

Abstract
Malaria remains a global public health threat, with half of the world’s population at risk. Despite numerous efforts in the past decade to develop new antimalarial drugs to surmount increasing resistance to common therapies, challenges remain in the expansion of the current antimalarial arsenal for the elimination of this disease. The requirement of prophylactic and radical cure activities for the next generation of antimalarial drugs demands that new research models be developed to support the investigation of the elusive liver stage of the malaria parasite. In this Review, we revisit current antimalarial therapies and discuss recent advances for in vitro and in vivo malaria research models of the liver stage and their importance in probing parasite biology and the discovery of novel drug candidates.
Introduction
Malaria remains a major health concern with half of the world population at risk of contracting the disease and 584,000 deaths tallied in 2013, mostly among children under the age of 5 in sub-Saharan Africa (1). Transmitted by infected female Anopheles mosquitoes, malaria is caused by protozoans of the Plasmodium genus. These Plasmodium parasites exhibit a complex life cycle composed of an asexual stage in the human host and a sexual stage in the vector mosquito (Figure 1).
Figure 1. The Plasmodium parasite life cycle in humans.

Mosquitoes carrying Plasmodium deposit parasites into the skin of the mammalian host. Parasites travel through the bloodstream to the liver, where they traverse several hepatocytes before establishing an infection. Within the final hepatocyte the parasite undergoes morphogenesis and then asexually divides to become over 10,000 blood-infective parasites, called merozoites. In the case of P. vivax, P. ovale, and P. cynomolgi, the parasites can remain dormant in the liver in forms called hypnozoites. The merozoites are released back into the bloodstream, where they begin the cyclical blood stage. Within erythrocytes, a small fraction of parasites differentiate into male or female gametocytes. These gametocytes are ingested by the mosquito during blood feeding, where they will reproduce sexually, eventually leading to the formation of sporozoites.
Among the Plasmodium species responsible for malaria, P. falciparum is responsible for the largest number of deaths, though the less deadly P. vivax causes the greatest morbidity worldwide due to its extensive distribution outside of sub-Saharan Africa (1). In all species of Plasmodium, the parasites enter a human host as sporozoites. This parasite form preferentially travels to the liver, where each sporozoite will invade a hepatocyte and undergo asexual replication, giving rise to a schizont that contains 10,000 to 30,000 erythrocyte-infective parasites termed merozoites (2). The asymptomatic liver stage takes between 2 and 16 days depending on the Plasmodium species (3), and precedes the cyclical blood-stage infection that results in the clinical manifestation of malaria. Within red blood cells, a small fraction of parasites differentiate into gametocytes, sexual parasite forms that enable transmission from the human host to the mosquito vector. In the case of P. vivax and P. ovale infections, relapse is often seen as a result of hypnozoites, dormant forms of the parasites in hepatic cells that can reactivate weeks or months after the primary infection (4). Targeting the development of liver-stage Plasmodium parasites represents a promising strategy for the development of malaria prophylaxis and presents a route to address disease eradication (5–7). Malaria eradication requires transmission-blocking strategies, and by intervening during the liver stage the development of both merozoites and gametocytes is prevented. The targeting of dormant hypnozoites is also important for disease eradication. Thus, targeting malaria’s liver stages offers a route to significantly decrease morbidity risks and address disease elimination. However, our limited understanding of the molecular processes during Plasmodium liver stages remains a major hindrance to developing novel antimalarials.
Though P. falciparum and P. vivax have the greatest impact on global health, investigations of the Plasmodium liver stage have been predominantly advanced by the use of rodent-infective parasites such as P. berghei and P. yoelii (3). First, we will review the most clinically relevant antimalarial therapies and resistance mechanisms associated with the use of these models. Then, we will focus on malaria research models that will help advance efforts to understand the biology of human-infective Plasmodium species, which are crucial to improve malaria control efforts.
Challenges and current state of malaria drug development
Current malaria therapies and drug resistance.
Spurred by many global initiatives to eradicate malaria (8), the last decade has seen major progress in the landscape of antimalarial drug discovery. Nonetheless, the treatment of malaria remains largely dominated by four major classes of drugs developed in the 20th century. The discovery of antimalarials with novel targets is especially important because the emergence of drug-resistant Plasmodium strains threatens the effectiveness of current treatments. Here, we highlight the major antimalarial drug classes, which are used as either treatment or curative options, and discuss their modes of action.
Quinoline derivatives.
Members of the quinoline family include chloroquine, mefloquine, and primaquine. Chloroquine (CQ), a 4-aminoquinoline, remains one of the most successful antimalarial drugs to date, considering cost, toxicity, and efficacy (9). However, the widespread drug resistance in Plasmodium strains has hindered its use for treatment. Quinoline derivatives are thought to act by inhibiting the digestion of heme in blood-infective parasites. Specifically, CQ inhibits the polymerization of heme to hemozoin in the parasite’s food vacuole, which contributes to its accumulation and toxic effects (10). While P. falciparum exhibits a widespread resistance to CQ, P. vivax remains sensitive to this treatment in most endemic regions. Mefloquine (MQ) is a quinine derivative agent that is active against the blood stages of P. falciparum and P. vivax infection (11). Its mechanism of action is still poorly understood; however, it is proposed to act through inhibition of hemoglobin endocytosis in Plasmodium parasites (12). In sensitive strains, quinoline derivatives such as CQ and MQ can be successfully used as clinical cures that clear blood-stage parasites. Resistance to quinoline drugs is primarily due to mutation in parasite transporter proteins. In P. falciparum, resistance to CQ is conferred through the K76T mutation in the chloroquine resistance transporter gene (Pfcrt) (13). Additionally, mutations identified in the P. falciparum multidrug resistance (Pfmdr), Na+/H+ exchanger (Pfnhe), and multidrug resistance–associated protein (Pfmrp1 and Pfmrp2) genes are known to contribute to quinoline resistance mechanisms (14–16).
Lastly, primaquine is distinct from CQ and MQ in that it is the only clinically approved drug for treatment of relapsing malaria caused primarily by P. vivax hypnozoites. In addition to its liver-stage activity, primaquine exhibits blood-stage and gametocidal antimalarial activities in vitro (17). Unfortunately primaquine has a high potential for hemolysis, which restricts its use. It is contraindicated in pregnant women and individuals with glucose-6-phosphate dehydrogenase deficiency, which is common among endemic populations (18). Despite its discovery almost six decades ago and prevalent use for the treatment of relapsing malaria, the mode of action of primaquine remains to be fully elucidated. Although it is a member of the same chemical family as CQ and MQ, the varying biological activity of primaquine against hypnozoites suggests it has an alternative mode of action. One proposed mode of action is that a primaquine metabolite impairs parasitic mitochondrial respiration (19). In support of the proposal that primaquine biotransformation is essential to elicit activity, a recent clinical study found that the metabolism of primaquine by the cytochrome P-450 2D6 may contribute to the production of an active metabolite responsible for hypnozoite killing (19, 20). It is currently thought that resistance to primaquine is almost nonexistent, although this assessment is confounded by the difficulties in defining and quantifying primaquine resistance (21). To surmount the emergence of drug-resistant P. vivax strains, the WHO recommends a regimen of artemisinin and primaquine combination-based therapies for radical cure in regions with CQ resistance (21, 22), but this regimen cannot be widely administered because of the restrictions mentioned above.
Sulfadoxine-pyrimethamine.
The folate inhibitors sulfadoxine and pyrimethamine, which target dihydropteroate synthetase (DHPS) and dihydrofolate reductase (DHFR) activities, respectively, act synergistically when administered in combination to impair parasite growth (23). Sulfadoxine-pyrimethamine combination therapy was introduced in the late 1960s to surmount the rapid emergence of drug resistance that occurred as a result of monotherapy with a single antifolate. This combination effectively inhibits malaria’s liver and blood stages. Unfortunately, resistance to sulfadoxine-pyrimethamine is now widespread, and this resistance results from the accumulation of mutations in the Pfdhps and Pfdhfr genes. In Africa, resistance to sulfadoxine-pyrimethamine treatment is associated with Pfdhfr mutations (N51I, C59R, and S108N), as well as the most commonly found Pfdhps mutations, A437G and K540E (24). In Southeast Asia and South America, addition of an I164L mutation in Pfdhfr along with the above mutations is responsible for a highly sulfadoxine-pyrimethamine–resistant Plasmodium strain (25).
Artemisinin.
Artemisinin combination therapies are recommended by the WHO as the first line of treatments against uncomplicated P. falciparum malaria (26). Artemisinin, a natural product, was widely recognized for its impact on reducing malaria rates worldwide with the 2015 Nobel Prize in Physiology or Medicine being awarded to Youyou Tu for its discovery. Artemisinin and its derivatives (artesunate, artemether, dihydroartemisinin) are fast-acting endoperoxide compounds, and potent antimalarials in the elimination of blood-stage Plasmodium parasites. However, despite its discovery in the late 1970s and popular use today, the mechanism by which artemisinin induces parasite death has been a pressing question. Recent studies have suggested that the iron-artemisinin adduct impairs the P. falciparum calcium ATPase 6 (27) and P. falciparum mitochondrial functions (28) to elicit parasite death. It has also been shown that the P. falciparum PI3K (PfPI3K), which phosphorylates phosphatidylinositol (PI), is a potential target of dihydroartemisinin, the active form of artemisinin during Plasmodium early ring stages (29). Importantly, artemisinin resistance was associated with increased levels of phosphatidylinositol 3-phosphate (PI3P) — the phospholipid product of PI phosphorylation — in infected erythrocytes, providing compelling evidence that the mode of action involves PfPI3K. Work is still under way to better understand PfPI3K and the link between PI3P levels and artemisinin drug resistance.
Atovaquone.
Atovaquone is a potent antimalarial that is active against Plasmodium blood and liver stages, but not gametocytes or hypnozoites (30). The drug acts through the inhibition of the Plasmodium cytochrome bc1 complex (cyt bc1), and, to date, it is the only clinically used drug targeting this complex (31). Cyt bc1, a membrane protein of the Plasmodium mitochondria, plays a critical role in the electron transport chain, and thus parasite survival. The Plasmodium mitochondrial electron transport chain generates a membrane potential across the inner mitochondrial membrane for transport, and regenerates ubiquinone, an essential molecule for the dihydroorotate dehydrogenase (DHODH), which is critical for the pyrimidine biosynthesis pathway (32). Atovaquone acts through competitive inhibition of the quinol oxidation site Q0, one of the two quinone binding sites of cyt bc1 (33, 34). A consequence of mitochondrial inhibition by atovaquone is the impairment of the purine salvage pathway (35). Atovaquone is currently used in combination with proguanil for the treatment of uncomplicated malaria and as a prophylactic agent (36, 37). Clinical resistance to atovaquone is known to arise from mutations of cyt bc1 (38–40). One of the most prominent mutations is at position 268 (Y268S), which reduces atovaquone binding and catalytic turnover (34, 41). The recent cocrystallization of atovaquone bound to the yeast cyt bc1 provides novel insights into the binding modes of this drug (33). Future studies aimed at inhibiting cyt bc1 Y268S, and other drug-resistant mutations, could be useful to overcome the emergence of atovaquone drug resistance.
Progress toward a multistage drug target.
The renewed resources and efforts in the last decade for malaria eradication have contributed to a significant expansion of the antimalarial drug portfolio (42). To promote the eradication of malaria, the Malaria Eradication Research Agenda initiative suggested that drug development efforts should concentrate on Single Encounter Radical Cure and Prophylaxis (SERCaP), which encompasses drugs with multistage activities (including liver and transmission-blocking activity), relapse prevention, and long-lasting effects to provide prophylactic treatment (43, 44). Recent projects aiming to identify novel chemical scaffolds for malaria drug discovery have relied on high-throughput screening strategies (42), which focus on phenotypic- and target-based screens. One strategy used an in silico virtual screening approach to identify novel type II fatty acid biosynthesis inhibitors with multistage antimalarial activity (45). Other strategies have focused on phenotypic-based screens for the discovery of new lead molecules with liver- and blood-stage activities (7, 46–49). The use of these phenotypic-based screens has contributed to the identification of various preclinical small-molecule compounds such as imidazopyrazines (e.g., KAI407), imidazolopiperazine (e.g., KAF156), Plasmodium phosphatidylinositol-4-OH kinase inhibitors, spiroindolones (e.g., KAE609), P. falciparum P-type ATPase 4 inhibitors, and DDD107498, a translation elongation factor 2 inhibitor (50–54). Target-based screens have also been important tools for generating new antimalarial leads. As opposed to phenotypic-based screens, the use of target-based drug approaches for developing novel antimalarials possesses the clear advantage of concentrating optimization efforts and safety assessments of preclinical drugs on a clearly defined target. However, this approach requires the knowledge of a validated or well-characterized molecular target. For example, the recent efforts to develop the first Plasmodium DHODH inhibitor led to the rational design and clinical development of a novel compound with liver- and blood-stage activity, DSM265 (55, 56).
While these efforts are encouraging in the fight against malaria, discovering and designing new and improved drugs is essential to surmount the challenges associated with the emergence of drug resistance. In fact, Burrows et al. have shown that for every 18 preclinical candidates discovered with blood-stage activity, only one will result in a new antimalarial drug (57). This number of preclinical candidates might be even higher considering the development of transmission-blocking and antirelapse agents. The recent development of novel high-throughput assays for discovering compounds with transmission-blocking activity should help address this pressing need (58–60). To date, tafenoquine, an 8-aminoquinoline derivative, is only the second developed drug, after primaquine, with proven clinical activity against hypnozoites (61, 62). A list of small molecules in ongoing clinical studies is available on the Medicines for Malaria Venture website (http://www.mmv.org) (Table 1). The dearth of antimalarial drugs with antihypnozoite activity remains a major hurdle to overcome in the fight for malaria eradication. Nonetheless, targeting the Plasmodium liver stage offers several opportunities when it comes to developing new prophylactic and curative drugs, as well as vaccines against malaria, because of the lower number of parasites present in the liver as compared with the blood stage (6, 63). However, this stage remains largely unexplored because of the scarcity of research models to investigate the underlying mechanisms of infection of human hepatocytes by Plasmodium species. One approach to tackle this problem is to develop new research platforms that will accelerate drug discovery efforts, while also providing tools to further probe the biology of the Plasmodium parasite.
Table 1. Current antimalarial clinical candidates.

Current in vitro and in vivo models to study the Plasmodium liver stage
Liver-stage malaria models have been continuously evolving since the discovery of this life stage. Early work relied on the use of monkeys with the primate-infective P. cynomolgi (64), but for ethical reasons monkey models have been more cautiously utilized. Fortunately, other in vivo and in vitro models emerged in the later half of the 20th century using the rodent-infective P. berghei or P. yoelii to infect hepatoma cell lines, primary hepatocytes, or mice (65–69). While P. berghei remains the most commonly used species for liver-stage studies, research developments in recent years have focused on generating much-needed models capable of supporting the establishment of hypnozoites for investigating relapses of malaria, one of the least understood aspects of Plasmodium infections.
Plasmodium hypnozoites arrest in hepatocytes for weeks or months before reactivating to infect red blood cells and cause disease. These dormant forms of the parasites are responsible for relapses seen in patients infected with P. vivax and P. ovale and are of great public health importance considering that P. vivax is responsible for the greatest number of malaria infections in the world (70). This dormant form of the parasite was first discovered in rhesus macaques infected with P. cynomolgi in the early 1980s (71–73), and this system was the only model known to support hypnozoite development until recent years. Owing to ethical concerns of monkey studies and the large costs associated with them, few researchers had access to this model and the study of hypnozoites had been nearly impossible. Fortunately, the last 5 years have brought impressive developments in both in vivo and in vitro models that are capable of establishing small liver-stage forms.
In vitro models.
Various in vitro models that rely on the infection of hepatoma cell lines or primary hepatocytes by rodent-infective species of Plasmodium have been successfully used to study the Plasmodium liver stage (Table 2) (68, 69, 74). The relatively low cost of hepatoma cell lines and the lower safety requirements of working with rodent-infective parasites, as they do not infect humans, make these in vitro models appealing for large-scale screens (7, 46–49, 75, 76). Other models also exist to support the development of human-infective Plasmodium species, such as P. vivax or P. falciparum, in HepG2 and HC04 hepatoma cell lines, respectively (77, 78). However, such studies are more challenging because of the lower infection rates of these parasites (79, 80) and the restricted accessibility of these Plasmodium species, requiring access to facilities able to breed mosquitoes infected with these human-infective Plasmodium parasites or cryopreserved Plasmodium species sporozoites (81–83). Indeed, few research institutes have insectaries that are able to breed human-infective parasites in mosquitoes. The importance of these in vitro models has been widely discussed in the scientific community (84, 85), but these infections do not entirely recapitulate the liver-stage infections in humans. Most notably, none of these models are able to sustain the development of hypnozoites. Here, we will discuss recent advances toward the goal of generating in vitro hypnozoite models.
Table 2. The most commonly used models to study liver-stage malaria.
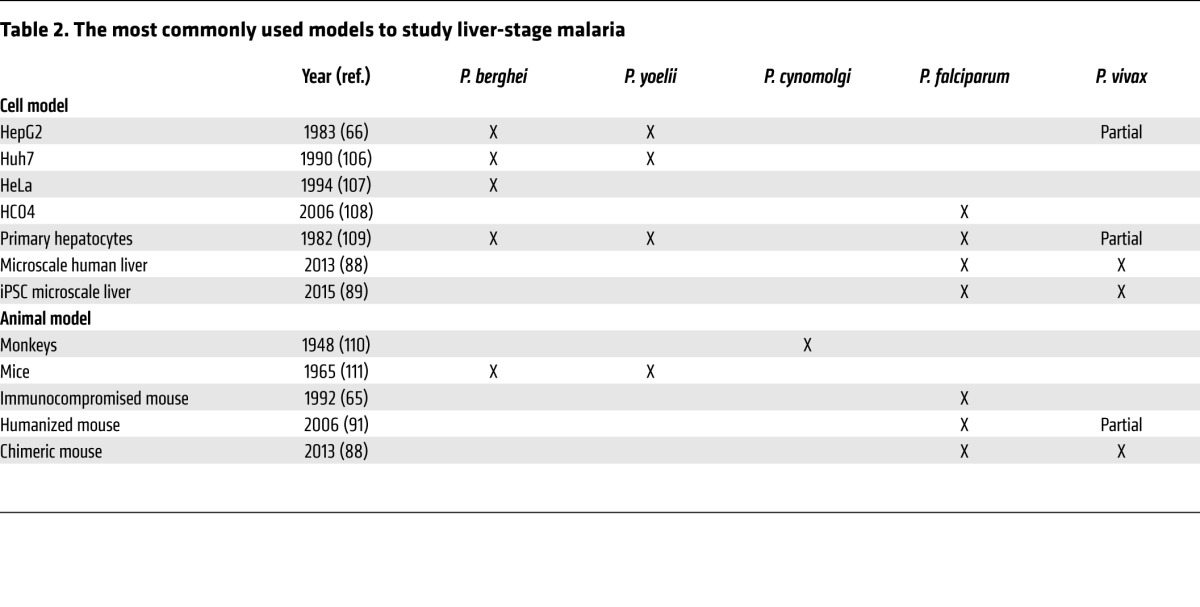
The first challenge in recapitulating hypnozoite development in vitro is the necessity to maintain cultured cell lines for several weeks, as hypnozoites remain dormant for extended periods of time. By coculturing hepatocytes with nonparenchymal cells, researchers found that hepatocyte cultures can be maintained for several weeks while maintaining liver-specific functions (86). With this technology, a “microscale of human liver cells” has been developed that takes into account higher-order processes absent in cell lines, such as interactions with neighboring cell types, extracellular matrices, and physical forces (87). In this microscale liver model, hepatocytes are seeded in a micropatterned arrangement and surrounded by stromal cells, which recapitulate some of the liver structures and help maintain their phenotypic functions for several weeks.
More recently, this microscale human liver cell culture was used to demonstrate the development of P. falciparum and P. vivax liver stage. Using cryopreserved primary human hepatocytes and cryopreserved P. falciparum, March et al. (88) achieved higher infection efficiency than with the HC04 cell line infected with freshly harvested P. falciparum, currently the only hepatoma cell line supporting the full development of this species in vitro. Microscale human liver cells have the added advantage of being able to model infections originating from the diverse genetic makeup of human populations, as they are not limited to primary hepatocytes sourced from a small pool of donors (i.e., patients with liver cancer). Most importantly, unlike primary hepatocytes, the micropatterned hepatocytes can be maintained for several weeks, allowing for the study of hypnozoite biology. The Bhatia group further developed this microscale culture technology by demonstrating that this tissue engineering of the human liver can also be derived from induced pluripotent stem cells (iPSCs) (89). A similar coculturing strategy of cynomolgus monkey primary hepatocytes with another hepatoma cell line was used to support the development of hypnozoites from P. cynomolgi (90). The advantage of this in vitro model compared with the iPSC model is the use of P. cynomolgi parasites, which are more readily accessible for experimentation than P. vivax. Undoubtedly, these newly established models will be critical for investigating hypnozoite formation, and the reactivation of these liver forms will be a critical validation for these models. While not currently suitable for high-throughput screening, the adaptation of these systems for economical and robust systematic searches for compounds with antihypnozoite activity will be transformative for malaria drug discovery efforts.
In vivo models.
In recent years, in vivo models that are able to support the development of human-infective Plasmodium species have evolved (Table 2). While these in vitro models are advancing to more accurately reconstitute the human liver, they lack key host-parasite characteristics such as the interaction of Plasmodium parasites with Kupffer cells and the host immune system. One of the greatest advances in liver-stage malaria research has been the development of a humanized mouse that can be infected by P. falciparum and P. vivax to more accurately recapitulate human infections. The development of P. falciparum in an immunocompromised mouse was demonstrated first in 1992 (65), and later in humanized mice in 2006 (91). Several years later, liver-chimeric mice that were immunocompromised and fumarylacetoacetate hydrolase–deficient were engrafted with human hepatocytes (FRGhuHep) to generate a humanized mouse with actual human liver tissue (92). This mouse model is T, B, and NK cell–deficient and is repopulated with greater than 90% human hepatocytes. As a proof of concept, it was demonstrated that primaquine treatment is able to clear hypnozoites in this model, making it the closest model to human infections to date.
The FRGhuHep mouse model can facilitate various aspects of malaria research. For example, these mice were used to create Plasmodium genetic crosses that, until now, have been extremely difficult to perform (93). These genetic crosses are powerful tools for understanding drug resistance and host specificity (94, 95), but few of them have been successfully performed to date (96). FRGhuHep mice were also used to show that host-based prophylaxis is a promising strategy for the treatment of malaria (97). Because human- and rodent-infective Plasmodium parasites have overlapping host hepatocyte requirements (98, 99), the FRGhuHep mouse model infected with P. falciparum or P. vivax represents the closest model to actual human infections. This will be an immensely useful resource in testing various treatments to predict human efficacy and toxicology before moving to higher organisms such as monkeys, which will save invaluable time and resources in the pursuit of new antimalarial drugs (100).
Limitations of current models.
Newly developed liver-stage models are greatly advancing malaria therapeutic development, yet some limitations remain. For instance, the microscale human liver platform is not yet readily available to the research community, and the high cost of cryopreserved sporozoites is prohibitive. Furthermore, the development of hypnozoites in this model is yet to be fully characterized. Improving access to such in vitro models, possibly through commercialization or shared resources, would greatly accelerate drug discovery efforts for molecules that are active against the dormant liver stage of malaria. Moreover, the humanized chimeric mice are still being developed and are currently prohibitively expensive, preventing their use in the wider malaria research community. With broader efforts in these systems it is likely that these humanized-liver mouse models will soon support cerebral and placental malaria as well. Despite these restrictions in use, both in vitro and in vivo models will be essential for evaluating promising therapeutics and understanding the biology of Plasmodium parasites.
Conclusions and future directions
The fight against malaria is markedly affected by the rise of Plasmodium strains resistant to mainstay therapies. While efforts to develop new drugs have greatly improved in the past decade (101), the discovery of new molecules remains critical to ensure effective treatment of the disease and its eradication. However, expanding our clinical arsenal requires the development of new research tools to facilitate the rapid progression of preclinical drugs to clinical settings. In addition, these new molecules could follow SERCaP guidelines for transmission-blocking activity and prevention of malaria relapse. To help in addressing the latter issue, the development of novel research models permitting the investigation of the Plasmodium liver stage is already making significant contributions to drug discovery efforts. One of the most important mysteries that has yet to be solved in the biology of hypnozoites is the identification of trigger signals that contribute either to dormancy or to reactivation in liver cells. The in vitro and in vivo models that have been developed will be crucial in deciphering these cellular events. Another exciting aspect of these novel in vitro and in vivo models is the identification and validation of biomarkers for the detection of Plasmodium infection.
The development of functional assays to investigate essential molecular mechanisms during malaria’s liver stage is critical for the validation of new targets to drive antimalarial drug discovery. One underexplored avenue is the manipulation of essential host targets to kill or block Plasmodium parasites. Functional genomic tools such as RNA interference and the CRISPR-Cas9 genome editing system will greatly benefit efforts to identify possible host targets and map host-parasite interactions, both of which are compatible with genome-wide screening. Additionally, the CRISPR-Cas9 system can target the Plasmodium parasite (102–104), facilitating studies to validate parasite targets. Renowned for its simplicity and versatility, CRISPR-Cas9 offers unique opportunities (e.g., introduction of point mutations, gene knockout) to investigate key host-parasite interactions during liver-stage malaria in vitro, and most importantly in vivo. This technology may also be applied to hypnozoite models in the future. The transcriptomic and/or proteomic interaction networks refined through such studies would be advantageous to research efforts aimed at understanding the biological processes involved in liver stage development. From a scientific perspective, our understanding of relapsing malaria is still nascent, and these innovative research models may help fill the gap to develop better diagnosis strategies and curative treatments for elimination of this pernicious disease.
Acknowledgments
This work was supported by the NIH (GM099796) and the Ralph E. Powe Junior Faculty Enhancement Award (to E.R. Derbyshire), and NIH F32AI118294 for fellowship support (to R. Raphemot). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2016;126(6):2013–2020. doi:10.1172/JCI82981.
Contributor Information
Rene Raphemot, Email: rene.raphemot@duke.edu.
Dora Posfai, Email: dora.posfai@duke.edu.
References
- 1. WHO. World Malaria Report 2014. Geneva, Switzerland: World Health Organization; 2014. [Google Scholar]
- 2.Bejon P, et al. Calculation of liver-to-blood inocula, parasite growth rates, and preerythrocytic vaccine efficacy, from serial quantitative polymerase chain reaction studies of volunteers challenged with malaria sporozoites. J Infect Dis. 2005;191(4):619–626. doi: 10.1086/427243. [DOI] [PubMed] [Google Scholar]
- 3.Prudencio M, Rodriguez A, Mota MM. The silent path to thousands of merozoites: the Plasmodium liver stage. Nat Rev Microbiol. 2006;4(11):849–856. doi: 10.1038/nrmicro1529. [DOI] [PubMed] [Google Scholar]
- 4.Mueller I, et al. Key gaps in the knowledge of Plasmodium vivax, a neglected human malaria parasite. Lancet Infect Dis. 2009;9(9):555–566. doi: 10.1016/S1473-3099(09)70177-X. [DOI] [PubMed] [Google Scholar]
- 5.Mazier D, Renia L, Snounou G. A pre-emptive strike against malaria’s stealthy hepatic forms. Nat Rev Drug Discov. 2009;8(11):854–864. doi: 10.1038/nrd2960. [DOI] [PubMed] [Google Scholar]
- 6.Rodrigues T, Prudencio M, Moreira R, Mota MM, Lopes F. Targeting the liver stage of malaria parasites: a yet unmet goal. J Med Chem. 2012;55(3):995–1012. doi: 10.1021/jm201095h. [DOI] [PubMed] [Google Scholar]
- 7.Derbyshire ER, Prudencio M, Mota MM, Clardy J. Liver-stage malaria parasites vulnerable to diverse chemical scaffolds. Proc Natl Acad Sci U S A. 2012;109(22):8511–8516. doi: 10.1073/pnas.1118370109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roberts L, Enserink M. Malaria. Science. 2007;318(5856):1544–1545. doi: 10.1126/science.318.5856.1544. [DOI] [PubMed] [Google Scholar]
- 9.Sudre P, Breman JG, McFarland D, Koplan JP. Treatment of chloroquine-resistant malaria in African children: a cost-effectiveness analysis. Int J Epidemiol. 1992;21(1):146–154. doi: 10.1093/ije/21.1.146. [DOI] [PubMed] [Google Scholar]
- 10.Sullivan DJ. On the molecular mechanism of chloroquine’s antimalarial action. Proc Natl Acad Sci U S A. 1996;93(21):11865–11870. doi: 10.1073/pnas.93.21.11865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maguire JD, Marwoto H, Richie TL, Fryauff DJ, Baird JK. Mefloquine is highly efficacious against chloroquine-resistant Plasmodium vivax malaria and Plasmodium falciparum malaria in Papua, Indonesia. Clin Infect Dis. 2006;42(8):1067–1072. doi: 10.1086/501357. [DOI] [PubMed] [Google Scholar]
- 12.Hoppe HC, van Schalkwyk DA, Wiehart UI, Meredith SA, Egan J, Weber BW. Antimalarial quinolines and artemisinin inhibit endocytosis in Plasmodium falciparum. Antimicrob Agents Chemother. 2004;48(7):2370–2378. doi: 10.1128/AAC.48.7.2370-2378.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wellems TE, Plowe CV. Chloroquine-resistant malaria. J Infect Dis. 2001;184(6):770–776. doi: 10.1086/322858. [DOI] [PubMed] [Google Scholar]
- 14.Vezmar M, Georges E. Direct binding of chloroquine to the multidrug resistance protein (MRP): possible role for MRP in chloroquine drug transport and resistance in tumor cells. Biochem Pharmacol. 1998;56(MRP):733–742. doi: 10.1016/s0006-2952(98)00217-2. [DOI] [PubMed] [Google Scholar]
- 15.Veiga MI, et al. Complex polymorphisms in the Plasmodium falciparum multidrug resistance protein 2 gene and its contribution to antimalarial response. Antimicrob Agents Chemother. 2014;58(12):7390–7397. doi: 10.1128/AAC.03337-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ursing J, Zakeri S, Gil JP, Bjorkman A. Quinoline resistance associated polymorphisms in the pfcrt, pfmdr1 and pfmrp genes of Plasmodium falciparum in Iran. Acta Trop. 2006;97(3):352–356. doi: 10.1016/j.actatropica.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 17.Cabrera M, Cui L. In vitro activities of primaquine-schizonticide combinations on asexual blood stages and gametocytes of Plasmodium falciparum. Antimicrob Agents Chemother. 2015;59(12):7650–7656. doi: 10.1128/AAC.01948-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hill DR, Baird JK, Parise ME, Lewis LS, Ryan ET, Magill AJ. Primaquine: report from CDC expert meeting on malaria chemoprophylaxis I. Am J Trop Med Hyg. 2006;75(3):402–415. [PubMed] [Google Scholar]
- 19.Vale N, Moreira R, Gomes P. Primaquine revisited six decades after its discovery. Eur J Med Chem. 2009;44(3):937–953. doi: 10.1016/j.ejmech.2008.08.011. [DOI] [PubMed] [Google Scholar]
- 20.Bennett JW, et al. Primaquine failure and cytochrome P-450 2D6 in Plasmodium vivax malaria. N Engl J Med. 2013;369(14):1381–1382. doi: 10.1056/NEJMc1301936. [DOI] [PubMed] [Google Scholar]
- 21.Fernando D, Rodrigo C, Rajapakse S. Primaquine in vivax malaria: an update and review on management issues. Malar J. 2011;10: doi: 10.1186/1475-2875-10-351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. WHO. Guidelines for the Treatment of Malaria. Geneva, Switzerland: World Health Organization; 2015. [Google Scholar]
- 23.Chulay JD, Watkins WM, Sixsmith DG. Synergistic antimalarial activity of pyrimethamine and sulfadoxine against Plasmodium falciparum in vitro. Am J Trop Med Hyg. 1984;33(3):325–330. doi: 10.4269/ajtmh.1984.33.325. [DOI] [PubMed] [Google Scholar]
- 24.Sridaran S, McClintock SK, Syphard LM, Herman KM, Barnwell JW, Udhayakumar V. Anti-folate drug resistance in Africa: meta-analysis of reported dihydrofolate reductase (dhfr) and dihydropteroate synthase (dhps) mutant genotype frequencies in African Plasmodium falciparum parasite populations. Malar J. 2010;9: doi: 10.1186/1475-2875-9-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Menard D, et al. Dihydrofolate reductase I164L mutation in Plasmodium falciparum, Madagascar. Emerg Infect Dis. 2008;14(7):1166–1167. doi: 10.3201/eid1407.071498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. WHO. Guidelines for the Treatment of Malaria. Geneva, Switzerland: World Health Organization; 2010. [Google Scholar]
- 27.Shandilya A, Chacko S, Jayaram B, Ghosh I. A plausible mechanism for the antimalarial activity of artemisinin: a computational approach. Sci Rep. 2013;3: doi: 10.1038/srep02513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang J, et al. Artemisinin directly targets malarial mitochondria through its specific mitochondrial activation. PLoS One. 2010;5(3): doi: 10.1371/journal.pone.0009582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mbengue A, et al. A molecular mechanism of artemisinin resistance in Plasmodium falciparum malaria. Nature. 2015;520(7549):683–687. doi: 10.1038/nature14412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baggish AL, Hill DR. Antiparasitic agent atovaquone. Antimicrob Agents Chemother. 2002;46(5):1163–1173. doi: 10.1128/AAC.46.5.1163-1173.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nixon GL, et al. Antimalarial pharmacology and therapeutics of atovaquone. J Antimicrob Chemother. 2013;68(5):977–985. doi: 10.1093/jac/dks504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Painter HJ, Morrisey JM, Mather MW, Vaidya AB. Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum. Nature. 2007;446(7131):88–91. doi: 10.1038/nature05572. [DOI] [PubMed] [Google Scholar]
- 33.Birth D, Kao WC, Hunte C. Structural analysis of atovaquone-inhibited cytochrome bc1 complex reveals the molecular basis of antimalarial drug action. Nat Commun. 2014;5: doi: 10.1038/ncomms5029. [DOI] [PubMed] [Google Scholar]
- 34.Fisher N, et al. Cytochrome b mutation Y268S conferring atovaquone resistance phenotype in malaria parasite results in reduced parasite bc1 catalytic turnover and protein expression. J Biol Chem. 2012;287(13):9731–9741. doi: 10.1074/jbc.M111.324319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bulusu V, Jayaraman V, Balaram H. Metabolic fate of fumarate, a side product of the purine salvage pathway in the intraerythrocytic stages of Plasmodium falciparum. J Biol Chem. 2011;286(11):9236–9245. doi: 10.1074/jbc.M110.173328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Osei-Akoto A, Orton L, Owusu-Ofori SP. Atovaquone-proguanil for treating uncomplicated malaria. Cochrane Database Syst Rev. 2005;(4): doi: 10.1002/14651858.CD004529.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakato H, Vivancos R, Hunter PR. A systematic review and meta-analysis of the effectiveness and safety of atovaquone proguanil (Malarone) for chemoprophylaxis against malaria. J Antimicrob Chemother. 2007;60(5):929–936. doi: 10.1093/jac/dkm337. [DOI] [PubMed] [Google Scholar]
- 38.Musset L, Bouchaud O, Matheron S, Massias L, Le Bras J. Clinical atovaquone-proguanil resistance of Plasmodium falciparum associated with cytochrome b codon 268 mutations. Microbes Infect. 2006;8(11):2599–2604. doi: 10.1016/j.micinf.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 39.Musset L, Le Bras J, Clain J. Parallel evolution of adaptive mutations in Plasmodium falciparum mitochondrial DNA during atovaquone-proguanil treatment. Mol Biol Evol. 2007;24(8):1582–1585. doi: 10.1093/molbev/msm087. [DOI] [PubMed] [Google Scholar]
- 40.Cottrell G, Musset L, Hubert V, Le Bras J, Clain J, Atovaquone-Proguanil Treatment Failure Study Group Emergence of resistance to atovaquone-proguanil in malaria parasites: insights from computational modeling and clinical case reports. Antimicrob Agents Chemother. 2014;58(8):4504–4514. doi: 10.1128/AAC.02550-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Korsinczky M, Chen N, Kotecka B, Saul A, Rieckmann K, Cheng Q. Mutations in Plasmodium falciparum cytochrome b that are associated with atovaquone resistance are located at a putative drug-binding site. Antimicrob Agents Chemother. 2000;44(8):2100–2108. doi: 10.1128/AAC.44.8.2100-2108.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Flannery EL, Chatterjee AK, Winzeler EA. Antimalarial drug discovery — approaches and progress towards new medicines. Nat Rev Microbiol. 2013;11(12):849–862. doi: 10.1038/nrmicro3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alonso PL, et al. A research agenda to underpin malaria eradication. PLoS Med. 2011;8(1): doi: 10.1371/journal.pmed.1000406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burrows JN, et al. Antimalarial drug discovery - the path towards eradication. Parasitology. 2014;141(1):128–139. doi: 10.1017/S0031182013000826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schrader FC, et al. Novel type II fatty acid biosynthesis (FAS II) inhibitors as multistage antimalarial agents. ChemMedChem. 2013;8(3):442–461. doi: 10.1002/cmdc.201200407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Derbyshire ER, et al. Dihydroquinazolinone inhibitors of proliferation of blood and liver stage malaria parasites. Antimicrob Agents Chemother. 2014;58(3):1516–1522. doi: 10.1128/AAC.02148-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Derbyshire ER, et al. Chemical interrogation of the malaria kinome. Chembiochem. 2014;15(13):1920–1930. doi: 10.1002/cbic.201400025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.da Cruz FP, et al. Drug screen targeted at Plasmodium liver stages identifies a potent multistage antimalarial drug. J Infect Dis. 2012;205(8):1278–1286. doi: 10.1093/infdis/jis184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meister S, et al. Imaging of Plasmodium liver stages to drive next-generation antimalarial drug discovery. Science. 2011;334(6061):1372–1377. doi: 10.1126/science.1211936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zeeman AM, et al. KAI407, a potent non-8-aminoquinoline compound that kills Plasmodium cynomolgi early dormant liver stage parasites in vitro. Antimicrob Agents Chemother. 2014;58(3):1586–1595. doi: 10.1128/AAC.01927-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McNamara CW, et al. Targeting Plasmodium PI(4)K to eliminate malaria. Nature. 2013;504(7479):248–253. doi: 10.1038/nature12782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baragana B, et al. A novel multiple-stage antimalarial agent that inhibits protein synthesis. Nature. 2015;522(7556):315–320. doi: 10.1038/nature14451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rottmann M, et al. Spiroindolones, a potent compound class for the treatment of malaria. Science. 2010;329(5996):1175–1180. doi: 10.1126/science.1193225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuhen KL, et al. KAF156 is an antimalarial clinical candidate with potential for use in prophylaxis, treatment, and prevention of disease transmission. Antimicrob Agents Chemother. 2014;58(9):5060–5067. doi: 10.1128/AAC.02727-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Phillips MA, et al. A long-duration dihydroorotate dehydrogenase inhibitor (DSM265) for prevention and treatment of malaria. Sci Transl Med. 2015;7(296): doi: 10.1126/scitranslmed.aaa6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Phillips MA, Rathod PK. Plasmodium dihydroorotate dehydrogenase: a promising target for novel anti-malarial chemotherapy. Infect Disord Drug Targets. 2010;10(3):226–239. doi: 10.2174/187152610791163336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Burrows JN, van Huijsduijnen RH, Mohrle JJ, Oeuvray C, Wells TN. Designing the next generation of medicines for malaria control and eradication. Malar J. 2013;12: doi: 10.1186/1475-2875-12-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Plouffe DM, et al. High-throughput assay and discovery of small molecules that interrupt malaria transmission. Cell Host Microbe. 2016;19(1):114–126. doi: 10.1016/j.chom.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lucantoni L, Fidock DA, Avery VM. Luciferase-based, high-throughput assay for screening and profiling transmission-blocking compounds against Plasmodium falciparum gametocytes. Antimicrob Agents Chemother. 2016;60(4):2097–2107. doi: 10.1128/AAC.01949-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.D’Alessandro S, et al. A chemical susceptibility profile of the Plasmodium falciparum transmission stages by complementary cell-based gametocyte assays. J Antimicrob Chemother. doi: 10.1093/jac/dkv493. [published online ahead of print February 16, 2016]. doi: 10.1093/jac/dkv493. [DOI] [PubMed] [Google Scholar]
- 61.Rajapakse S, Rodrigo C, Fernando SD. Tafenoquine for preventing relapse in people with Plasmodium vivax malaria. Cochrane Database Syst Rev. 2015;4: doi: 10.1002/14651858.CD010458.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Beck HP, et al. Estimation of the antirelapse efficacy of Tafenoquine, using Plasmodium vivax genotyping. J Infect Dis. 2016;213(5):794–799. doi: 10.1093/infdis/jiv508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Derbyshire ER, Mota MM, Clardy J. The next opportunity in anti-malaria drug discovery: the liver stage. PLoS Pathog. 2011;7(9): doi: 10.1371/journal.ppat.1002178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tsay CJ. Julius Wagner-Jauregg and the legacy of malarial therapy for the treatment of general paresis of the insane. Yale J Biol Med. 2013;86(2):245–254. [PMC free article] [PubMed] [Google Scholar]
- 65.Sacci JB. Mouse model for exoerythrocytic stages of Plasmodium falciparum malaria parasite. Proc Natl Acad Sci U S A. 1992;89(9):3701–3705. doi: 10.1073/pnas.89.9.3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hollingdale MR, Leland P, Schwartz AL. In vitro cultivation of the exoerythrocytic stage of Plasmodium berghei in a hepatoma cell line. Am J Trop Med Hyg. 1983;32(4):682–684. doi: 10.4269/ajtmh.1983.32.682. [DOI] [PubMed] [Google Scholar]
- 67.Hollingdale MR, Leland P, Sigler C. In vitro cultivation of the exoerythrocytic stage of Plasmodium berghei in irradiated hepatoma cells. Am J Trop Med Hyg. 1985;34(1):21–23. doi: 10.4269/ajtmh.1985.34.21. [DOI] [PubMed] [Google Scholar]
- 68.Mota MM, Rodriguez A. Plasmodium yoelii: efficient in vitro invasion and complete development of sporozoites in mouse hepatic cell lines. Exp Parasitol. 2000;96(4):257–259. doi: 10.1006/expr.2000.4570. [DOI] [PubMed] [Google Scholar]
- 69.Hollingdale MR, Leland P, Schwartz AL. In vitro cultivation of the exoerythrocytic stage of Plasmodium berghei in a hepatoma cell line. Am J Trop Med Hyg. 1983;32(4):682–684. doi: 10.4269/ajtmh.1983.32.682. [DOI] [PubMed] [Google Scholar]
- 70.Guerra CA, et al. The international limits and population at risk of Plasmodium vivax transmission in 2009. PLoS Negl Trop Dis. 2010;4(8): doi: 10.1371/journal.pntd.0000774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Krotoski WA, et al. Relapses in primate malaria: discovery of two populations of exoerythrocytic stages. Br Med J. 1980;280(6208):153–154. doi: 10.1136/bmj.280.6208.153-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Krotoski WA, et al. Observations on early and late post-sporozoite tissue stages in primate malaria. Am J Trop Med Hyg. 1982;31(2):211–225. [PubMed] [Google Scholar]
- 73.Krotoski WA, et al. Observations on early and late post-sporozoite tissue stages in primate malaria. Am J Trop Med Hyg. 1982;31(1):24–35. [PubMed] [Google Scholar]
- 74.Prudencio M, Rodrigues CD, Ataide R, Mota MM. Dissecting in vitro host cell infection by Plasmodium sporozoites using flow cytometry. Cell Microbiol. 2008;10(1):218–224. doi: 10.1111/j.1462-5822.2007.01032.x. [DOI] [PubMed] [Google Scholar]
- 75.Prudencio M, et al. Kinome-wide RNAi screen implicates at least 5 host hepatocyte kinases in Plasmodium sporozoite infection. PLoS Pathog. 2008;4(11): doi: 10.1371/journal.ppat.1000201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rodrigues CD, et al. Host scavenger receptor SR-BI plays a dual role in the establishment of malaria parasite liver infection. Cell Host Microbe. 2008;4(3):271–282. doi: 10.1016/j.chom.2008.07.012. [DOI] [PubMed] [Google Scholar]
- 77.Hollingdale MR, Collins WE, Campbell CC, Schwartz AL. In vitro culture of two populations (dividing and nondividing) of exoerythrocytic parasites of Plasmodium vivax. Am J Trop Med Hyg. 1985;34(2):216–222. doi: 10.4269/ajtmh.1985.34.216. [DOI] [PubMed] [Google Scholar]
- 78.Sattabongkot J, et al. Establishment of a human hepatocyte line that supports in vitro development of the exo-erythrocytic stages of the malaria parasites Plasmodium falciparum and P. vivax. Am J Trop Med Hyg. 2006;74(5):708–715. [PubMed] [Google Scholar]
- 79.Karnasuta C, et al. Complete development of the liver stage of Plasmodium falciparum in a human hepatoma cell line. Am J Trop Med Hyg. 1995;53(6):607–611. doi: 10.4269/ajtmh.1995.53.607. [DOI] [PubMed] [Google Scholar]
- 80.Smith JE, Meis JF, Ponnudurai T, Verhave JP, Moshage HJ. In-vitro culture of exoerythrocytic form of Plasmodium falciparum in adult human hepatocytes. Lancet. 1984;2(8405):757–758. doi: 10.1016/s0140-6736(84)92670-9. [DOI] [PubMed] [Google Scholar]
- 81.Sheehy SH, et al. Optimising controlled human malaria infection studies using cryopreserved parasites administered by needle and syringe. PLoS One. 2013;8(6): doi: 10.1371/journal.pone.0065960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shekalaghe S, et al. Controlled human malaria infection of Tanzanians by intradermal injection of aseptic, purified, cryopreserved Plasmodium falciparum sporozoites. Am J Trop Med Hyg. 2014;91(3):471–480. doi: 10.4269/ajtmh.14-0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gomez-Perez GP, et al. Controlled human malaria infection by intramuscular and direct venous inoculation of cryopreserved Plasmodium falciparum sporozoites in malaria-naive volunteers: effect of injection volume and dose on infectivity rates. Malar J. 2015;14: doi: 10.1186/s12936-015-0817-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Janse CJ, Waters AP. Plasmodium berghei: the application of cultivation and purification techniques to molecular studies of malaria parasites. Parasitol Today. 1995;11(4):138–143. doi: 10.1016/0169-4758(95)80133-2. [DOI] [PubMed] [Google Scholar]
- 85.Langhorne J, et al. The relevance of non-human primate and rodent malaria models for humans. Malar J. 2011;10: doi: 10.1186/1475-2875-10-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bhatia SN, Balis UJ, Yarmush ML, Toner M. Effect of cell-cell interactions in preservation of cellular phenotype: cocultivation of hepatocytes and nonparenchymal cells. FASEB J. 1999;13(14):1883–1900. doi: 10.1096/fasebj.13.14.1883. [DOI] [PubMed] [Google Scholar]
- 87.Khetani SR, Bhatia SN. Microscale culture of human liver cells for drug development. Nat Biotechnol. 2008;26(1):120–126. doi: 10.1038/nbt1361. [DOI] [PubMed] [Google Scholar]
- 88.March S, et al. A microscale human liver platform that supports the hepatic stages of Plasmodium falciparum and vivax. Cell Host Microbe. 2013;14(1):104–115. doi: 10.1016/j.chom.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ng S, et al. Human iPSC-derived hepatocyte-like cells support Plasmodium liver-stage infection in vitro. Stem Cell Reports. 2015;4(3):348–359. doi: 10.1016/j.stemcr.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dembele L, et al. Persistence and activation of malaria hypnozoites in long-term primary hepatocyte cultures. Nat Med. 2014;20(3):307–312. doi: 10.1038/nm.3461. [DOI] [PubMed] [Google Scholar]
- 91.Morosan S, et al. Liver-stage development of Plasmodium falciparum, in a humanized mouse model. J Infect Dis. 2006;193(7):996–1004. doi: 10.1086/500840. [DOI] [PubMed] [Google Scholar]
- 92.Vaughan AM, et al. Complete Plasmodium falciparum liver-stage development in liver-chimeric mice. J Clin Invest. 2012;122(10):3618–3628. doi: 10.1172/JCI62684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vaughan AM, et al. Plasmodium falciparum genetic crosses in a humanized mouse model. Nat Methods. 2015;12(7):631–633. doi: 10.1038/nmeth.3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hayton K, et al. Erythrocyte binding protein PfRH5 polymorphisms determine species-specific pathways of Plasmodium falciparum invasion. Cell Host Microbe. 2008;4(1):40–51. doi: 10.1016/j.chom.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wellems TE, et al. Chloroquine resistance not linked to mdr-like genes in a Plasmodium falciparum cross. Nature. 1990;345(6272):253–255. doi: 10.1038/345253a0. [DOI] [PubMed] [Google Scholar]
- 96.Vaughan AM, et al. Plasmodium falciparum genetic crosses in a humanized mouse model. Nat Methods. 2015;12(7):631–633. doi: 10.1038/nmeth.3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Douglass AN, et al. Host-based prophylaxis successfully targets liver stage malaria parasites. Mol Ther. 2015;23(5):857–865. doi: 10.1038/mt.2015.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kaushansky A, Kappe SH. The crucial role of hepatocyte growth factor receptor during liver-stage infection is not conserved among Plasmodium species. Nat Med. 2011;17(10):1180–1181. doi: 10.1038/nm.2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Silvie O, et al. Hepatocyte CD81 is required for Plasmodium falciparum and Plasmodium yoelii sporozoite infectivity. Nat Med. 2003;9(1):93–96. doi: 10.1038/nm808. [DOI] [PubMed] [Google Scholar]
- 100.Schulz-Utermoehl T, et al. Evaluation of the pharmacokinetics, biotransformation and hepatic transporter effects of troglitazone in mice with humanized livers. Xenobiotica. 2012;42(6):503–517. doi: 10.3109/00498254.2011.640716. [DOI] [PubMed] [Google Scholar]
- 101.Wells TN, Hooft van Huijsduijnen R, Van Voorhis WC. Malaria medicines: a glass half full? Nat Rev Drug Discov. 2015;14(6):424–442. doi: 10.1038/nrd4573. [DOI] [PubMed] [Google Scholar]
- 102.Ghorbal M, Gorman M, Macpherson CR, Martins RM, Scherf A, Lopez-Rubio JJ. Genome editing in the human malaria parasite Plasmodium falciparum using the CRISPR-Cas9 system. Nat Biotechnol. 2014;32(8):819–821. doi: 10.1038/nbt.2925. [DOI] [PubMed] [Google Scholar]
- 103.Wagner JC, Platt RJ, Goldfless SJ, Zhang F, Niles JC. Efficient CRISPR-Cas9-mediated genome editing in Plasmodium falciparum. Nat Methods. 2014;11(9):915–918. doi: 10.1038/nmeth.3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhang C, et al. Efficient editing of malaria parasite genome using the CRISPR/Cas9 system. MBio. 2014;5(4):e01414–e01414. doi: 10.1128/mBio.01414-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Medicines for Malaria Venture Interactive R&D portfolio. [April 12, 2016];MMV Web site. http://www.mmv.org/research-development/interactive-rd-portfolio
- 106.Sinden RE, Suhrbier A, Davies CS, Fleck SL, Hodivala K, Nicholas JC. The development and routine application of high-density exoerythrocytic-stage cultures of Plasmodium berghei. Bull World Health Organ. 1990;68(suppl):115–125. [PMC free article] [PubMed] [Google Scholar]
- 107.Calvo-Calle JM, Moreno A, Eling WM, Nardin EH. In vitro development of infectious liver stages of P. Exp Parasitol. 1994;79(3):362–373. doi: 10.1006/expr.1994.1098. [DOI] [PubMed] [Google Scholar]
- 108.Sattabongkot J, et al. Establishment of a human hepatocyte line that supports in vitro development of the exo-erythrocytic stages of the malaria parasites Plasmodium falciparum and P. vivax. Am J Trop Med Hyg. 2006;74(5):708–715. [PubMed] [Google Scholar]
- 109.Mazier D, et al. [In vitro infection of adult Thamnomys hepatocytes by sporozoites of Plasmodium yoelii: development of schizonts and release of infective merozoites] Ann Parasitol Hum Comp. 1982;57(1):99–100. [PubMed] [Google Scholar]
- 110.Shortt HE, Garnham PC. Demonstration of a persisting exo-erythrocytic cycle in Plasmodium cynomolgi and its bearing on the production of relapses. Br Med J. 1948;1(4564):1225–1228. doi: 10.1136/bmj.1.4564.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yoeli M, Most H. Pre-erythrocytic development of Plasmodium berghei. Nature. 1965;205:715–716. doi: 10.1038/205715a0. [DOI] [PubMed] [Google Scholar]