

Abstract
Objective:
To identify the genetic basis of a recessive syndrome characterized by prenatal hyperechogenic brain foci, congenital microcephaly, hypothalamic midbrain dysplasia, epilepsy, and profound global developmental disability.
Methods:
Identification of the responsible gene by whole exome sequencing and homozygosity mapping.
Results:
Ten patients from 4 consanguineous Palestinian families manifested in utero with hyperechogenic brain foci, microcephaly, and intrauterine growth retardation. Postnatally, patients had progressive severe microcephaly, neonatal seizures, and virtually no developmental milestones. Brain imaging revealed dysplastic elongated masses in the midbrain–hypothalamus–optic tract area. Whole exome sequencing of one affected child revealed only PCDH12 c.2515C>T, p.R839X, to be homozygous in the proband and to cosegregate with the condition in her family. The allele frequency of PCDH12 p.R839X is <0.00001 worldwide. Genotyping PCDH12 p.R839X in 3 other families with affected children yielded perfect cosegregation with the phenotype (probability by chance is 2.0 × 10−12). Homozygosity mapping revealed that PCDH12 p.R839X lies in the largest homozygous region (11.7 MB) shared by all affected patients. The mutation reduces transcript expression by 84% (p < 2.4 × 10−13). PCDH12 is a vascular endothelial protocadherin that promotes cellular adhesion. Endothelial adhesion disruptions due to mutations in OCLN or JAM3 also cause congenital microcephaly, intracranial calcifications, and profound psychomotor disability.
Conclusions:
Loss of function of PCDH12 leads to recessive congenital microcephaly with profound developmental disability. The phenotype resembles Aicardi-Goutières syndrome and in utero infections. In cases with similar manifestations but no evidence of infection, our results suggest consideration of an additional, albeit rare, cause of congenital microcephaly.
Congenital microcephaly can be the outcome of genetic lesions, metabolic disorders, or intrauterine infections. We describe 4 consanguineous families with a congenital microcephaly syndrome characterized by prenatal perithalamic hyperechogenicity, congenital progressive microcephaly with hypothalamus-midbrain dysplasia, early-onset intractable epilepsy, and profound psychomotor disability. This phenotype overlaps with certain manifestations of in utero infections and Aicardi-Goutières syndrome (AGS), both of which are mediated by exposure of the developing brain to toxic levels of interferon-α.1,2 This exposure induces extensive microangiopathy with calcifications associated with blood vessels.3,4 Direct endothelial interruption due to mutations in occludin (OCLN5–7) or in junctional adhesion molecule 3 (JAM38,9) also results in congenital microcephaly, brain calcifications, and profound developmental disability.10 In the patients we report, there was no evidence of intrauterine infection and the phenotype was not consistent with any known genetic syndrome that mimics congenital infection. We therefore undertook genome-wide analysis to determine the genetic cause of this microcephaly syndrome.
METHODS
Patients.
Families were seen at the Medical Genetics and Pediatric Neurology clinics at Shaare Zedek Medical Center, Jerusalem, Israel.
Standard protocol approvals, registrations, and patient consent.
Families were enrolled and blood or amniotic fluid samples were obtained after written consent. The study was approved by the Institutional Review Board of Shaare Zedek Medical Center and the Israel National Ethics Committee for Genetic Studies.
Genomic analyses.
Genomic DNA was extracted from peripheral blood mononuclear cells or amniocytes. Whole exome sequencing11 and homozygosity mapping12 were performed as previously described. Mutation and haplotype analyses were performed using primers listed in table e-1 on the Neurology® Web site at Neurology.org. For evaluation of transcript expression, total RNA was extracted from peripheral blood leucocytes drawn into Tempus Blood RNA tubes (ABI, Chiba, Japan), then reverse-transcribed and converted to cDNA using random hexamers in the presence of RNase inhibitor (rRNasin; Promega, Madison, WI). A cDNA segment of PCDH12 exons 1 and 2 was amplified by PCR and Sanger sequenced.
TA cloning.
Reverse transcription PCR products were cloned using the pGEM-T Easy Vector System (Promega) and transformed into JM109 high-efficiency competent cells (Promega). Blue/white color screening was used in order to select successful cloning of the insert. Selected clones were amplified with PCR Ready-Mix (Bio-Lab, Jerusalem, Israel) using T7 promoter primer and SP6 promoter primer (table e-1), and Sanger sequenced using BigDye Terminator V.1.1.
RESULTS
Clinical features.
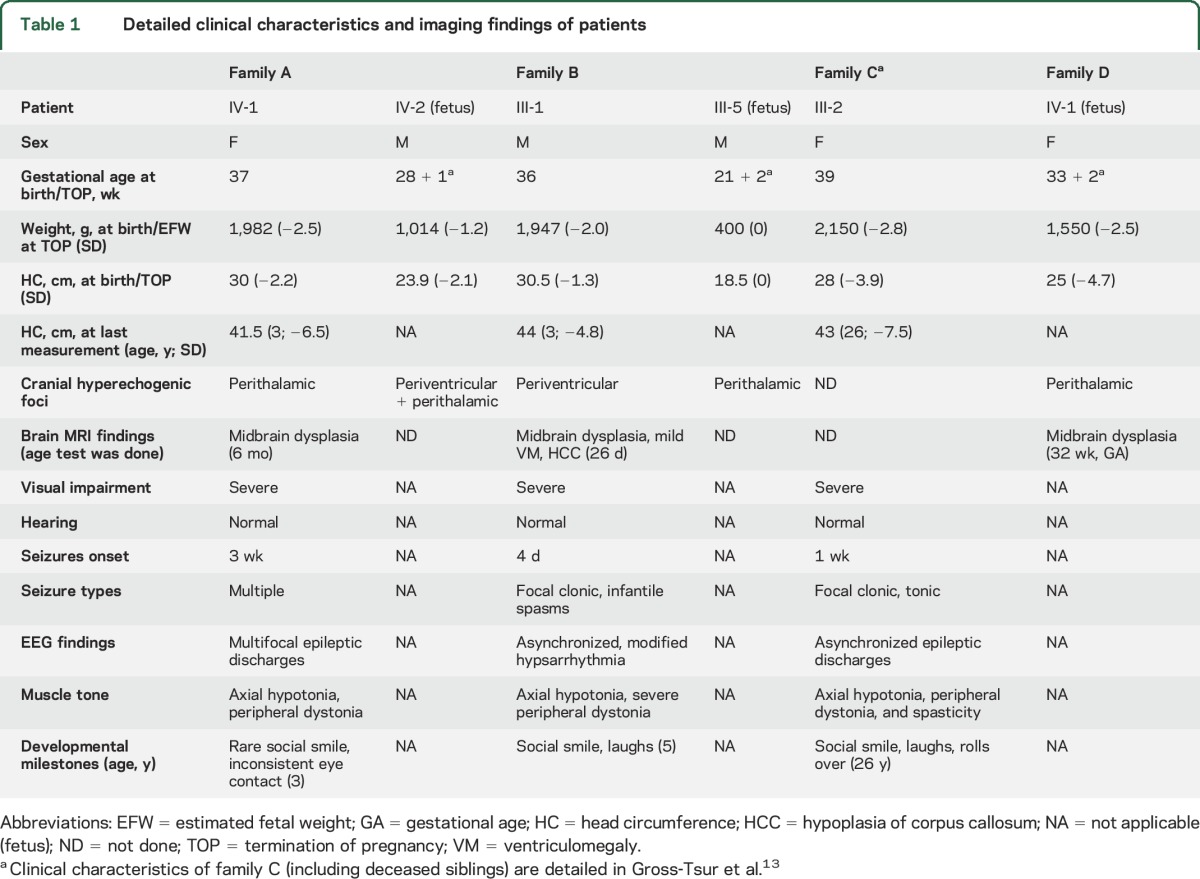
Four consanguineous families of Palestinian origin included 10 individuals with the disorder (figures 1 and 2, table 1). All families are from the same town of ∼7,000 residents. In family A, index patient IV-1 presented with low birthweight, congenital microcephaly, and perithalamic hyperechogenic foci. PCR for cytomegalovirus (CMV) in urine and serology for Toxoplasma in blood were negative. Refractory tonic and multifocal clonic seizures appeared at age 3 weeks. The patient had no dysmorphism and her neurologic examination was remarkable for axial hypotonia and peripheral dystonia with brisk deep tendon reflexes, severe visual impairment, and profound global developmental disability. Brain MRI revealed dysplastic, elongated midbrain with poor distinction between the crus cerebri of midbrain, hypothalamus, and optic tract (figure 2A.d). Auditory brainstem response, ophthalmologic examination, and electroretinography were normal. At 1 year, visual evoked potential demonstrated bilateral markedly reduced amplitudes; however, repeat evaluation at 25 months was significantly better. At age 3 years, the patient died unexpectedly during sleep. The parents refused autopsy.
Figure 1. Families and PCDH12 genotypes.

Families with recessive congenital progressive microcephaly with prenatal perithalamic hyperechogenicity and hypothalamus-midbrain dysplasia. Genotypes of PCDH12 c.2515 C>T, p.R839X, are indicated below each sampled individual. PCDH12 c.2515C>T is homozygous in all affected individuals and segregates as expected for a recessive disease in all 4 families. TOP = termination of pregnancy.
Figure 2. Brain imaging of patients.

Columns 1–3 illustrate prenatal ultrasounds in coronal, axial, and parasagittal planes; column 4 illustrates T2-axial sections of brain MRIs. Participants are as follows: top row, age-matched controls; A.a, patient A.IV-2 (26w2d); A.b, A.c, patient A.IV-2 (27w6d); A.d, patient A.IV-1 (6 months); B.a-B.c, patient B.III-5 (21w2d); B.d, patient B.III-1 (1 month); and C1–C4, patient D.IV-2 (33w2d). On prenatal ultrasounds, regions with thick perithalamic hyperechogenicity are indicated by white arrows. Brain MRIs show midbrain-hypothalamus dysplasia. The normal structure resembles a black arrow, with optic tracts as arrowhead and hypothalami separated by third ventricle as 2 arrowtails, hanging above a Mickey Mouse ears–shaped midbrain. In patients, the midbrain is elongated and indistinguishable from the hypothalamic and optic tracts. ^ = Cavum septi pellucidi above 3rd ventricle; A = anterior; H = hypothalamus; LV = lateral ventricles; M = midbrain; OT = optic tract; P = posterior; T = thalamus.
Table 1.
Detailed clinical characteristics and imaging findings of patients
At the first appointment with the index patient, her mother was in week 28 of a second pregnancy (IV-2). Prenatal ultrasound revealed microcephaly and hyperechogenic foci in the perithalamic and periventricular areas (figure 2, A.a–A.c). The parents chose to terminate the pregnancy and refused autopsy.
In family B, the oldest child (III-1), with symmetric intrauterine growth retardation (IUGR), significant periventricular hyperechogenicity, and suspected maternal CMV seroconversion (positive immunoglobulin G and immunoglobulin M), was initially suspected to have congenital infection. However, PCR for urine CMV and serology for Toxoplasma in blood and CSF were negative, and liver span, hepatic enzymes, and blood thrombocytes were normal. Brief clonic seizures began at age 4 days and evolved by age 8 months to multiple seizure types including infantile spasms. Metabolic workup (including lactate, pyruvate, pH, ammonia, aminoacids, and acylcarnitines in blood and urine organic acids) was normal. Chromosomal microarray analysis revealed no abnormalities. CSF lymphocyte count (2 cells/μL), aminoacids, and lactate were normal. The patient had severe microcephaly with no additional dysmorphism, severe visual impairment, dystonia, and profound developmental disability. Brain MRI revealed midbrain-hypothalamus dysplasia similar to that found in family A (figure 2B.d). Based on marked clinical and imaging resemblance to the index case, and the shared geographic origin, we suspected that this child has the same syndrome observed in family A. The fetus of the fifth pregnancy in family B (III-5) was found in utero to have cranial hyperechogenic foci on prenatal sonography and negative toxoplasmosis, other (syphilis, varicella-zoster, parvovirus b19), rubella, cytomegalovirus, and herpes (TORCH) serology (figure 2, B.a–B.c). The parents elected to terminate the pregnancy and refused autopsy, but consented to examination of the placenta, which revealed the placenta to be small (10th percentile) with vascular ectasia of the fetal blood vessels in the stem and intermediate villi.
Family C was identified from a previous report13 as likely having the same syndrome, and was found to originate from the same town as families A and B. Briefly, affected children in family C presented with congenital microcephaly, refractory epilepsy of neonatal onset, severe visual impairment, and profound global disability. The only surviving patient is III-2, now 26 years old. Her 4 older siblings died at ages 9, 3, 12, and 6 years from pulmonary aspiration. No DNA was available for the deceased children.
Family D was referred in pregnancy with IV-2 after visualization of perithalamic hyperechogenic foci and progressive symmetric IUGR (figure 2, C.a–C.c). At 24 weeks, microcephaly (−1.6 SD) was noticed; at 26 weeks, reduced abdominal circumference (−1.6 SD) emerged. TORCH serology was negative. The microcephaly progressed to −4.7 SD, and fetal MRI revealed the same midbrain-hypothalamus dysplasia described above (Figure 2C.d). The parents elected to terminate the pregnancy and refused autopsy, but consented to pathology examination of the placenta, which revealed the placenta to be small (5th percentile) without clear vascular pathology.
All patients had fetal presentation of IUGR, microcephaly, and distinctive hyperechogenic foci in the perithalamic area (table 1, figure 2). Serology tests for intrauterine infections were normal for all. Postnatally, patients had neonatal seizures that became refractory to antiepileptic medications, profound global developmental disability, and severe visual impairment. Neurologic examinations revealed severe microcephaly with no other dysmorphism, axial hypotonia, and peripheral hypertonia (mostly dystonia). Brain MRI demonstrated unique midbrain dysplasia with undistinguished boundaries among the elongated midbrain, hypothalamus, and optic tracts. Chromosomal microarray analysis for genomic rearrangements and metabolic tests were normal.
Gene discovery.
Whole exome sequencing of genomic DNA from patient IV-1 in family A (figure 1A) revealed 7 variants that were rare (<1% allele frequency in world populations), homozygous in the patient, and potentially functional (i.e., nonsense, frameshift, at a splice site, or altered a completely conserved aminoacid) (table e-2). These 7 variants were genotyped in the other affected and unaffected relatives of family A. Only one variant segregated with the disease in the family under a recessive model for inheritance of the microcephaly phenotype: PCDH12 c.2515C>T, p.R839X, at chr5:141334902 G>A (hg19, NM_016580, NP_057664) (figures 1A and 3A). This nonsense mutation occurs at codon 839 of the 1,184-aminoacid PCDH12 protein. The mutation appeared as a heterozygote in 1 of 60,684 persons (121,368 alleles) in public databases (exac.broadinstitute.org), yielding an allele frequency <0.00001. In contrast, the mutation was present in 3 of 83 unrelated healthy individuals from the same town as family A, yielding an estimated frequency of 0.018 (3/166) for the mutant allele in the family's hometown. No homozygotes were observed among local healthy controls. The mutation was not present in 200 ethnically matched (Muslim Arab) controls from other geographic locales.
Figure 3. Genetics and expression analysis.

(A) Sanger sequences of PCDH12 c.2515C>T in genomic DNA of a control with wild-type (WT) sequence, and of individuals from family A who are heterozygous (HET) and homozygous (HOM) for the mutation. Nucleotides and expected aminoacid sequence are shown above the chromatograms. (B) Haplotype analysis of the genomic region flanking PCDH12 c.2515C>T. Markers used were short tandem repeat markers (numbers indicate amplicon size, bp) and single nucleotide polymorphisms (dbSNP build 142, letters A or B indicate genotype). Names and genomic positions (hg19) of tested markers on chromosome 5 are indicated, with PCDH12 c.2515C>T indicated in red. The disease-linked haplotype shared in all families is shaded in green. (C) Homozygosity mapping of patients, analyzed by Chromosome Analysis Suite. Homozygous regions for each individual are indicated by a colored block; genomic location is shown beneath. The shared region of homozygosity on chromosome 5 from 129,673,384 to 141,354,220 (hg19) is indicated by a black rectangle. The position of PCDH12 is indicated with a red line. (D) Expression of PCDH12 WT and mutant transcripts. Sanger sequencing of genomic DNA (gDNA) shows equal peak heights for WT and mutant DNA sequences, both at PCDH12 c.2515C>T and at a nearby polymorphism, rs2434322. Allele G of rs2434322 is in cis with PCDH12 c.2515C>T. In contrast, Sanger sequencing of cDNA reveals much lower expression of the mutant compared to the WT transcript: TA cloning showed a 14:88 ratio (see Results).
Next, PCDH12_R839X was genotyped in all participating relatives of families B, C, and D, none of whom had been evaluated by exome sequencing. Cosegregation of PCDH12_R839X with the syndrome perfectly fit a recessive model, with homozygosity for the mutation in all affected individuals and no unaffected individuals (figure 1). Given the allele frequency of 0.018 in the patients' hometown, the likelihood of this extent of cosegregation of genotype and phenotype by chance in families B, C, and D is 2.0 × 10−12.
As an independent approach, genome-wide homozygosity mapping was carried out for 6 affected individuals and their 8 parents in the 4 families (figure 3B). Only one genomic region greater than 3 MB was homozygous in all 6 affected individuals and in none of the parents: a region of 11.7 MB on chromosome 5 from 129,673,384 to 141,354,220 (hg19; figure 2C). This region harbors 220 genes (table e-3), including the entire chromosome 5 protocadherin cluster and hence PCDH12. To confirm that PCDH12_R839X was the only likely pathogenic variant in the shared 11.7 MB homozygous region, 25 short coding segments that were not covered by exome sequencing were individually evaluated by Sanger sequencing. No other candidate variants were identified. Analysis of microsatellites and single nucleotide polymorphisms in the 430 kb region immediately surrounding PCDH12 suggests that PCDH12_R839X has a common origin in all 4 families (figure 3B).
If translated, the PCDH12_R839X protein would be expected to lack most of the protocadherin cytoplasmic domain (figure e-1). However, the mutant transcript appears subject to nonsense-mediated decay. Sanger sequencing of cDNA from a heterozygote carrier revealed higher peak height for the wild-type vs mutant allele at the site of the mutation (figure 3D), and cloned cDNA from a heterozygous carrier yielded 88 wild-type and 14 mutant transcripts (p < 2.4 × 10−13 for deviation from the expected 1:1 ratio).
DISCUSSION
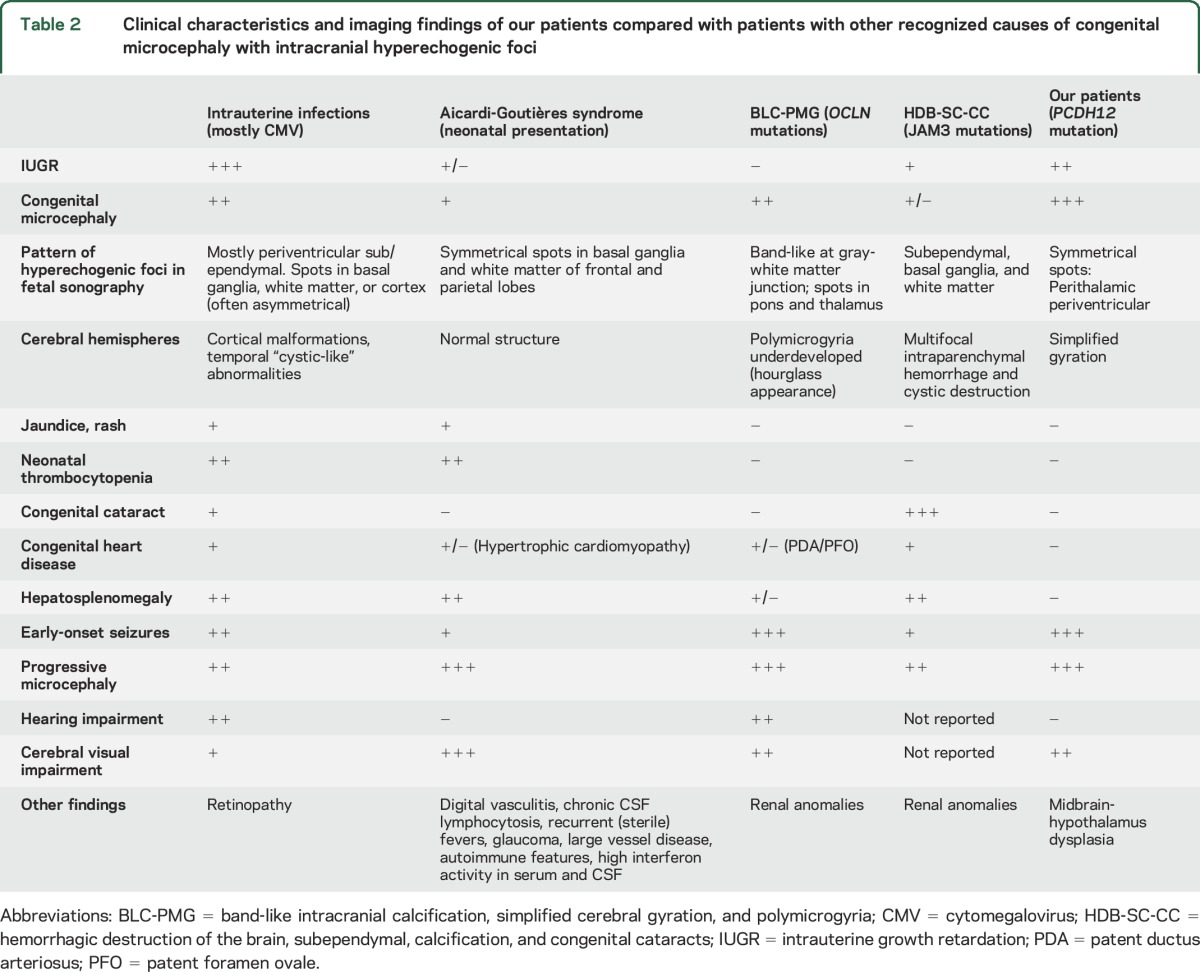
Congenital microcephaly with intracranial hyperechogenicity is associated primarily with congenital infection, particularly by CMV. However, several genetic disorders resemble congenital CMV infection (table 2). In 1984, Aicardi and Goutières14 reported 8 children with familial progressive encephalopathy manifested by progressive microcephaly, spasticity, dystonia, basal ganglia calcifications, neonatal thrombocytopenia, and hepatomegaly. The clinical similarities between these children and those with congenital infections, and the observation in these children of chronic lymphocytosis in CSF, led to the discovery of increased CSF levels of the antiviral cytokine interferon-α in patients with AGS.2 AGS is genetically heterogeneous. In patients with the syndrome, mutations in 7 genes have been identified to date: TREX1 (MIM 606609), RNASEH2B (MIM 610326), RNASEH2C (MIM 610330), RNASEH2A (MIM 606034), SAMHD1 (MIM 606754), ADAR (MIM 146920), and IHIF1 (MIM 606951). AGS is inherited as a recessive condition in all patients except those with mutations in TREX1. The function of all AGS genes is related to nucleic acid metabolism.15 It appears that either host-mediated, as in the case of AGS, or virus-encoded nucleic acids signaling can induce overexpression of proinflammatory cytokines such as interferon-α that damage the developing brain. Among the clinical symptoms of AGS are development of recurrent (sterile) fever, chilblain-like skin lesions, and glaucoma,15 none of which was found in our patients (tables 1 and 2). Cystic leukoencephalopathy due to RNASET2 mutations can also mimic congenital CMV infection. However, cystic leukoencephalopathy is not associated with severe progressive microcephaly.16
Table 2.
Clinical characteristics and imaging findings of our patients compared with patients with other recognized causes of congenital microcephaly with intracranial hyperechogenic foci
PCDH12 is a member of the nonclustered protocadherin family of calcium-dependent cell adhesion proteins. Protocadherins are classified as clustered and nonclustered based on their genomic structure. More than 70 protocadherin proteins have been identified, most of which have been reported to play an important role in the regulation of brain development and function.17–19 Several nonclustered PCDH genes have been implicated previously in neurologic disorders, including PCDH19 in female-restricted epilepsy and mental retardation,20 PCDH15 in Usher syndrome type 1F,21 PCDH21 in retinal degeneration,22 PCDH8 in autism,23 and PCDH7 in epilepsy.24
PCDH12 promotes endothelial adhesion.25,26 Other disruptions of endothelial adhesion, due to mutations in occludin (OCLN)5–7 or junctional adhesion molecule 3 (JAM3),8,9 lead to intracranial calcifications, severe and progressive congenital microcephaly, early-onset seizures, and profound developmental disability. However, unlike patients with the PCDH12 mutation, patients with OCLN mutations also have underdeveloped cortex with bilateral polymicrogyria and patients with JAM3 mutations also have intracerebral hemorrhage with cystic destruction (tables 1 and 2).
In our patients, brain imaging illustrated distinctive characteristics: thick perithalamic hyperechogenicity on prenatal ultrasound and unique hypothalamic-midbrain dysplasia in prenatal and postnatal brain MRI (figure 2). Similar midbrain dysplasia has been reported previously in 6 patients with postnatal progressive microcephaly, but no information on prenatal imaging was available.27 The nature of the hyperechogenic foci in our patients is distinctive. The foci do not cast an acoustic shadow as might be expected for calcifications of similar size; thus, they may possibly represent multiple tortuous vessels with multiple echographic interfaces. As PCDH12 expression is not limited to the perithalamic-midbrain brain area, the intriguing associations among PCDH12 mutations, perithalamic microangiopathy, hypothalamic-midbrain dysplasia, and profound microcephaly and brain impairment are yet to be discovered.28–31
Studies of the biological consequences of PCDH12 mutations may require use of cerebral organoids, rather than model organisms, as microcephaly syndromes are difficult to recapitulate in mice.32 Indeed, mice null for PCDH12 demonstrate only mild vascular (primarily placental) deficit without clear neurologic involvement.25,26,33 Similarly, mice null for TREX1, SAMHD1, IFIH1, or OCLN have neither structural nor functional neurologic deficits.34,35
A likely explanation for the lack of phenotypic correlation between the human disease and the corresponding murine knockouts is differing tissue specificity in expression patterns of the various protocadherins in mice and humans,36 especially since precise spatial and temporal protocadherin expression is critical for vertebrate brain development.28,30,37 In the mouse, PCDH12 is expressed almost exclusively in the placenta; its expression in brain is 500-fold lower than in placenta. In humans, PCDH12 is expressed at similar levels in many tissues, including brain and placenta (biogps.org). The same distinction holds for TREX1, IFIH1, and OCLN (but not SAMHD1); their relative expression in brain is much lower in mouse than in humans. Notably, the vascular pathology in the placenta demonstrated in PCDH12 knockout mice26 may be reflected in the vascular ectasia of the fetal blood vessels of the placenta from fetus III-5 in family B.
Mutations leading to loss of function of protocadherins are implicated in other neurodevelopmental disorders, most notably PCDH19 mutations in female-restricted mental retardation and epilepsy. We add to these observations 4 families with a severely debilitating syndrome including congenital and progressive microcephaly with midbrain dysplasia and profound developmental disability due to a loss-of-function mutation in PCDH12. While it is formally possible that this syndrome is caused by a mutation that is not detectable by exome sequencing and that lies in a region of homozygosity less than 3 MB, cosegregation of homozygosity for the allele with the phenotype in 4 extended families and the loss of expression of the mutant PCDH12 transcript strongly, and independently, suggest that PCDH12 p.R839X is the causal allele. Identification of other patients with similar phenotypes and different mutations in PCDH12 would provide additional confirmation for these findings.
Finally, discovery of PCDH12 as a genetic basis for congenital microcephaly and intracranial hyperechogenic foci has practical clinical implications. In families with subsequently identified genetic causes for congenital microcephaly and intracranial calcifications, the first affected child was frequently misdiagnosed with congenital TORCH infections. Such misdiagnoses may result in familial recurrence of these devastating syndromes. Because each of these syndromes is rare, they are easily overlooked, but in the absence of serologic, molecular or epidemiologic evidence of infection, genetic causes of congenital microcephaly and intracranial calcifications are worthy of consideration. Given unique imaging findings in fetal sonography and brain MRI and a known genetic basis, this syndrome can be identified in utero. Mutations in PCDH12 also should be considered in unresolved cases of congenital microcephaly and intracranial calcifications.10
Supplementary Material
ACKNOWLEDGMENT
The authors thank Tzvia Rosen for laboratory assistance and the patients and their families for cooperation and participation.
GLOSSARY
- AGS
Aicardi-Goutières syndrome
- CMV
cytomegalovirus
- IUGR
intrauterine growth retardation
- TORCH
toxoplasmosis, other (syphilis, varicella-zoster, parvovirus b19), rubella, cytomegalovirus, and herpes
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Adi Aran assisted in conceptualizing the study, identified and recruited patients and family members, examined the participants, collected DNA samples and clinical data, wrote the first draft with N.R., and approved final manuscript version. Nuphar Rosenfeld assisted in conceptualizing the study, preformed most of the genetic testing and analysis, wrote the first draft with A.A., and approved revision of the manuscript. Ranit Jaron and Reeval Segel examined the participants, assisted in collection and interpretation of clinical data, and approved revision of the manuscript. Paul Renbaum assisted in conceptualizing the study, provided insights about the hypothesis, and directed genetic testing and analysis. Sharon Zeligson performed homozygosity mapping. Shachar Zuckerman and Hila Fridman identified and recruited patients, counseled participating families, and collected DNA samples. Yoav Kohn, Moein Kanaan, and Lara Kamal obtained and analyzed control DNA samples. Yoram Segev interpreted brain MRIs. Eyal Mazaki, Ron Rabinowitz, and Ori Shen examined pregnant patients, performed prenatal ultrasounds, obtained prenatal samples, recorded and interpreted clinical data, and revised and approved the manuscript. Ming K. Lee performed genomic analysis and interpreted results. Mary-Claire King directed genomic analysis, interpreted results, and critically revised the manuscript. Tom Walsh and Suleyman Gulsuner performed genomic analysis, interpreted results, and wrote sections of the manuscript. Ephrat Levy-Lahad initiated and conceptualized the study, obtained funding, directed genetic testing, analysis, and interpretation of the results, and critically revised the manuscript.
STUDY FUNDING
This study was supported by the US Agency for International Development program for Middle East Regional Cooperation (award TA-MOU-10-M30-021 to E. Levy-Lahad and M. Kanaan) and a gift from the Hassenfeld family (to Shaare Zedek Medical Center).
DISCLOSURE
A. Aran, N. Rosenfeld, and R. Jaron report no disclosures relevant to the manuscript. P. Renbaum reports grant funding from the Israel Science Fund. S. Zuckerman, H. Fridman, S. Zeligson, and R. Segel report no disclosures relevant to the manuscript. Y. Kohn reports grant funding from NIH, NARSAD, US-Israel Binational Science Foundation BSF, National Institute for Psychobiology in Israel, and the Rosenbaum Milton Endowment Fund. L. Kamal reports no disclosures relevant to the manuscript. M. Kanaan reports grant funding from USAID-MERC (Middle East Regional Cooperation). Y. Segev, E. Mazaki, R. Rabinowitz, O. Shen, and M. Lee report no disclosures relevant to the manuscript. T. Walsh reports grant funding from NIH/NIMH R01MH083989. M. King reports grant funding from NIH/NIMH R01MH083989. S. Gulsuner reports grant funding from the Brain and Behavior Research Foundation. E. Levy-Lahad reports grant funding from USAID MERC (Middle East Regional Cooperation). Grants for other projects include funding from the Breast Cancer Research Foundation (NY), Israel Science Fund, Israel Cancer Research Fund, and Israel Cancer Association. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Gresser I, Morel-Maroger L, Riviere Y, et al. Interferon-induced disease in mice and rats. Ann NY Acad Sci 1980;350:12–20. [DOI] [PubMed] [Google Scholar]
- 2.Lebon P, Badoual J, Ponsot G, Goutieres F, Hemeury-Cukier F, Aicardi J. Intrathecal synthesis of interferon-alpha in infants with progressive familial encephalopathy. J Neurol Sci 1988;84:201–208. [DOI] [PubMed] [Google Scholar]
- 3.Klok MD, Bakels HS, Postma NL, van Spaendonk RM, van der Knaap MS, Bugiani M. Interferon-alpha and the calcifying microangiopathy in Aicardi-Goutières syndrome. Ann Clin Transl Neurol 2015;2:774–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cantey JB, Sisman J. The etiology of lenticulostriate vasculopathy and the role of congenital infections. Early Hum Dev 2015;91:427–430. [DOI] [PubMed] [Google Scholar]
- 5.Rajab A, Aldinger KA, El-Shirbini HA, Dobyns WB, Ross ME. Recessive developmental delay, small stature, microcephaly and brain calcifications with locus on chromosome 2. Am J Med Genet A 2009;149A:129–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O'Driscoll MC, Daly SB, Urquhart JE, et al. Recessive mutations in the gene encoding the tight junction protein occludin cause band-like calcification with simplified gyration and polymicrogyria. Am J Hum Genet 2010;87:354–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elsaid MF, Kamel H, Chalhoub N, et al. Whole genome sequencing identifies a novel occludin mutation in microcephaly with band-like calcification and polymicrogyria that extends the phenotypic spectrum. Am J Med Genet A 2014;164A:1614–1617. [DOI] [PubMed] [Google Scholar]
- 8.Mochida GH, Ganesh VS, Felie JM, et al. A homozygous mutation in the tight-junction protein JAM3 causes hemorrhagic destruction of the brain, subependymal calcification, and congenital cataracts. Am J Hum Genet 2010;87:882–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Akawi NA, Canpolat FE, White SM, et al. Delineation of the clinical, molecular and cellular aspects of novel JAM3 mutations underlying the autosomal recessive hemorrhagic destruction of the brain, subependymal calcification, and congenital cataracts. Hum Mutat 2013;34:498–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Livingston JH, Stivaros S, Warren D, Crow YJ. Intracranial calcification in childhood: a review of aetiologies and recognizable phenotypes. Dev Med Child Neurol 2014;56:612–626. [DOI] [PubMed] [Google Scholar]
- 11.Navon Elkan P, Pierce SB, Segel R, et al. Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. N Engl J Med 2014;370:921–931. [DOI] [PubMed] [Google Scholar]
- 12.Weinberg-Shukron A, Renbaum P, Kalifa R, et al. A mutation in the nucleoporin-107 gene causes XX gonadal dysgenesis. J Clin Invest 2015;125:4295–4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gross-Tsur V, Joseph A, Blinder G, Amir N. Familial microcephaly with severe neurological deficits: a description of five affected siblings. Clin Genet 1995;47:33–37. [DOI] [PubMed] [Google Scholar]
- 14.Aicardi J, Goutieres F. A progressive familial encephalopathy in infancy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis. Ann Neurol 1984;15:49–54. [DOI] [PubMed] [Google Scholar]
- 15.Crow YJ, Chase DS, Lowenstein Schmidt J, et al. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am J Med Genet A 2015;167A:296–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henneke M, Diekmann S, Ohlenbusch A, et al. RNASET2-deficient cystic leukoencephalopathy resembles congenital cytomegalovirus brain infection. Nat Genet 2009;41:773–775. [DOI] [PubMed] [Google Scholar]
- 17.Garrett AM, Weiner JA. Control of CNS synapse development by {gamma}-protocadherin-mediated astrocyte-neuron contact. J Neurosci 2009;29:11723–11731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Keeler AB, Molumby MJ, Weiner JA. Protocadherins branch out: multiple roles in dendrite development. Cell Adh Migr 2015;9:214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seong E, Yuan L, Arikkath J. Cadherins and catenins in dendrite and synapse morphogenesis. Cell Adh Migr 2015;9:202–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dibbens LM, Tarpey PS, Hynes K, et al. X-linked protocadherin 19 mutations cause female-limited epilepsy and cognitive impairment. Nat Genet 2008;40:776–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahmed ZM, Riazuddin S, Bernstein SL, et al. Mutations of the protocadherin gene PCDH15 cause Usher syndrome type 1F. Am J Hum Genet 2001;69:25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Henderson RH, Li Z, Abd El Aziz MM, et al. Biallelic mutation of protocadherin-21 (PCDH21) causes retinal degeneration in humans. Mol Vis 2010;16:46–52. [PMC free article] [PubMed] [Google Scholar]
- 23.Butler MG, Rafi SK, Hossain W, Stephan DA, Manzardo AM. Whole exome sequencing in females with autism implicates novel and candidate genes. Int J Mol Sci 2015;16:1312–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.International League Against Epilepsy Consortium on Complex Epilepsies. Genetic determinants of common epilepsies: a meta-analysis of genome-wide association studies. Lancet Neurol 2014;13:893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rampon C, Prandini MH, Bouillot S, et al. Protocadherin 12 (VE-cadherin 2) is expressed in endothelial, trophoblast, and mesangial cells. Exp Cell Res 2005;302:48–60. [DOI] [PubMed] [Google Scholar]
- 26.Philibert C, Bouillot S, Huber P, Faury G. Protocadherin-12 deficiency leads to modifications in the structure and function of arteries in mice. Pathol Biol 2012;60:34–40. [DOI] [PubMed] [Google Scholar]
- 27.Zaki MS, Saleem SN, Dobyns WB, et al. Diencephalic-mesencephalic junction dysplasia: a novel recessive brain malformation. Brain 2012;135:2416–2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Biswas S, Emond MR, Jontes JD. Protocadherin-19 and N-cadherin interact to control cell movements during anterior neurulation. J Cell Biol 2010;191:1029–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoshina N, Tanimura A, Yamasaki M, et al. Protocadherin 17 regulates presynaptic assembly in topographic corticobasal ganglia circuits. Neuron 2013;78:839–854. [DOI] [PubMed] [Google Scholar]
- 30.Biswas S, Emond MR, Duy PQ, Hao le T, Beattie CE, Jontes JD. Protocadherin-18b interacts with Nap1 to control motor axon growth and arborization in zebrafish. Mol Biol Cell 2014;25:633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coughlin GM, Kurrasch DM. Protocadherins and hypothalamic development: do they play an unappreciated role? J Neuroendocrinol 2015;27:544–555. [DOI] [PubMed] [Google Scholar]
- 32.Lancaster MA, Renner M, Martin CA, et al. Cerebral organoids model human brain development and microcephaly. Nature 2013;501:373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rampon C, Bouillot S, Climescu-Haulica A, et al. Protocadherin 12 deficiency alters morphogenesis and transcriptional profile of the placenta. Physiol Genomics 2008;34:193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crow YJ, Manel N. Aicardi-Goutières syndrome and the type I interferonopathies. Nat Rev Immunol 2015;15:429–440. [DOI] [PubMed] [Google Scholar]
- 35.Saitou M, Furuse M, Sasaki H, et al. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol Biol Cell 2000;11:4131–4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frank M, Ebert M, Shan W, et al. Differential expression of individual gamma-protocadherins during mouse brain development. Mol Cell Neurosci 2005;29:603–616. [DOI] [PubMed] [Google Scholar]
- 37.Cooper SR, Emond MR, Duy PQ, Liebau BG, Wolman MA, Jontes JD. Protocadherins control the modular assembly of neuronal columns in the zebrafish optic tectum. J Cell Biol 2015;211:807–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.