

Abstract
A unique chemo- and regioselective α- and γ- arylation of palladium azapentadienyl intermediates is presented. Two distinct catalysts and sets of conditions successfully controlled the regioselectivity of the arylation. These methods provide the first umpolung C−H functionalization of azapentadienyl palladium intermediates and enable the divergent synthesis of allylic amine and enamine derivatives, which are of significant interest in the pharmaceutical industry.
Keywords: allylic amines, arylation, divergent synthesis, enamines, regioselectivity
Chemo- and regioselective allylic C−H functionalization of organic compounds by transition-metal-based catalysts has attracted considerable recent attention.[1] Beautiful applications of this approach to organic synthesis continue to emerge.[2] In contrast, the analogous C−H functionalization of azaallyl derivatives remains largely unexplored. Given that allylic amines and enamines[3] are valuable building blocks and are prevalent in bioactive compounds[4] (e.g. naftifine[4d] and flunarizine[4e]), the C−H functionalization of azaallyl systems has significant untapped potential.
Early studies with azaallyl palladium intermediates were reported by O’Donnell et al.[5] and involved relatively sensitive hemiaminal precursors (Scheme 1A). In 2015, Trost et al.[6] reported the use of readily available N-allyl imines in oxidative C−H functionalization reactions in the presence of 2,6-dimethylbenzoquinone (Scheme 1B). This groundbreaking chemistry appears to generate equilibrating π-allyl intermediates that can be regioselectively trapped with nucleophiles with differing steric demand.
Scheme 1.
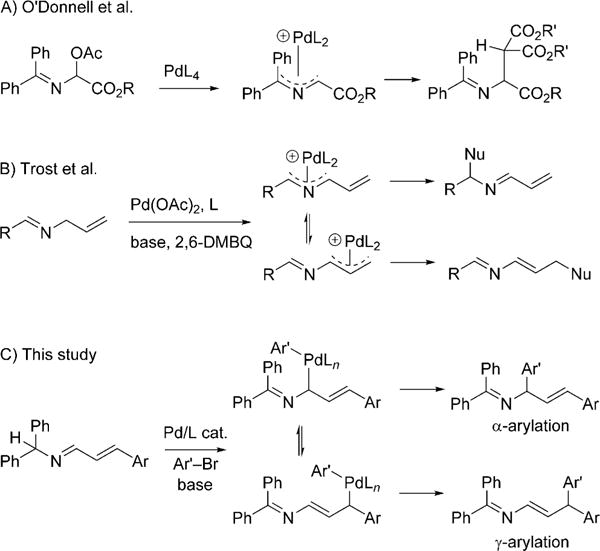
Arylation of azaallyl anions.
Owing to our interest in allylic C−H functionalization reactions[7] and the arylation of nitrogen-containing compounds,[8,9] we were inspired to investigate an umpolung approach to generate palladium azapentadienyl intermediates (Scheme 1C). Our goal was to control the regioselectivity in the C−C bond-forming reductive-elimination step. If successful, such a method would enable access to arylated allylic amines and enamines. Herein, we disclose the first examples of umpolung allylic arylation that proceed with high levels of regioselectivity.
We initiated our studies with aldimine 1a and bromobenzene (2a) by microscale screening (0.01 mmol) with 24 electronically diverse mono- and bidentate phosphine ligands and two solvents [cyclopentyl methyl ether (CPME) and THF] at two temperatures (23 and 60°C) with Pd(OAc)2 and KN(SiMe3)2 (Scheme 2; see the Supporting Information for details). P(tBu)3HBF4 and Xantphos were identified as ligands favoring α-arylation to generate allylic amine derivative 3aa. In sharp contrast, NiXantphos,[10] developed by van Leeuwen and co-workers, generated the γ-arylation product, enamine 4aa. Cyclization of the α-arylation product led to by-product 5aa in up to 50% yield.
Scheme 2.
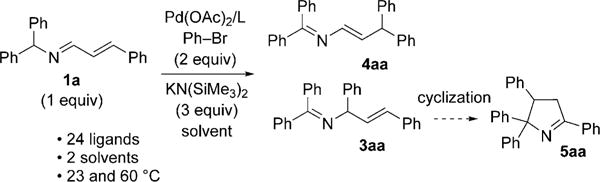
Reaction development and initial optimization.
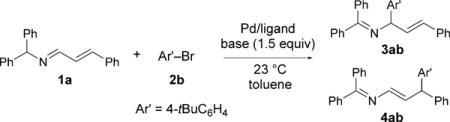
We suspected we could minimize the formation of the cyclic by-product 5aa by using less of the base at the lower temperature (23°C). With this in mind, we performed a series of microscale experiments (0.02 mmol) with 1.5 equivalents of the base, ligands PtBu3HBF4, Xantphos, and NiXantphos, palladium complexes [Pd(dba)2], Pd(OAc)2, [{ClPd(2-methallyl)}2], and [{ClPd(allyl)}2], four solvents (THF, CPME, DME, toluene), and two bases [NaN(SiMe3)2 and KN-(SiMe3)2; see the Supporting Information for details]. The most promising α-arylation screening result was obtained with [{ClPd(2-methallyl)}2], PtBu3HBF4, and KN(SiMe3)2 in toluene. Laboratory-scale optimization (0.1 mmol) led to α-arylation to give 3ab in 84% assay yield (AY; Table 1, entry 1). At the optimal concentration of 0.05M, excellent selectivity was observed (> 20:1), and the product was isolated in 90% yield (Table 1, entry 2).
Table 1.
Optimization of arylation α and γ to the nitrogen atom of 1a with 2b.a
| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| Entry | Pd (mol%)/L (mol%) | M in MN(SiMe3)2 | t [h] | Conc. [M] | Yield [%]b | |
| 3ab | 4ab | |||||
| 1 | [{ClPd(2-methallyl)}2] (5)/PtBu3 (15) | K | 10 | 0.1 | 84 | <5 |
| 2 | [{ClPd(2-methallyl)}2] (5)/PtBu3 (15) | K | 10 | 0.05 | 93 (90)c | <5 |
| 3 | [Pd(dba)2] (10)/NiXantphos (20) | Na | 10 | 0.1 | <5 | 72 |
| 4 | [Pd(dba)2] (10)/NiXantphos (20) | Na | 10 | 0.05 | <5 | 75 |
| 5 | precatalyst 6 (10) | Na | 10 | 0.05 | <5 | 82 |
| 6 | precatalyst 7 (10) | Na | 10 | 0.05 | <5 | 85 |
| 7 | precatalyst 7 (10) | Na | 6 | 0.05 | <5 | 89 (88)c |
| 8 | precatalyst 7 (10) | K | 6 | 0.05 | <5 | 67 |
| 9 | precatalyst 7 (10) | Li | 6 | 0.05 | 4 | 63d |
Reactions were conducted on a 0.1 mmol scale.
Assay yield determined by 1H NMR spectroscopy of the crude reaction mixture.
Yield of the isolated product after chromatographic purification.
The reaction was carried out at 60°C. dba= dibenzylideneacetone.
The leading screening result for the γ-arylation was with [Pd(dba)2], NiXantphos, and NaN(SiMe3)2 in toluene, which gave the product in 72% AY on a laboratory scale (0.1 mmol; Table 1, entry 3). A slight increase in AY was observed at 0.05M (75% assay yield; Table 1, entry 4). Buchwald precatalysts[8b,c] facilitate catalyst generation, often lead to increased activity, and allow a lower ligand loading. Therefore, we employed Buchwald-type[11] precatalysts 6 and 7 (with L = NiXantphos; Table 1, entries 5 and 6). The use of precatalyst 7 led to the γ-arylated product 4ab in 85% AY with high selectivity (> 20:1). A reduction in the reaction time to 6 h enabled the isolation of 4ab in 88% yield (Table 1, entry 7). The assay yield dropped to 67% when we used KN(SiMe3)2 instead of NaN(SiMe3)2, but the regioselectivity was > 20:1 (Table 1, entry 8). No reaction was observed with LiN(SiMe3)2 at room temperature, but at 60°C γ-arylation predominated and afforded the product in 63% assay yield with 16:1 selectivity (Table 1, entry 9).
Having optimized the reaction conditions, we investigated the scope of the α-arylation with respect to the aryl bromide substrate (Scheme 3). High regioselectivity (α/γ 9:1 to > 20:1) was observed in all cases. Bromobenzene and 1-bromo-4-tert-butylbenzene underwent coupling in excellent yield (85 and 90%, respectively). Efficient coupling of electron-donating 4-bromoanisole and 4-bromo-N,N-dimethylaniline afforded allylic amine derivatives 3ac and 3ad in 81 and 82% yield with 10:1 and 14:1 selectivity, respectively. Coupling reactions with sterically hindered 1-bromonaphthylene and 2-bromotoluene were more efficient with NaN(SiMe3)2 in CPME, under which conditions the products 3ae and 3af were obtained in 68 and 74% yield, respectively. Heterocyclic 5-bromobenzofuran and 5-bromo-N-methylindole were also good substrates: Products 3ag and 3ah were formed in 70 and 80% yield, respectively, both with excellent regioselectivity. The meta-substituted aryl bromides 3-bromotoluene and 3-bromo-N,N-dimethylaniline underwent coupling with 1a in 80% yield with 9:1 and 10:1 selectivity, respectively. The coupling of 3-bromoanisole afforded 3ak in 71% yield, and that of 2-bromonaphthalene afforded 3al in 72% yield; in both cases, the regioselectivity was higher than 20:1. Unfortunately, the reaction with electron-withdrawing 1-bromo-4-fluorobenzene exhibited low selectivity.[12] To probe the scalability of the reaction, we carried out the synthesis of 3ab on a 3 mmol scale with a catalyst loading of 5 mol%. The desired product was isolated in 86% yield (1.1 g, > 20:1 regioselectivity).
Scheme 3.
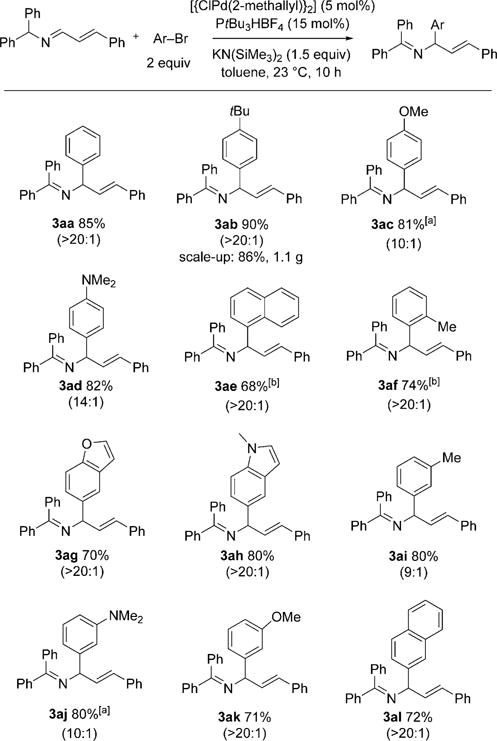
Scope of the α-arylation with respect to the aryl bromide substrate. Reactions were conducted on a 0.1 mmol scale at 0.05M. Yields are for the isolated product after chromatographic purification. The 3/4 product ratio was determined by 1H NMR spectroscopy of the crude reaction mixture. [a] CPME was used as the solvent. [b] The reaction was carried out with NaN(SiMe3)2 in CPME.
Having demonstrated the scope of the α-selective arylation with respect to the aryl bromide substrate, we next turned to the γ-arylation. High regioselectivity (> 20:1) was observed in most cases with the NiXantphos-based catalyst (Scheme 4). Bromobenzene and 1-bromo-4-tert-butylbenzene provided products 4aa and 4ab in 86 and 88% yield, respectively. Aryl bromides with electron-withdrawing substituents, such as 4-F, 4-CF3, and 3-CF3, all underwent coupling in about 80% yield with > 20:1 regioselectivity. The coupling of 1a with aryl bromides containing electron-donating groups at the 3-position resulted in decreased yields (60–63%), partially as a result of the only moderate selectivity (7:1 and 3:1, respectively). Sterically more demanding 1-bromonaphthalene also exhibited catalyst-controlled reactivity and afforded the γ-arylated product in 68% yield with excellent regioselectivity. Finally, the coupling of 1a with heterocyclic 5-bromobenzofuran gave the desired product in 72% yield. The reaction with 1-bromo-3-(trifluoromethyl)benzene was scaled up to 3 mmol with a catalyst loading of 5 mol%. The product was isolated in 70% yield (0.93 g, > 20:1 regioselectivity).
Scheme 4.
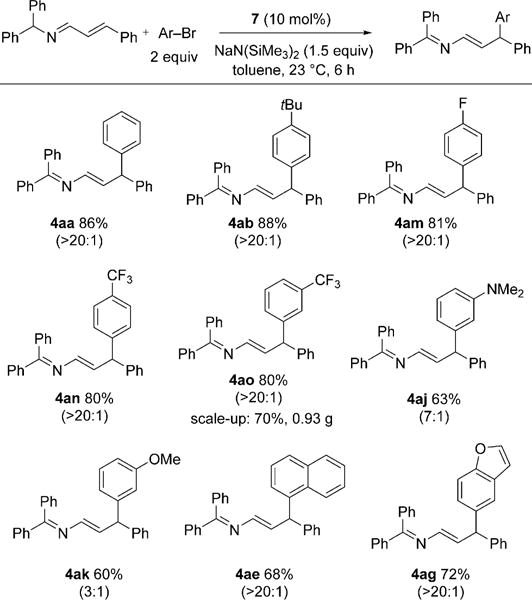
Scope of the γ-arylation with respect to the aryl bromide substrate. Reactions were conducted on a 0.1 mmol scale at 0.05M. Yields are for the isolated product after chromatographic purification. The 4/3 product ratio was determined by 1H NMR spectroscopy of the crude reaction mixture.
We next surveyed a few cinnamaldehyde-derived aldimines (Scheme 5). The α-arylation of aldimines 1b (Ar = 4-OMeC6H4), 1c (Ar = 4-ClC6H4), 1d (Ar = 4-FC6H4) with 1-bromo-4-tert-butylbenzene and the PtBu3-based catalyst afforded the corresponding products in 80–90% yield; the regioselectivity of the reactions was not affected by the different electronic properties of the substrates. Likewise, high regioselectivity (> 20:1) and good yields (70–88%) were observed in the γ-arylation with the NiXantphos-based catalyst (Scheme 5).
Scheme 5.
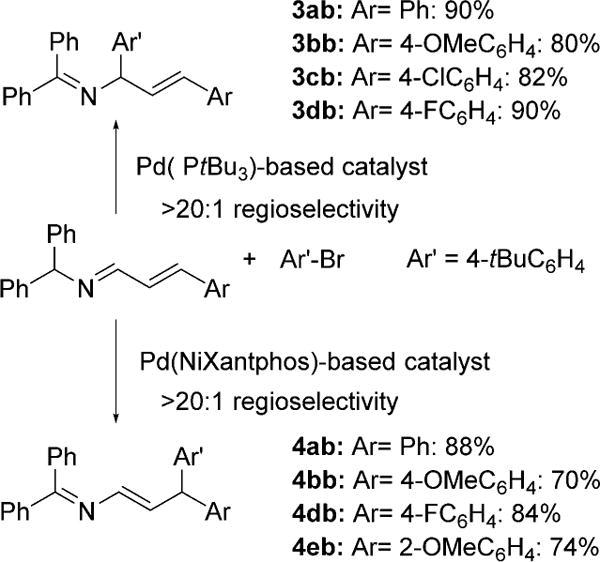
Scope of the regioselective α- and γ-arylation reactions with respect to the aldimine substrate (see the Supporting Information for details). Reaction conditions for the α-arylation: 1 (1 equiv), aryl bromide (2 equiv), [{ClPd(2-methallyl)}2] (5 mol%), PtBu3HBF4 (15 mol%), KN(SiMe3)2 (1.5 equiv), toluene, 0.1 mmol scale, 0.05M, 23°C, 10 h. Reaction conditions for the γ-arylation: 1 (1 equiv), ArBr (2 equiv), precatalyst 7 (10 mol%), NaN(SiMe3)2 (1.5 equiv), toluene,0.1 mmol scale, 0.05M, 23°C, 6 h. Yields are for the isolated product after chromatographic purification. Selectivity was determined by 1H NMR spectroscopy of the crude reaction mixture.
Hydrolysis of the α-arylated product 3ab gave the allylic amine (99% yield, Scheme 6). Similarly, the enamine derivative 4aj was hydrolyzed to the aldehyde in 95% yield. As compared with traditional 1,2- or 1,4-addition reactions,[13] our method provides the first umpolung disconnection for such aldehydes.
Scheme 6.
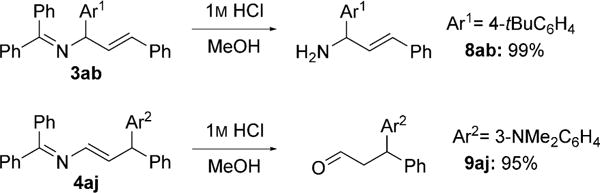
Hydrolysis to afford allylic amine and aldehyde products. See the Supporting Information for optimization and experimental details. Yields are for the isolated product after chromatographic purification.
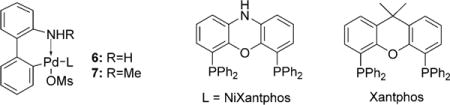
Although a mechanistic study has not yet been performed, the ligand-screening results provide some food for thought. In particular, of the 24 ligands screened, 22 favor α-arylation, thus establishing the α position as the substrate-controlled arylation site. Only NiXantphos and bulky P(2-tolyl)3 gave γ-arylation. Additionally, there is an intriguing difference in selectivity between the Xantphos- and NiXantphos-based catalysts in this arylation reaction. Although both ligands are structurally similar (see Table 1), NiXantphos possesses an acidic N−H group (pKa ≈ 21), which we have shown to be deprotonated in other catalytic reactions[14] in the presence of silylamide bases, MN(SiMe3)2, to give a heterobimetallic catalyst. When the Pd(Xantphos) system was employed, the α-arylation product was favored over the γ-product (1.6:1, Scheme 7). In contrast, the NiXantphos-based catalyst provided exclusively the γ-arylation product. Moreover, when the NiXantphos N−H group was replaced with N−Bn, the resulting catalyst was unselective (Scheme 7). To rationalize this surprising shift in selectivity, we propose that the cation of the bimetallic NiXantphos-based catalyst forms a cation–π interaction with the aryl ring of the substrate (Figure 1),[15] thus forcing reductive elimination at the γ-position to form the enamine product with high selectivity. Our working model has a bridging NaN(SiMe3)2 moiety, and is thus similar to related crystal structures of deprotonated NiXantphos complexes.[14]
Scheme 7.
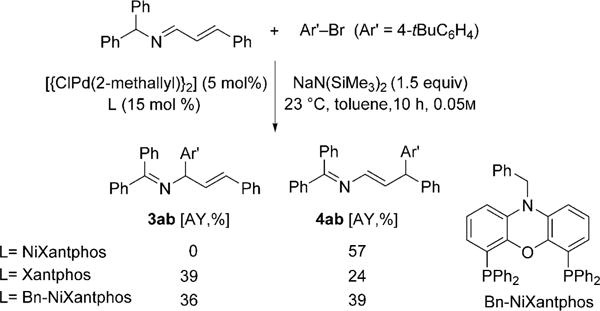
Comparison of Pd complexes of NiXantphos, Xantphos, and Bn-NiXantphos. Bn = benzyl.
Figure 1.
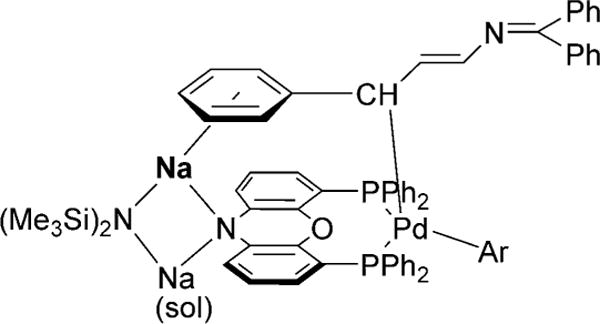
Proposed cation–π interaction guiding regioselectivity.
In summary, we have developed a remarkable catalyst-controlled chemo- and regioselective α- and γ-arylation of azaallyl anions. This umpolung C−H functionalization of azapentadienyl palladium intermediates enables the synthesis of allylic amine and enamine derivatives from common, readily accessible precursors. We propose that the high selectivity observed for the γ-arylation is the result of a cation–π interaction between the heterobimetallic Pd/Na NiXantphos-based catalyst and the substrate.
Supplementary Material
Acknowledgments
We thank the National Science Foundation (CHE-1464744) and the National Institutes of Health (NIGMS 104349) for financial support. We thank Dr. Sonia Montel of the University of Pennsylvania for helpful discussions.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201509757.
Contributor Information
Minyan Li, Roy and Diana Vagelos Laboratories, Penn/Merck Laboratory for High-Throughput Experimentation, Department of Chemistry, University of Pennsylvania, 231 South 34th Street, Philadelphia, PA (USA).
María González-Esguevillas, Roy and Diana Vagelos Laboratories, Penn/Merck Laboratory for High-Throughput Experimentation, Department of Chemistry, University of Pennsylvania, 231 South 34th Street, Philadelphia, PA (USA).
Dr. Simon Berritt, Roy and Diana Vagelos Laboratories, Penn/Merck Laboratory for High-Throughput Experimentation, Department of Chemistry, University of Pennsylvania, 231 South 34th Street, Philadelphia, PA (USA)
Prof. Xiaodong Yang, Roy and Diana Vagelos Laboratories, Penn/Merck Laboratory for High-Throughput Experimentation, Department of Chemistry, University of Pennsylvania, 231 South 34th Street, Philadelphia, PA (USA) Key Laboratory of Medicinal Chemistry for Natural Resource, School of Chemical Science and Technology, Yunnan University, Kunming, 650091 (P.R. China).
Dr. Ana Bellomo, Roy and Diana Vagelos Laboratories, Penn/Merck Laboratory for High-Throughput Experimentation, Department of Chemistry, University of Pennsylvania, 231 South 34th Street, Philadelphia, PA (USA)
Prof. Dr. Patrick J. Walsh, Roy and Diana Vagelos Laboratories, Penn/Merck Laboratory for High-Throughput Experimentation, Department of Chemistry, University of Pennsylvania, 231 South 34th Street, Philadelphia, PA (USA).
References
- 1.For early examples, see:; a) Hansson S, Heumann A, Rein T, Aakermark B. J Org Chem. 1990;55:975–984. [Google Scholar]; b) Grennberg H, Simon V, Bäckvall JE. J Chem Soc Chem Commun. 1994:265–266. [Google Scholar]; c) Chen MS, White MC. J Am Chem Soc. 2004;126:1346–1347. doi: 10.1021/ja039107n. [DOI] [PubMed] [Google Scholar]; d) Chen MS, Prabagaran N, Labenz NA, White MC. J Am Chem Soc. 2005;127:6970–6971. doi: 10.1021/ja0500198. [DOI] [PubMed] [Google Scholar]; for a recent example, see:; e) Liron F, Oble J, Lorion MM, Poli G. Eur J Org Chem. 2014:5863–5883. [Google Scholar]; “Allylic Oxidations: Palladium-Catalyzed Allylic Oxidation of Olefins”:; f) Grennberg H, Bäckvall J-E. In: Transition Metals for Organic Synthesis. 2nd. Beller M, Bolm C, editors. Vol. 2. Wiley-VCH; Weinheim: 2004. pp. 243–255. [Google Scholar]
- 2.For selected examples, see:; a) Stang EM, White MC. Nat Chem. 2009;1:547–551. doi: 10.1038/nchem.351. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Stang EM, White MC. Angew Chem Int Ed. 2011;50:2094–2097. doi: 10.1002/anie.201007309. Angew. Chem.2011, 123, 2142–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Luzung MR, Lewis CA, Baran PS. Angew Chem Int Ed. 2009;48:7025–7029. doi: 10.1002/anie.200902761. Angew. Chem.2009, 121, 7159–7163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Negri G, Kascheres C, Kascheres AJ. J Heterocycl Chem. 2004;41:461–491. [Google Scholar]; b) Liu D, Xie F, Zhang W. Tetrahedron Lett. 2007;48:7591–7594. [Google Scholar]; c) Mukherjee S, Yang JW, Hoffmann S, List B. Chem Rev. 2007;107:5471–5569. doi: 10.1021/cr0684016. [DOI] [PubMed] [Google Scholar]; d) Maji B, Lakhdar S, Mayr H. Chem Eur J. 2012;18:5732–5740. doi: 10.1002/chem.201103519. [DOI] [PubMed] [Google Scholar]; e) Xiao J. ChemCatChem. 2012;4:612–615. [Google Scholar]; f) Gigant N, Chausset-Boissarie L, Gillaizeau I. Chem Eur J. 2014;20:7548–7564. doi: 10.1002/chem.201402070. [DOI] [PubMed] [Google Scholar]
- 4.a) Stütz A. Angew Chem Int Ed Engl. 1987;26:320–328. Angew. Chem.1987, 99, 323–331. [Google Scholar]; b) Stütz A, Georgopoulos A, Granitzer W, Petranyi G, Berney D. J Med Chem. 1986;29:112–125. doi: 10.1021/jm00151a019. [DOI] [PubMed] [Google Scholar]; c) Nanavati SM, Silverman RB. J Am Chem Soc. 1991;113:9341–9349. [Google Scholar]; d) Petranyi G, Ryder NS, Stutz A. Science. 1984;224:1239–1241. doi: 10.1126/science.6547247. [DOI] [PubMed] [Google Scholar]; e) Olesen J. J Neurol. 1991;238:S23–S27. doi: 10.1007/BF01642902. [DOI] [PubMed] [Google Scholar]
- 5.O’Donnell MJ, Yang X, Li M. Tetrahedron Lett. 1990;31:5135–5138. [Google Scholar]
- 6.Trost BM, Mahapatra S, Hansen M. Angew Chem Int Ed. 2015;54:6032–6036. doi: 10.1002/anie.201501322. Angew. Chem.2015, 127, 6130–6134. [DOI] [PubMed] [Google Scholar]
- 7.Hussain N, Frensch G, Zhang J, Walsh PJ. Angew Chem Int Ed. 2014;53:3693–3697. doi: 10.1002/anie.201309084. Angew. Chem.2014, 126, 3767–3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) McGrew GI, Stanciu C, Zhang J, Carroll PJ, Dreher SD, Walsh PJ. Angew Chem Int Ed. 2012;51:11510–11513. doi: 10.1002/anie.201201874. Angew. Chem.2012, 124, 11678–11681. [DOI] [PubMed] [Google Scholar]; b) Li M, Berritt S, Walsh PJ. Org Lett. 2014;16:4312–4315. doi: 10.1021/ol502043j. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Li M, Yucel B, Adrio J, Bellomo A, Walsh PJ. Chem Sci. 2014;5:2383–2391. doi: 10.1039/C3SC53526F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.For other examples of the use of azaallyl anions in palladium catalysis, see:; a) Burger EC, Tunge JA. J Am Chem Soc. 2006;128:10002–10003. doi: 10.1021/ja063115x. [DOI] [PubMed] [Google Scholar]; b) Fields WH, Chruma JJ. Org Lett. 2010;12:316–319. doi: 10.1021/ol902651j. [DOI] [PubMed] [Google Scholar]; c) Fields WH, Khan AK, Sabat M, Chruma JJ. Org Lett. 2008;10:5131–5134. doi: 10.1021/ol801986m. [DOI] [PubMed] [Google Scholar]; d) Yeagley AA, Lowder MA, Chruma JJ. Org Lett. 2009;11:4022–4025. doi: 10.1021/ol901745x. [DOI] [PubMed] [Google Scholar]; e) Wolf G, Würthwein EU. Tetrahedron Lett. 1988;29:3647–3650. [Google Scholar]; f) Zhu Y, Buchwald SL. J Am Chem Soc. 2014;136:4500–4503. doi: 10.1021/ja501560x. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Niwa T, Yorimitsu H, Oshima K. Org Lett. 2008;10:4689–4691. doi: 10.1021/ol802070d. [DOI] [PubMed] [Google Scholar]; h) Niwa T, Suehiro T, Yorimitsu H, Oshima K. Tetrahedron. 2009;65:5125–5131. [Google Scholar]
- 10.vander Veen LA, Keeven PH, Schoemaker GC, Reek JNH, Kamer PCJ, Leeuwen PWNMvan, Lutz M, Spek AL. Organometallics. 2000;19:872–883. [Google Scholar]
- 11.a) Bruno NC, Tudge MT, Buchwald SL. Chem Sci. 2013;4:916–920. doi: 10.1039/C2SC20903A. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bruno NC, Niljianskul N, Buchwald SL. J Org Chem. 2014;79:4161–4166. doi: 10.1021/jo500355k. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
12.Reaction with 1-bromo-4-fluorobenzene:
- 13.Das K, Shibuya R, Nakahara Y, Germain N, Ohshima T, Mashima K. Angew Chem Int Ed. 2012;51:150–154. doi: 10.1002/anie.201106737. Angew. Chem.2012, 124, 154–158. [DOI] [PubMed] [Google Scholar]
- 14.Zhang J, Bellomo A, Trongsiriwat N, Jia T, Carroll PJ, Dreher SD, Tudge MT, Yin H, Robinson JR, Schelter EJ, Walsh PJ. J Am Chem Soc. 2014;136:6276–6287. doi: 10.1021/ja411855d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) Kumpf R, Dougherty D. Science. 1993;261:1708–1710. doi: 10.1126/science.8378771. [DOI] [PubMed] [Google Scholar]; b) Meyer EA, Castellano RK, Diederich F. Angew Chem Int Ed. 2003;42:1210–1250. doi: 10.1002/anie.200390319. Angew. Chem.2003, 115, 1244–1287. [DOI] [PubMed] [Google Scholar]; c) Raju RK, Bloom JW, An Y, Wheeler SE. ChemPhysChem. 2011;12:3116–3130. doi: 10.1002/cphc.201100542. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.