemotherapy enhances tumor cell susceptibility to CTL-mediated killing during cancer immunotherapy in mice. J Clin Invest. 2010;120:1111–1124. doi: 10.1172/JCI40269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma WL, Lai HC, Yeh S, Cai X, Chang C. Androgen receptor roles in hepatocellular carcinoma, fatty liver, cirrhosis and hepatitis. Endocr Relat Cancer. 2014;21:R165–R182. doi: 10.1530/ERC-13-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu MH, Ma WL, Hsu CL, Chen YL, Ou JH, Ryan CK, et al. Androgen receptor promotes hepatitis B virus-induced hepatocarcinogenesis through modulation of hepatitis B virus RNA transcription. Sci Transl Med. 2010;2:32ra35. doi: 10.1126/scitranslmed.3001143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma WL, Hsu CL, Yeh CC, Wu MH, Huang CK, Jeng LB, et al. Hepatic androgen receptor suppresses hepatocellular carcinoma metastasis through modulation of cell migration and anoikis. Hepatology. 2012;56:176–185. doi: 10.1002/hep.25644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma WL, Jeng LB, Lai HC, Liao PY, Chang C. Androgen receptor enhances cell adhesion and decreases cell migration via modulating beta1-integrin-AKT signaling in hepatocellular carcinoma cells. Cancer Lett. 2014;351:64–71. doi: 10.1016/j.canlet.2014.05.017. [DOI] [PubMed] [Google Scholar]
- 14.He D, Li L, Zhu G, Liang L, Guan Z, Chang L, et al. ASC-J9 suppresses renal cell carcinoma progression by targeting an androgen receptor-dependent HIF2alpha/VEGF signaling pathway. Cancer Res. 2014;74:4420–4430. doi: 10.1158/0008-5472.CAN-13-2681. [DOI] [PubMed] [Google Scholar]
- 15.Kudo M. Treatment of advanced hepatocellular carcinoma with emphasis on hepatic arterial infusion chemotherapy and molecular targeted therapy. Liver Cancer. 2012;1:62–70. doi: 10.1159/000342402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Horie-Inoue K, Bono H, Okazaki Y, Inoue S. Identification and functional analysis of consensus androgen response elements in human prostate cancer cells. Biochem Biophys Res Commun. 2004;325:1312–1317. doi: 10.1016/j.bbrc.2004.10.174. [DOI] [PubMed] [Google Scholar]
- 17.Chen Z, Lan X, Thomas-Ahner JM, Wu D, Liu X, Ye Z, et al. Agonist and antagonist switch DNA motifs recognized by human androgen receptor in prostate cancer. EMBOJ. 2015;34:502–516. doi: 10.15252/embj.201490306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bhatt K, Zhou L, Mi QS, Huang S, She JX, Dong Z. MicroRNA-34a is induced via p53 during cisplatin nephrotoxicity and contributes to cell survival. Mol Med. 2010;16:409–416. doi: 10.2119/molmed.2010.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ostling P, Leivonen SK, Aakula A, Kohonen P, Makela R, Hagman Z, et al. Systematic analysis of microRNAs targeting the androgen receptor in prostate cancer cells. Cancer Res. 2011;71:1956–1967. doi: 10.1158/0008-5472.CAN-10-2421. [DOI] [PubMed] [Google Scholar]
- 20.Abedini MR, Muller EJ, Bergeron R, Gray DA, Tsang BK. Akt promotes chemoresistance in human ovarian cancer cells by modulating cisplatin-induced, p53-dependent ubiquitination of FLICE-like inhibitory protein. Oncogene. 2010;29:11–25. doi: 10.1038/onc.2009.300. [DOI] [PubMed] [Google Scholar]
- 21.Dantuma NP, Groothuis TA, Salomons FA, Neefjes J. A dynamic ubiquitin equilibrium couples proteasomal activity to chromatin remodeling. J Cell Biol. 2006;173:19–26. doi: 10.1083/jcb.200510071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marusyk A, Polyak K. Tumor heterogeneity: causes and consequences. Biochim Biophys Acta. 2010;1805:105–117. doi: 10.1016/j.bbcan.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Whiteside TL. The tumor microenvironment and its role in promoting tumor growth. Oncogene. 2008;27:5904–5912. doi: 10.1038/onc.2008.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abou-Alfa GK. Sorafenib use in hepatocellular carcinoma: more questions than answers. Hepatology. 2014;60:15–18. doi: 10.1002/hep.27044. [DOI] [PubMed] [Google Scholar]
- 25.Kudo M, Izumi N, Kokudo N, Matsui O, Sakamoto M, Nakashima O, et al. Management of hepatocellular carcinoma in Japan: consensus-based clinical practice guidelines proposed by the Japan Society of Hepatology (JSH) 2010 updated version. Dig Dis. 2011;29:339–364. doi: 10.1159/000327577. [DOI] [PubMed] [Google Scholar]
- 26.Ricke J, Seidensticker M, Mohnike K. Noninvasive diagnosis of hepatocellular carcinoma in cirrhotic liver: current guidelines and future prospects for radiological imaging. Liver Cancer. 2012;1:51–58. doi: 10.1159/000339020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chiang KC, Tsui KH, Chung LC, Yeh CN, Feng TH, Chen WT, et al. Cisplatin modulates B-cell translocation gene 2 to attenuate cell proliferation of prostate carcinoma cells in both p53-dependent and p53-independent pathways. Sci Rep. 2014;4:5511. doi: 10.1038/srep05511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heinemann A, Zhao F, Pechlivanis S, Eberle J, Steinle A, Diederichs S, et al. Tumor suppressive microRNAs miR-34a/c control cancer cell expression of ULBP2, a stress-induced ligand of the natural killer cell receptor NKG2D. Cancer Res. 2012;72:460–471. doi: 10.1158/0008-5472.CAN-11-1977. [DOI] [PubMed] [Google Scholar]
- 29.Yu MW, Cheng SW, Lin MW, Yang SY, Liaw YF, Chang HC, et al. Androgen-receptor gene CAG repeats, plasma testosterone levels, and risk of hepatitis B-related hepatocellular carcinoma. J Natl Cancer Inst. 2000;92:2023–2028. doi: 10.1093/jnci/92.24.2023. [DOI] [PubMed] [Google Scholar]
- 30.Cai C, He HH, Chen S, Coleman I, Wang H, Fang Z, et al. Androgen receptor gene expression in prostate cancer is directly suppressed by the androgen receptor through recruitment of lysine-specific demethylase 1. Cancer Cell. 2011;20:457–471. doi: 10.1016/j.ccr.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grosse A, Bartsch S, Baniahmad A. Androgen receptor-mediated gene repression. Mol Cell Endocrinol. 2012;352:46–56. doi: 10.1016/j.mce.2011.06.032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.