

Abstract
Endogenous retroviruses (ERVs) are abundant in mammalian genomes and contain sequences modulating transcription. How ERV propagation impacts the evolution of gene regulation remains poorly understood. Here we show that ERVs have shaped the evolution of a transcriptional network underlying the interferon (IFN) response, a major branch of innate immunity. We found that lineage-specific ERVs have dispersed numerous IFN-inducible enhancers independently in diverse mammalian genomes. CRISPR-Cas9 deletion of a subset of these ERV elements in the human genome impaired expression of adjacent IFN-induced genes and revealed their involvement in the regulation of essential immune functions, including activation of the AIM2 inflammasome. While these regulatory sequences likely arose in ancient viruses, they now constitute a dynamic reservoir of IFN-inducible enhancers fueling genetic innovation in mammalian immune defenses.
Changes in gene regulatory networks underlie many biological adaptations, but the mechanisms promoting their emergence are not well understood. Transposable elements (TEs), including endogenous retroviruses (ERVs), have been proposed to facilitate regulatory network evolution because they contain regulatory elements, and can amplify in number and/or move throughout the genome (1-3). Genomic studies support this model (4), revealing that a substantial fraction of TE-derived noncoding sequences evolve under selective constraint (3, 5), are frequently bound by transcription factors (6-10), and often exhibit cell-type specific chromatin states consistent with regulatory activity (11, 12). These observations implicate TEs as a potential source of lineage-specific cis-elements capable of rewiring regulatory networks, but the adaptive consequences of this process for specific physiological functions remain largely unexplored.
We investigated the evolution of gene regulatory networks induced by the pro-inflammatory cytokine interferon gamma (IFNG). Interferons are pro-inflammatory signaling molecules that are released upon infection to promote transcription of innate immunity factors, collectively defined as IFN-stimulated genes (ISGs) (13). ISGs are regulated by cis-regulatory elements that are bound by interferon regulatory factor (IRF) and signal transducer and activator of transcription (STAT) transcription factors upon activation of IFN signaling pathways (13). Although innate immune signaling pathways are conserved among mammals, the transcriptional outputs of these pathways differ across species (14, 15), likely reflecting lineage-specific adaptation in response to independent host-pathogen conflicts. Thus, these pathways provide useful systems that allow us to investigate if TE-derived regulatory elements influence biological outcomes.
To explore the influence of TEs on IFNG-inducible regulatory networks, we examined their contribution to IRF1 and STAT1 binding sites using ChIP-Seq data published for three human cell lines treated with IFNG: K562 myeloid-derived cells, HeLa epithelial-derived cells, and primary CD14+ macrophages (16, 17). Our initial analysis revealed 27 TE families enriched within IFNG-induced binding peaks in at least one of the datasets examined (18) (Table S1, Fig S1A-B), and included TEs previously predicted to be cis-regulatory elements (11, 19). These sequences contain evolutionarily young to ancient TE families, of which the majority (20 out of 27) originated from Long Terminal Repeat (LTR) promoter regions of ERVs (Fig 1A). These data suggest that ERVs, which arose from ancient retroviral infections and currently constitute 8% of the human genome (20), represent a source of novel binding sites bound by IFNG-inducible transcription factors.
Fig. 1. Dispersion of IFNG-inducible regulatory elements by ERVs.
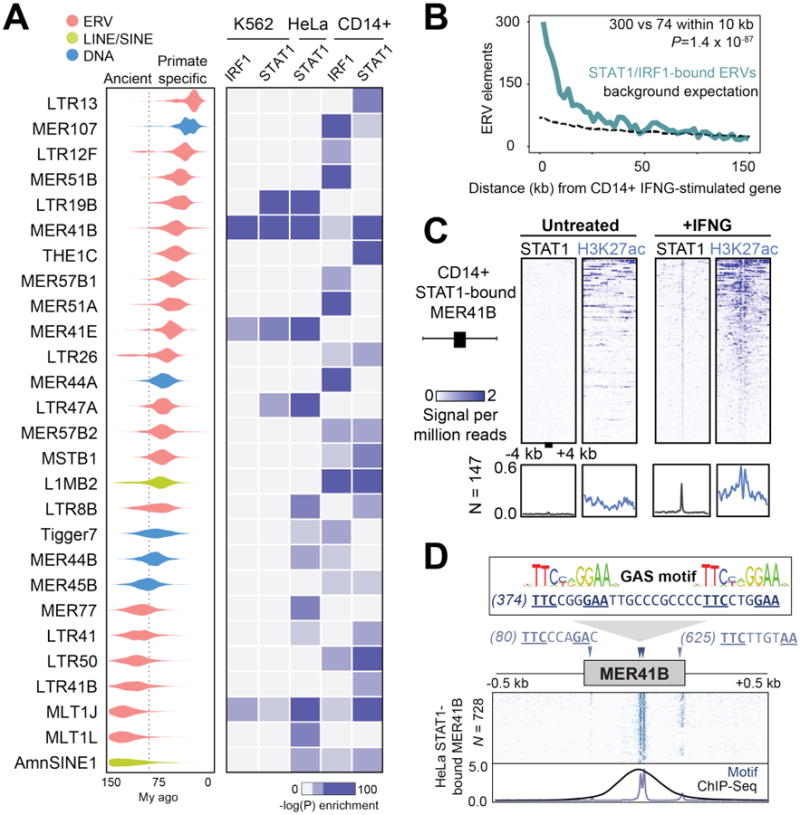
A) Age distribution (left) and enrichment within ChIP-Seq datasets (right) of 27 TE families that were enriched within binding sites for IFNG-stimulated cells (18). Estimated primate/rodent divergence time (82 My) from (34). B) Frequency histogram of absolute distances from each ERV to the nearest ISG, for CD14+ cells. The background expectation is from the genome-wide ERV distribution (18). Statistical significance of the observed enrichment within the first 10 kb of the nearest ISG assessed by binomial test. C) Heatmap of CD14+ ChIP-Seq signals centered across STAT1 peak summits within MER41B elements. Bottom metaprofiles represent average normalized ChIP signal across bound elements. D) Schematic of the MER41B LTR consensus sequence. Triangles indicate Gamma Activated Site (GAS; TTCNNNGAA) motifs predicted to bind STAT1 in response to IFNG (13). Heatmap depicts the presence of GAS motifs across 728 extant STAT1-bound MER41B copies in HeLa cells (18). Bottom metaprofile represents average presence of STAT1 motifs relative to the MER41 consensus sequence, overlain with normalized STAT1 ChIP-Seq density across the same elements.
We next investigated whether these ERVs may contribute to IFNG-inducible regulation of adjacent cellular genes. ERVs bound by STAT1 and/or IRF1 were strongly enriched near ISGs (binomial test, P=1.4×10-87, Figs 1B, S2), based on a matched RNA-Seq dataset from CD14+ macrophages (Table S2) (18, 21). A complementary approach using the genomic regions enrichment of annotations tool (GREAT) (22) revealed enrichment of CD14+ STAT1/IRF1-bound ERVs near genes annotated with immune functions (Fig S3A-B). These findings suggest a potentially widespread role for ERVs in the regulation of the human IFNG response.
MER41 is an endogenized gammaretrovirus that invaded the genome of an anthropoid primate ancestor ∼45-60 million years ago with 7,190 LTR elements, from 6 subfamilies (MER41A-MER41G), now fixed in the human genome (Fig S4A). Our analysis revealed the primate-specific MER41 family of ERVs as a source of IFNG-inducible binding sites (Fig S4B), with nearly 1,000 copies in humans (N=962) bound by STAT1 and/or IRF1 in at least one cell type (Table S3, Fig S4C). In CD14+ macrophages, STAT1-bound MER41 elements exhibit stereotyped induction of H3K27ac upon IFNG stimulation, a hallmark of cis-regulatory enhancer activity (23) (Fig 1C).
Consistent with this ERV family affecting IFNG-inducible regulation, MER41B sequences were identified as enriched within STAT1 ChIP-Seq peaks in IFNG-stimulated HeLa cells (19). A tandem pair of predicted STAT1 binding sites coincides with STAT1 ChIP-Seq peak localization (Fig 1D). These sites also occur in the ancestral (consensus) sequence of the MER41B subfamily (Fig 1D) but not in the MER41A subfamily, which is characterized by a 43 bp deletion that has eliminated these binding sites (Fig S5). MER41A sequences show no enrichment within IFNG-inducible binding sites despite otherwise sharing 99% sequence identity with MER41B (Figs S4B, S5). Together these data suggest that many MER41 elements are directly bound by STAT1 upon IFNG treatment, likely owing to the presence of ancestral STAT1 binding motifs within their LTR sequences.
Next we focused on the MER41.AIM2 ERV which is located 220 bp upstream of the gene Absent in Melanoma 2 (AIM2), an ISG that encodes a sensor of foreign cytosolic DNA and activates an inflammatory response response (24). Importantly, while AIM2 is IFNG-inducible in humans, it is constitutively transcribed in mice (24). In humans, MER41.AIM2 appears to provide the only STAT1 binding site within 50 kb of the AIM2 gene and the element gains H3K27 acetylation upon IFNG stimulation (Fig 2A). Therefore, the regulation of AIM2 has undergone evolutionary divergence across mammalian lineages, suggesting that the transposition of MER41 upstream of AIM2 may have conferred regulation by IFN signaling in anthropoid primates.
Fig. 2. A MER41 element is essential for AIM2 inflammasome activation.
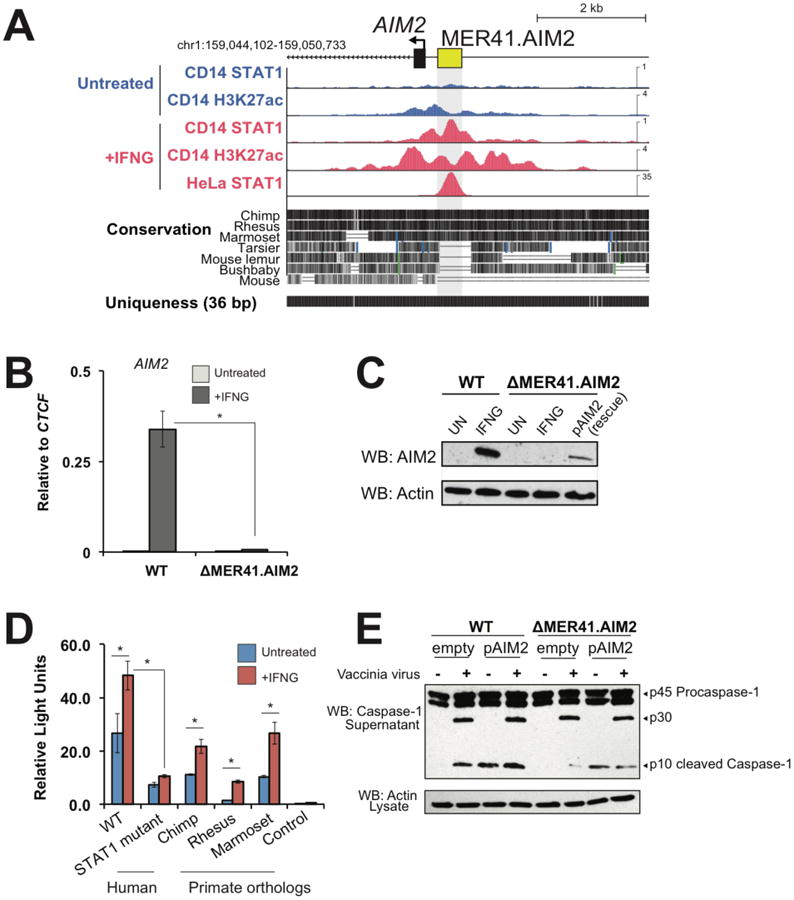
A) Genome browser view of AIM2. ChIP-Seq tracks are normalized per million reads. The “Uniqueness” track displays genome-wide short-read alignability. B) qPCR of AIM2 levels in wild-type and ΔMER41.AIM2 HeLa cells after 24 hrs IFNG treatment. C) Western blot of AIM2 in wild-type and ΔMER41.AIM2 cells after IFNG treatment. D) Luciferase reporter assays of MER41.AIM2, MER41.AIM2 with mutations in the predicted STAT1 sites, and primate orthologs of MER41.AIM2 (see Fig S7A). E) Western blot of caspase-1 from supernatants of wild-type and ΔMER41.AIM2 cells infected with vaccinia virus (18). * p < 0.05, Student's t-test.
We used the CRISPR-Cas9 system to delete the MER41.AIM2 element in HeLa cells (Fig S6) (18). Cells homozygous for the MER41.AIM2 deletion (ΔMER41.AIM2) failed to express AIM2 upon IFNG treatment, in contrast to control cells where AIM2 transcript levels were robustly induced by IFNG (Fig 2B). IFNG-induced AIM2 protein levels were undetectable in ΔMER41.AIM2 cells (Fig 2C), thus demonstrating that MER41.AIM2 is necessary for endogenous IFNG-inducible regulation of AIM2.
We further delineated the regulatory activity of MER41.AIM2 using luciferase reporter assays (18). MER41.AIM2 was sufficient to drive IFNG-inducible reporter expression in HeLa cells, and this activity was significantly diminished by point mutations ablating the predicted STAT1 binding motifs (Fig 2D). These binding sites are conserved across anthropoid primates (Fig S7A), and IFNG-inducible reporter activity was conserved across orthologous MER41.AIM2 elements cloned from chimpanzee, rhesus macaque, and marmoset (Fig 2D). We also confirmed that orthologs of AIM2 were all IFNG-inducible in primary fibroblasts from these species (Fig S7B). These results establish MER41.AIM2 as an IFNG-inducible enhancer and suggest that it was co-opted for AIM2 regulation in an ancestor of anthropoid primates.
The binding of AIM2 to cytoplasmic double-stranded DNA from intracellular bacteria and viruses promotes the assembly of a molecular platform known as an inflammasome, which initiates pyroptotic cell death by cleaving and activating caspase-1 (25). To test whether MER41.AIM2 is required for this response to infection, we infected ΔMER41.AIM2 cells with vaccinia virus (VACV) for 24 hrs and assayed secretion of the active cleaved form of caspase-1 (subunit p10) as the readout of inflammasome activity. Secreted levels of activated caspase-1 were markedly reduced in ΔMER41.AIM2 cells compared to wild type cells, and caspase-1 activation was restored by transient transfection with an AIM2 overexpression construct [pCMV-AIM2 plasmid (Fig 2E)]. Collectively these experiments demonstrate that MER41.AIM2 is likely a necessary element of the inflammatory response to infection.
The dispersion of cis-regulatory elements propagated by the same TE family might facilitate recruitment of multiple genes into the same regulatory network (3). We identified 3 additional MER41 elements within 20 kb of APOL1, IFI6, and SECTM1, which all are involved in human immunity (26-28) (Fig 3A). As with MER41.AIM2, we used CRISPR-Cas9 to generate genomic deletions of MER41.APOL1, MER41.IFI6, and MER41.SECTM1 in HeLa cells (Figs S8, S9). Upon treatment with IFNG, each mutant cell line exhibited significantly decreased transcript levels of the corresponding ISG relative to wild-type levels (Fig 3B) indicating that these MER41 elements have also been co-opted as IFNG-inducible enhancers. However, in contrast to AIM2, deletion of these MER41 elements did not completely abolish IFNG-induced transcript levels of these genes. This difference may be due to additional STAT1 binding sites located near these genes (Fig 3A). In such cases MER41 elements may contribute regulatory robustness as partially redundant or “shadow” enhancers (29).
Fig. 3. Multiple MER41 elements have been co-opted to regulate the IFNG response.

A) Genome browser views of MER41 elements located near APOL1, IFI6, and SECTM1. ChIP-Seq data is depicted as normalized signal per million reads. B) qPCR of each gene comparing IFNG-inducible levels in wild-type HeLa cells and MER41 deletion mutants. * p < 0.05, Student's t-test.
ERVs related to the primate-specific MER41 family (“MER41-like”) have been identified in most major mammalian lineages (30), raising the possibility of similar contributions to immune regulation. Further analysis, including cross-species genomic alignments, confirmed that multiple mammalian lineages were independently colonized by related MER41-like gammaretroviruses ∼50-75 My ago (Table S4). Remarkably, we found that the tandem STAT1 binding motifs present in anthropoid MER41 are conserved in MER41-like relatives found in lemuriformes, vesper bats, carnivores, and artiodactyls (Figs 4A, S10), suggesting that they might also have dispersed IFN-inducible enhancers in the genomes of these species. Consistent with this prediction, we found that reconstructed ancestral (consensus) sequences of MER41-like LTRs from dog and cow can drive robust IFNG-inducible reporter activity in HeLa cells (Fig 4B).
Fig. 4. IFNG-inducible ERVs are pervasive in mammalian genomes.
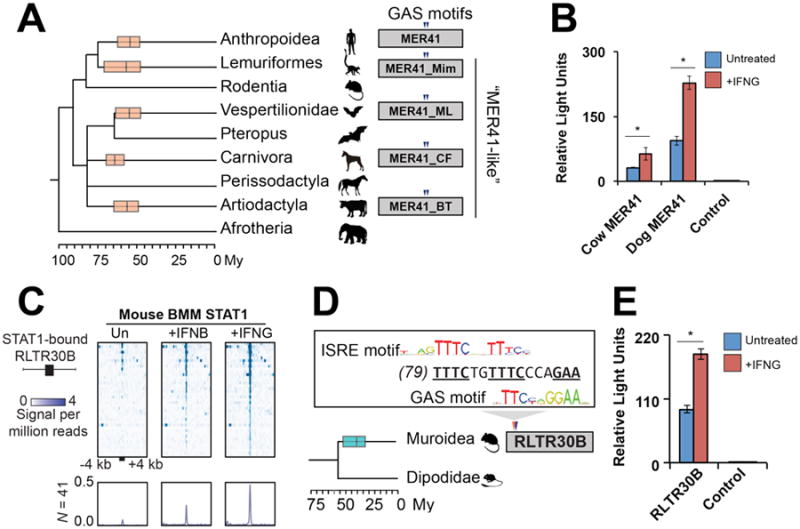
A) A consensus mammalian species phylogeny overlain with boxplots (median and 25th/75th percentiles) depicting the estimated age of MER41-like amplifications (18). Triangles depict conserved GAS motifs. B) Luciferase reporter assays of MER41-like LTR consensus sequences from cow and dog (18). C) Heatmap of ChIP-Seq signals centered across STAT1 peak summits within mouse RLTR30B elements. BMM: bone marrow-derived macrophages. Bottom metaprofiles represent average normalized ChIP signal across bound elements. D) Rodent phylogeny overlain with a boxplot depicting the amplification of RLTR30B, as in (A). ISRE: Interferon Stimulated Response Element motif (TTTCNNTTTC) predicted to bind STAT1 in response to IFNB (13). E) Luciferase reporter assay of RLTR30B consensus sequence, as in (B). Time-calibrated phylogenies in (A) and (D) are from (34). * p < 0.05, Student's t-test.
These results suggest that ERVs may have independently expanded the IFN regulatory network in multiple mammalian lineages. To further investigate this possibility, we analyzed a STAT1 ChIP-Seq dataset of IFNG- and IFN-Beta (IFNB)-stimulated primary macrophages from mouse (31), a species that lacks MER41-like elements but harbors a diverse repertoire of lineage-specific ERVs (30). Our analysis revealed a muroid-specific endogenous gammaretrovirus named RLTR30B enriched for both IFNG- and IFNB-inducible STAT1 binding events (Figs 4C, S11A), which coincide with overlapping motifs corresponding to both IFNG and IFNB-induced STAT1 binding sites located in the 5′ end of the LTR consensus sequence (Fig 4D). Reporter assays revealed that the consensus sequence of RLTR30B also provides IFNG-inducible enhancer activity in HeLa cells (Fig 4E). GREAT analysis also revealed significant enrichment of mouse STAT1-bound ERVs near functionally annotated immunity genes (Fig S11B).
Together our findings uncover IFN-inducible enhancers introduced and amplified by ERVs in many mammalian genomes. On occasion, these elements have been co-opted to regulate host genes encoding immunity factors. While we demonstrate that ERVs play a functional role regulating innate immune pathways in human HeLa cells, further studies will be necessary to extend our findings to primary hematopoietic cells and other species such as mouse. We speculate that the prevalence of IFN-inducible enhancers in the LTRs of these ancient retroviruses is not coincidental, but may reflect former viral adaptations to exploit immune signaling pathways promoting viral transcription and replication (32). Indeed, several extant viruses, including HIV, possess IFN-inducible cis-regulatory elements (33). It would be ironic if viral molecular adaptations had been evolutionarily recycled to fuel innovation and turnover of the host immune repertoire. Regardless of the original raison d'être of these sequences, our study illuminates how selfish genetic elements have contributed raw material that has been repurposed for cellular innovation.
Supplementary Material
Acknowledgments
Accession numbers for the published datasets analyzed in this study are available in Materials and Methods. We thank all members of the Elde and Feschotte labs for insightful discussions. We thank A. Kapusta, A. Lewis, D. Downhour, J. Carleton, and K. Cone for technical assistance, and D. Hancks and J. F. McCormick for their critical input. This work is supported by awards from the Pew Charitable Trusts and NIH to N.C.E. (GM082545 and GM114514), and to C.F. (GM112972). E.B.C. is a HHMI postdoctoral fellow of the Jane Coffin Childs Fund. N.C.E. is a Pew Scholar in the Biomedical Sciences and Mario R. Capecchi Endowed Chair in Genetics. The authors declare no financial conflicts of interest.
Footnotes
Supplementary Materials
www.sciencemag.org
Materials and Methods
Supplementary References (35-49)
References and Notes
- 1.Britten RJ, Davidson EH. Science. 1969:349–357. doi: 10.1126/science.165.3891.349. [DOI] [PubMed] [Google Scholar]
- 2.McClintock B. Proc Natl Acad Sci U S A. 1950;36:344–21175. doi: 10.1073/pnas.36.6.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feschotte C. Nat Rev Genet. 2008;9:397–405. doi: 10.1038/nrg2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rebollo R, Romanish MT, Mager DL. Annu Rev Genet. 2012;46:21–42. doi: 10.1146/annurev-genet-110711-155621. [DOI] [PubMed] [Google Scholar]
- 5.Lowe CB, Bejerano G, Haussler D. Proc Natl Acad Sci U S A. 2007;104:8005–8010. doi: 10.1073/pnas.0611223104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang T, et al. Proc Natl Acad Sci U S A. 2007;104:18613–18618. doi: 10.1073/pnas.0703637104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kunarso G, et al. Nat Genet. 2010;42:631–634. doi: 10.1038/ng.600. [DOI] [PubMed] [Google Scholar]
- 8.Schmidt D, et al. Cell. 2012;148:335–348. doi: 10.1016/j.cell.2011.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chuong EB, Rumi MAK, Soares MJ, Baker JC. Nat Genet. 2013;45:325–329. doi: 10.1038/ng.2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Notwell JH, Chung T, Heavner W, Bejerano G. Nat Commun. 2015;6:6644. doi: 10.1038/ncomms7644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jacques PÉ, Jeyakani J, Bourque G. PLoS Genet. 2013;9:e1003504. doi: 10.1371/journal.pgen.1003504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sundaram V, et al. Genome Research. 2014;24:1963–1976. doi: 10.1101/gr.168872.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Platanias LC. Nat Rev Immunol. 2005;5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 14.Barreiro LB, Marioni JC, Blekhman R, Stephens M, Gilad Y. PLoS Genet. 2010;6:e1001249. doi: 10.1371/journal.pgen.1001249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schroder K, et al. Proc Natl Acad Sci U S A. 2012;109:E944–53. doi: 10.1073/pnas.1110156109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gerstein MB, et al. Nature. 2012;489:91–100. doi: 10.1038/nature11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qiao Y, et al. Immunity. 2013;39:454–469. doi: 10.1016/j.immuni.2013.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Methods
- 19.Schmid CD, Bucher P. PLoS ONE. 2010;5:e11425. doi: 10.1371/journal.pone.0011425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lander ES, et al. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 21.Su X, et al. Nat Immunol. 2015;16:838–849. doi: 10.1038/ni.3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McLean CY, et al. Nat Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ostuni R, et al. Cell. 2013;152:157–171. doi: 10.1016/j.cell.2012.12.018. [DOI] [PubMed] [Google Scholar]
- 24.Hornung V, et al. Nature. 2009;458:514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fernandes-Alnemri T, et al. Nat Immunol. 2010;11:385–393. doi: 10.1038/ni.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vanhamme L, et al. Nature. 2003;422:83–87. doi: 10.1038/nature01461. [DOI] [PubMed] [Google Scholar]
- 27.Meyer K, et al. Sci Rep. 2015;5:9012. doi: 10.1038/srep09012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang T, et al. J Leukoc Biol. 2012;91:449–459. doi: 10.1189/jlb.1011498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lagha M, Bothma JP, Levine M. Trends in Genetics. 2012;28:409–416. doi: 10.1016/j.tig.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bao W, Kojima KK, Kohany O. Mob DNA. 2015;6:11. doi: 10.1186/s13100-015-0041-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ng SL, et al. Proc Natl Acad Sci U S A. 2011;108:21170–21175. doi: 10.1073/pnas.1119137109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Randall RE, Goodbourn S. Journal of General Virology. 2008;89:1–47. doi: 10.1099/vir.0.83391-0. [DOI] [PubMed] [Google Scholar]
- 33.Sgarbanti M, et al. J Virol. 2008;82:3632–3641. doi: 10.1128/JVI.00599-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meredith RW, et al. Science. 2011;334:521–524. doi: 10.1126/science.1211028. [DOI] [PubMed] [Google Scholar]
- 35.Li H, Durbin R. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu T, et al. Genome Biol. 2011;12:R83. doi: 10.1186/gb-2011-12-8-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramírez F, Dündar F, Diehl S, Grüning BA, Manke T. Nucleic Acids Res. 2014;42:W187–91. doi: 10.1093/nar/gku365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Edgar RC. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mathelier A, et al. Nucleic Acids Res. 2014;42:D142–7. doi: 10.1093/nar/gkt997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grant CE, Bailey TL, Noble WS. Bioinformatics. 2011;27:1017–1018. doi: 10.1093/bioinformatics/btr064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bulmer M, Wolfe KH, Sharp PM. PNAS. 1991;88:5974–5978. doi: 10.1073/pnas.88.14.5974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pace JK, II, Gilbert C, Clark MS, Feschotte C. PNAS. 2008;105:17023–17028. doi: 10.1073/pnas.0806548105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim D, Langmead B, Salzberg SL. Nat Meth. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pertea M, et al. Nat Biotechnol. 2015;33:290–295. doi: 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Trapnell C, et al. Nat Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ran FA, et al. Nat Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Adey A, et al. Nature. 2013;500:207–211. doi: 10.1038/nature12064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bertrand MJM, et al. Immunity. 2009;30:789–801. doi: 10.1016/j.immuni.2009.04.011. [DOI] [PubMed] [Google Scholar]
- 49.Romanish MT, Lock WM, van de Lagemaat LN, Dunn CA, Mager DL. PLoS Genet. 2007;3:e10. doi: 10.1371/journal.pgen.0030010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.