

Abstract
Although endocytosis maintains synaptic transmission, how endocytosis is initiated is unclear. We found that calcium influx initiated all forms of endocytosis at a single nerve terminal in rodents, including clathrin-dependent slow endocytosis, bulk endocytosis, rapid endocytosis and endocytosis overshoot (excess endocytosis), with each being evoked with a correspondingly higher calcium threshold. As calcium influx increased, endocytosis gradually switched from very slow endocytosis to slow endocytosis to bulk endocytosis to rapid endocytosis and to endocytosis overshoot. The calcium-induced endocytosis rate increase was a result of the speeding up of membrane invagination and fission. Pharmacological experiments suggested that the calcium sensor mediating these forms of endocytosis is calmodulin. In addition to its role in recycling vesicles, calcium/calmodulin-initiated endocytosis facilitated vesicle mobilization to the readily releasable pool, probably by clearing fused vesicle membrane at release sites. Our findings provide a unifying mechanism for the initiation of various forms of endocytosis that are critical in maintaining exocytosis.
After vesicle exocytosis, endocytosis generates new vesicles, which replenishes the vesicle pool and maintains exocytosis1. Five different kinetic forms of endocytosis have been reported. First, slow endocytosis, which takes tens of seconds, has been observed widely at synapses and non-neuronal secretory cells2–7. It is mediated by a classical, clathrin-dependent mechanism5,8–10. Second, rapid endocytosis, which takes a few seconds, has been observed at ribbon-type and calyx-type synapses2,6, but its existence is debated at small synapses1. Rapid endocytosis may be clathrin independent in chromaffin cells and goldfish ribbon-type synapses8,10. Third, bulk endocytosis, which forms large endosome-like structures by retrieving large pieces of membrane, has been seen at many synapses after strong stimulation (for examples, see refs. 11,12). Fourth, endocytosis overshoot, which retrieves more membrane than was exocytosed by the stimulation, has been observed in non-neuronal secretory cells13 and calyx-type synapses14. The mechanisms mediating bulk endocytosis and endocytosis overshoot are largely unclear. Finally, very slow or absent endocytosis has been observed15,16.
Because of these various forms, endocytosis in different conditions differs markedly in speed, amount and vesicle size. The mechanisms that initiate these different forms and rates of endocytosis are unclear. It has been proposed that the same mechanism underlies rapid and slow rates, with individual events occurring stochastically17. Another view is that different rates depend on the ratio between simultaneously occurring rapid and slow endocytosis pathways2,4,6,18, although what determines this ratio is somewhat controversial. For example, calcium facilitates rapid endocytosis at calyceal synapses6, but was reported to facilitate or inhibit rapid endocytosis in different studies at ribbon synapses2,4,18. It has been suggested that calcium regulates the endocytic rate at many nerve terminals3,19–25. Whether calcium initiates endocytosis remains unclear, as quantitative measurements did not reveal a complete block of endocytosis when calcium influx was reduced3,25. Because of the difficulty in identifying the endocytic trigger, it is often assumed that endocytosis follows exocytosis automatically, perhaps through molecular coupling.
Wee studied the mechanisms that initiate various forms of endocytosis with capacitance measurements at a large nerve terminal, the calyx of Held. We found that calcium influx initiated slow endocytosis, bulk endocytosis, rapid endocytosis and endocytosis overshoot with increasingly higher thresholds. Pharmacological experiments suggested that the calcium sensor mediating these forms of endocytosis was calmodulin. Our results may explain how exocytosis in single nerve terminals is maintained by various forms of endocytosis in various physiological conditions.
RESULTS
Calcium influx initiates slow endocytosis
Similar to trains of action potential-like stimuli6,14, a 20-ms depolarization (from −80 to +10 mV, unless indicated otherwise) at the calyx induced slow endocytosis with a time constant (τ) of 15.6 ± 2.1 s and an initial endocytosis rate (rateendo) of 39.2 ± 7.1 fF s−1 (n = 12; Fig. 1a) in control conditions, in which the extracellular calcium concentration ([Ca2+]o) was 2 mM and the pipette contained 50 μM BAPTA. Decreasing the [Ca2+]o from 5.5 to 0.5 mM decreased the calcium current charge (QICa) and the rateendo induced by the 20-ms depolarization (Fig. 1a). At a [Ca2+]o of 0.25 mM, the capacitance jump induced by a 20-ms depolarization was often too small to observe. However, 5–10 pulses of 10–20-ms depolarization at 0.5–1 Hz induced a clear capacitance jump (363 ± 114 fF, n = 4), which was followed by a rateendo of 2.0 ± 0.8 fF s−1 (n = 4 Fig. 1b). This rate was ~54-fold less than that induced after the same stimulus at a [Ca2+]o of 2 mM (107 ± 17 fF s−1, n = 4, P < 0.01; Fig. 1b) and ~20–49-fold less than that after a 20-ms depolarization at a [Ca2+]o of 2–5.5 mM (Fig. 1a). A similarly low rateendo was observed after a prolonged depolarization that induced no detectable Ca2+ current (Supplementary Fig. 1).
Figure 1.

Calcium influx triggers endocytosis. (a) The calcium channel current (ICa, upper) and the capacitance change (Cm, lower) induced by a 20-ms depolarization at an [Ca2+]o of 2, 5.5 or 0.5 mM. In all figures, the stimulus was applied before the capacitance jump; sampled traces are mostly single traces and occasionally an average of 2–3 traces. A summary is also shown (right, mean ± s.e., n in labels applies to other similar figures). (b) Sampled Cm induced by 5–10 pulses of 10–20-ms depolarization at 0.5–1 Hz with [Ca2+]o = 2 or 0.25 mM. (c) Sampled Cm induced by a 20-ms depolarization in control (50 μM BAPTA) or with 10 mM EGTA. (d) Sampled Cm induced by 20 pulses of 20-ms depolarization at 10 Hz in control or with 10 mM BAPTA. To reduce the capacitance noise during prolonged recordings, we used the software X-chart (HEKA) to average the capacitance value every ~0.3 s (applies only to this panel). (e) Sampled Cm induced by a 20-ms depolarization in the presence of DPF (1 mM) or DNF (1 mM).
The slow calcium chelator EGTA (10 mM in the pipette) reduced the rateendo after a 20-ms depolarization ~4.9-fold (EGTA: 8.0 ± 1.2 fF s−1, n = 18; control: 39.2 ± 7.1 fF s−1, n = 12; P < 0.01; Fig. 1c). With the fast calcium buffer BAPTA (10 mM in the pipette), the 20-ms depolarization induced no detectable capacitance jump. However, 10–20 pulses of 20-ms depolarization at 10 Hz induced a jump of 288 ± 32 fF (n = 10), followed by a rateendo of 0.2 ± 0.5 fF s−1 (n = 10; Fig. 1d). This rate (mean) was ~1,440-fold less than that after the same stimulus in control (50 μM BAPTA: 288 ± 31 fF s−1, n = 14; P < 0.01; Fig. 1d), and ~196-fold less than that induced by a 20=ms depolarization in control (39.2 ± 7.1 fF s−1, n = 12). Thus, 10 mM BAPTA essentially abolished endocytosis. The same result was observed in P13–14 rats with more mature calyces (n = 7, data not shown)26. These results suggest that calcium influx is required for endocytosis.
Two pieces of evidence suggest that the slow endocytosis that we observed is clathrin dependent, which would be consistent with studies at other synapses5,9,10. First, the proline-rich domain peptide pp11, which interrupts the interaction between amphiphysin and dynamin, an important step in clathrin-dependent endocytosis9,10, blocked slow endocytosis27,28. Second, a 12mer containing the DNF motif of amphiphysin I (1 mM), which disrupts the interaction between amphiphysin and the AP2 adaptor complex10, reduced rateendo after a 20-ms depolarization to 43 ± 12% (n = 7, P < 0.01), as compared with its control peptide (DPF, 1 mM, 100 ± 23%, n = 8, rateendo normalized to the mean of control; Fig. 1e).
Our results (Fig. 1) predicted a low rateendo at low intracellular calcium concentrations ([Ca2+]i). We tested this prediction by dialyzing 0.5 μM calcium into the calyx via the whole-cell pipette and simultaneously monitoring the calyx membrane capacitance and the miniature excitatory postsynaptic currents (mEPSCs) from the same synapses (Fig. 2a)29. We divided synapses into three groups corresponding to mEPSC frequencies of <5 Hz, 5–50 Hz and >50 Hz (mean = 22.9 ± 1.9 Hz, n = 135 synapses) and found that higher mEPSC frequencies were accompanied by larger capacitance increases (Fig. 2a–c).
Figure 2.

Very slow endocytosis at a [Ca2+]i of 0.5–0.75 μM. (a) Sampled mEPSCs (left) and the corresponding presynaptic Cm (right) from three synapses (red, blue and black) at 0.5 μM presynaptic [Ca2+]i. (b) The accumulated number of mEPSCs (NmEPSC, left, circles) and Cm (right, triangles) plotted versus the time after presynaptic break in (time 0) with a [Ca2+]i of 0.5 μM. Data were divided into three groups corresponding to mEPSC frequencies of 0–5 Hz (32 synapses, black), 5–50 Hz (89 synapses, blue) and >50 Hz (14 synapses, red). The Cm in calyces dialyzed with 0 calcium and 0.5 μM BoNT/C is also shown (eight synapses, brown triangles). Color codes apply to a–c. (c) The numbers of vesicles (Nves), calculated from the mEPSC and Cm traces in b, are plotted versus the time after presynaptic break in (NmEPSC, circles; NCm, triangles). To obtain NCm, we corrected the Cm traces in b for the baseline drift (subtracting the mean of the Cm trace obtained in 0.5 μM BoNT/C (brown triangles in b) from each Cm trace and divided by the mean vesicle’s capacitance, which is 65 aF29). The black curve is the predicted NCm based on the mean NmEPSC with an endocytosis τ of ~600 s (see Supplementary Data 1). (d) Sampled mEPSC (left) and Cm (right) from two synapses, one with 0.75 μM calcium (red) and the other with 0.75 μM calcium and 0.5 μM BoNT/C in the calyx (black). (e) NmEPSC (left, circles) and Cm (right, triangles) plotted versus the time after presynaptic break-in with a pipette containing 0.75 μM calcium (n = 14, red) or 0.75 μM calcium with 0.5 μM BoNT/C (n = 15, black). (f) Nves, calculated from NmEPSC and NCm, is plotted in the presence of 0.75 μM calcium. To obtain NCm, we corrected the Cm traces in e for the baseline drift (brown triangles in b) and divided by 65 aF. Color codes in e apply to f. The predicted NCm (black curve) was obtained as described in c with an endocytosis τ of 600 s.
Considering that a single vesicle’s capacitance is 0.065 fF29, a mEPSC frequency of <5 Hz (mean = 2.3 ± 1.4 Hz, n = 32 synapses) predicted a negligible increase in 330 s of recordings (2.3 Hz × 330 s × 0.065 fF = 49 fF). Thus, the capacitance change for the group with <5 Hz of mEPSCs largely reflected baseline drift (Fig. 2b). Consistent with this, a similar drift occurred in calyces dialyzed with pipette solution containing 0 calcium and with botulinum neurotoxin C (BoNT/C, 0.5 μM; Fig. 2b), which blocked exocytosis6. This drift implies that whole-cell break-in influences the normal membrane equilibrium on a very slow time scale. By correcting the capacitance drift and converting the capacitance increase into vesicle number (NCm; see Fig. 2c), we found that NCm approximately matched the accumulated mEPSC number (NmEPSC), particularly during the first 200 s of calcium dialysis (P > 0.2; Fig. 2c).
Compared with dialysis of 0.5 μM calcium, dialysis of 0.75 μM calcium caused a higher capacitance increase and mEPSC frequency (Fig. 2d–f), both of which were blocked by 0.5 μM BoNT/C (n = 15; Fig. 2d,e). Similar to 0.5 μM calcium, baseline-corrected NCm mostly overlapped with NmEPSC during the first 200 s of calcium dialysis (n = 14 synapses; Fig. 2f). This close overlap suggests that endocytosis was very slow (see Supplementary Data 1 for additional discussion). After 200 s of calcium dialysis, the NmEPSC continued to increase, whereas the NCm approached a plateau level at ~600 s of dialysis of 0.75 μM calcium (Fig. 2f), suggesting that very slow endocytosis was able to balance further exocytosis after the initial rise in capacitance. The measured mean NmEPSC with a mean endocytosis τ of ~600 s (~400–800 s) could well predict the observed NCm and thus account for the difference between NCm and NmEPSC (Fig. 2c,f and Supplementary Fig. 2; see Supplementary Data 1 for details). Simulations predict that a very slow endocytosis with a τ of 600 s will balance out continuous exocytosis in 1,200–1,800 s during a wide range of exocytosis frequencies (Supplementary Fig. 3, see Supplementary Data 1 for details). The rateendo decrease that we observed was independent of the amount of exocytosis (for detail, see Supplementary Data 1). Given that the amount of calcium influx determined whether efficient endocytosis occurred or not, we concluded that calcium influx is required to initiate endocytosis (see the Discussion).
Calcium influx regulates the endocytosis rate and amount
We previously showed that ten pulses of 20-ms depolarization delivered at 10 Hz activates rapid endocytosis and thus increased the rateendo6. To systematically study how increasing calcium influx affects endocytosis, we applied ten pulses of 2–50-ms depolarization at 10 Hz at a [Ca2+]o of 2 mM and ten pulses of 50-ms depolarization at a [Ca2+]o of 5.5 mM. Except for ten pulses of 2-ms depolarization, which induced only slow endocytosis, endocytosis after ten pulse stimuli was bi-exponential with a rapid (τ, ~1–4 s) and a slow component (τ, ~12–23 s) (Fig. 3a,b)6. Notably, we found that as QICa increased, rateendo and endocytosis overshoot gradually increased (Fig. 3a,b), with >80% of the rateendo increase being contributed by the rapid component, as a result of an increase in its amplitude and a decrease in its τ, from ~4 s to ~1.5 s (Fig. 3a,b). The rate of the slow component was also increased by calcium influx, though its contribution to the increased rateendo was minimal (Fig. 3b and Supplementary Fig. 4 and Supplementary Data 2). Rateendo could increase to 1,138 ± 172 fF s−1 (n = 7; Fig. 3a,b), corresponding to ~32 vesicles per s per active zone, considering that a calyx contains ~550 active zones and a vesicle’s capacitance is 65 aF29,30. Thus, differing from the general assumption, rapid endocytosis does not have a fixed speed or time constant but can be regulated in speed, time constant and relative contribution by calcium influx.
Figure 3.

Calcium influx increases rateendo and triggers rapid endocytosis and endocytosis overshoot. (a) ICa (upper) and Cm (lower) induced by ten pulses of 2-, 5-, 20- and 50-ms depolarization at 10 Hz in a [Ca2+]o of 2 mM and ten pulses of 50-ms depolarization at 10 Hz in a [Ca2+]o of 5.5 mM (n = 7–17). (b) Rateendo, the endocytosis amplitude (ampendo, normalized to the capacitance jump) and τ are plotted against QICa induced by the stimuli listed in a (QICa increased in the order of stimuli listed). The rateendo (left) and ampendo (middle left) of the rapid and slow component of endocytosis and their sum are shown. For τ, the rapid (τrapid, middle right) and the slow (τslow, right) components are shown separately. (c) Sampled Cm induced by ten pulses of 50-ms depolarization in control in 10 or 70 mM EGTA. (d) Sampled Cm induced by ten pulses of 50-ms depolarization in 10 mM EGTA with a [Ca2+]o of 5.5 mM. (e) At 34–36 °C, Cm was induced by 20, 200 and 500 pulses of 0.5-ms depolarization at 200 Hz and by 500 pulses with 10 mM EGTA. (f) Similar to b but with 20, 200 and 500 pulses of 0.5-ms depolarization at 100–300 Hz at 34–36 °C (n = 5–6).
The addition of EGTA decreased the rateendo after ten pulses of 50-ms depolarization in a concentration-dependent manner (control: 830 ± 177 fF s−1, n = 14; 10 mM EGTA: 184 ± 40 fF s−1, n 5; 70 mM EGTA: 74 ± 25 fF s−1, n = 3; Fig. 3c). The decrease was not a result of changes in the capacitance jump (for details, see ref. 6). EGTA (10–70 mM) also abolished the overshoot induced by ten pulses of 50-ms depolarization at a [Ca2+]o of 5.5 mM (n = 18, P < 0.01; Fig. 3d), which induced an overshoot with an amplitude 60% of the capacitance jump in control (50 μM BAPTA; Fig. 3a,b). We obtained similar results at near body temperature (34–36 °C) with trains of 0.5-ms depolarization to +10 mV that mimicked action potential trains in vivo31 (Fig. 3e,f). We concluded that increasing calcium influx increased rateendo by initiating rapid endocytosis and endocytosis overshoot.
Calcium triggers bulk endocytosis and speeds up fission
We previously found that bulk endocytosis is reflected as a brief downward capacitance shift (DCS) of ~20–500 fF32, the frequency of which increases with stronger stimulation (Supplementary Data 2)32. Consistent with these results, ten pulses of 50-ms depolarization at 10 Hz at a [Ca2+]o of 5.5 mM significantly increased the DCS frequency within 10 s of stimulation (0.14 ± 0.05 Hz, n = 12 calyces, P < 0.01; Fig. 4), indicating that rapid bulk endocytosis was occurring32. This frequency increase was much higher than that (<0.03 Hz)32 observed after ten pulses of 20-ms depolarization with a [Ca2+]o of 2 mM, and was reduced to near baseline level by adding 70 mM EGTA in the pipette (P = 0.02, n = 15 calyces; Fig. 4a–c). The DCS frequency change was not accompanied by significant changes in the DCS size (P > 0.6, see also ref. 32). Although 70 mM EGTA reduced the capacitance jump to ~57% of the control value (EGTA: 0.82 ± 0.09 pF, n = 15; control: 1.45 ± 0.15 pF, n = 12), the drop could not account for the nearly fourfold decrease in the DCS frequency (Fig. 4c). These results suggest that calcium influx initiates bulk endocytosis. Two pieces of evidence further support this suggestion. First, at a [Ca2+]i of 0.5–0.75 μM, at which endocytosis was very slow (Fig. 2), the DCS was rarely observed (n = 17, data not shown). Second, calmidazolium, an inhibitor of the calcium-binding protein calmodulin, essentially abolished the stimulation-induced increase of the DCS frequency but decreased the capacitance jump by less than 30%. Given that a 20-ms depolarization could induce DCSs32, but not rapid endocytosis (Fig. 1a), the QICa required to initiate bulk endocytosis must be lower than that required for rapid endocytosis.
Figure 4.
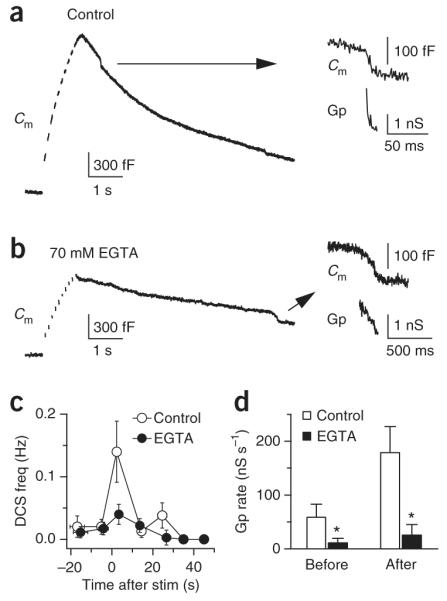
Calcium influx initiates bulk endocytosis and speeds up the fission pore closure rate. (a,b) Sampled Cm induced by ten pulses of 50-ms depolarization at 10 Hz in a [Ca2+]o of 5.5 mM in control conditions (a) and in 70 mM EGTA (b). For each trace, the DCSs (arrows) are enlarged and shown with the calculated Gp (right). (c) Frequency of DCSs, binned every 10 s, plotted in control conditions (n = 12 calyces) and in 70 mM EGTA (n = 15 calyces). At time 0, ten pulses of 50-ms depolarization at 10 Hz were applied. The [Ca2+]o was 5.5 mM. (d) The rate of the Gp decrease during DCSs before and within 10 s of stimulation in control conditions and in 70 mM EGTA. The stimulation was the same as that described in a–c.
During DCSs within 10 s of stimulation, the rate of the fission pore conductance (Gp) decrease (20–80%) was 182 ± 45 nS s−1 (n = 21 DCSs, Fig. 4a, see also Supplementary Fig. 5), corresponding to a decrease of the fission pore diameter at ~105 nm s−1. This rate was much higher than that before (P < 0.05; Fig. 4d) or >20 s after stimulation (data not shown) and was significantly reduced by 70 mM EGTA (P < 0.01; Fig. 4a,b,d), suggesting that calcium influx increases the fission pore closure rate.
Calmodulin involvement in endocytosis
It has been postulated that the calcium-binding proteins calmodulin and synaptotagmin (Syt) regulate endocytosis5,33. To test this, we measured endocytosis at ~4–10 min after break in with a pipette containing calmodulin inhibitors: the calmodulin binding–domain peptide (CBD, consisting of CAM kinase II residues 290–309, 300–500 μM), the myosin light chain kinase peptide (MLCK, 20 μM) or the organic small molecule inhibitor calmidazolium (10 μM in 0.1% DMSO)34. Compared with controls (scrambled CBD, mutated MLCK or 0.1% DMSO, respectively), these three inhibitors reduced the rateendo by >75% (P < 0.01; Fig. 5a), but did not significantly reduce the capacitance jump (P > 0.05) after a 20-ms depolarization (scrambled CBD: 100 ± 6%, n = 12; CBD: 13 ± 9%, n = 14; mutated MLCK: 100 ± 15%, n = 7; MLCK: 22 ± 9%, n = 11; 0.1% DMSO: 100 ± 15%, n = 12; calmidazolium: 10 ± 5%, n = 8; rateendo were normalized to the mean of each control group, respectively). A similar large reduction of rateendo was found in the presence of these inhibitors after ten pulses of 20-ms depolarization at 10 Hz (scrambled CBD: 100 ± 23%, n = 10; CBD: 20 ± 4%, n = 11; mutated MLCK: 100 ± 14%, n = 7; MLCK: 18 ± 5%, n = 12; 0.1% DMSO: 100 ± 19%, n = 12; calmidazolium: 21 ± 7%, n = 8; all P < 0.01; Fig. 5b). These inhibitors reduced the QICa by ~0–22% (Supplementary Fig. 6) and the capacitance jump induced by the ten pulse stimulus by <30%, which could not account for the observed >75% decrease in rateendo (see also Supplementary Data 1 and 2). Furthermore, calmidazolium essentially blocked overshoot (0.1% DMSO: 960 ± 90 fF overshoot, n = 20; calmidazolium: 280 ± 110 fF undershoot, n = 19; P < 0.01; endocytosis was not complete in the presence of calmidazolium) and significantly reduced the DCS frequency after ten pulses of 50-ms depolarization at a [Ca2+]o of 5.5 mM (P < 0.01; Fig. 5c,d). These results suggest that calmodulin may mediate calcium-triggered slow and rapid endocytosis, endocytosis overshoot and bulk endocytosis.
Figure 5.

Calmodulin blockers inhibit endocytosis. (a) Top, sampled Cm induced by a 20-ms depolarization in the presence of scrambled CBD (CBD-c, 500 μM, left) or CBD (500 μM, right). Middle, mutated MLCK peptide (MLCK-c, 20 μM) or MLCK (20 μM). Bottom, 0.1% DMSO (control) or calmidazolium (Calm, 10 μM with 0.1% DMSO). (b) Data are presented as in a but with Cm induced by ten pulses of 20-ms depolarization at 10 Hz. (c) Sampled Cm induced by ten pulses of 50-ms depolarization at 10 Hz at a [Ca2+]o of 5.5 mM in control conditions (0.1% DMSO, left) or in the presence of 20 μM calmidazolium (with 0.1% DMSO, right). The arrow denotes the DCS from the left sampled trace on a different scale (middle). (d) The frequency of DCSs (right) induced by ten pulses of 50-ms depolarization at 10 Hz at a [Ca2+]o of 5.5 mM in control conditions (0.1% DMSO) or with 20 μM calmidazolium.
The calmodulin proteins involved in endocytosis are probably immobile and membrane-bound, as endocytosis persisted during whole-cell dialysis of a control solution containing no calmodulin for longer than 10 min28. Furthermore, whole-cell dialysis of wild-type calmodulin protein (100–200 μM) did not significantly change the rateendo induced by ten pulses of 20-ms depolarization, as measured 5–10 min after whole-cell break in (calmodulin: 458 ± 111 fF s−1, n = 6; control: 394 ± 58 fF s−1, n = 10; P = 0.62).
Calyces do not contain Syt1, but do express the closely related Syt2, and genetic deletion of Syt2 abolishes synchronized release in calyces35. However, we were able to induce less synchronized release by a 20-ms depolarization (Fig. 6a) and ten pulses of 20-ms depolarization at 10 Hz (Fig. 6b). The rateendo after these two stimuli in Syt2−/− mice were similar to those from Syt2+/+ littermates (Fig. 6), suggesting that Syt2 is not critical for calcium-triggered endocytosis (20-ms pulse: Syt2+/+, 100 ± 10%, n = 6; Syt2−/−, 85 ± 15%, n = 7; P = 0.48; ten pulse train: Syt2+/+, 100 ± 17%, n = 10; Syt2−/−, 109 ± 25%, n = 11; P = 0.75; data were normalized to the mean in Syt2+/+ group).
Figure 6.
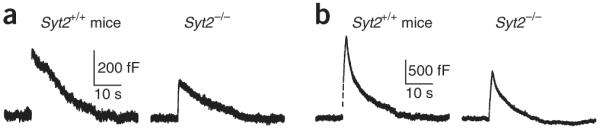
Syt2 is not critical in mediating endocytosis. (a) Sampled Cm induced by a 20-ms depolarization in a Syt2−/− mouse or a Syt2+/+ littermate. (b) Sampled Cm induced by ten pulses of 20-ms depolarization at 10 Hz in a Syt2−/− mouse or a Syt2+/+ littermate.
Endocytosis speeds up vesicle mobilization
Calcium/calmodulin speeded up both the rateendo (Figs. 1–5) and vesicle recruitment to the readily releasable pool (RRP)34, which implies that faster endocytosis may facilitate RRP replenishment. To test this possibility, we monitored the recovery of the RRP during ten pulses of 10–20 ms depolarization at 10 Hz, where each pulse depleted the RRP36. Consistent with previous observations34, the capacitance jump evoked by the second to the tenth depolarizing pulse, which reflected the RRP replenishment rate, was reduced by including CBD (300–500 μM), MLCK (20 μM, data not shown) or calmidazolium (10 μM) in the pipette (P < 0.01; Fig. 7a,b). We observed a similar reduction with dynasore (100 μM, P < 0.01; Fig. 7a,b), a dynamin inhibitor that inhibits rapid and slow endocytosis at calyces28. The decrease in the capacitance jump in the presence of CBD, calmidazolium or dynasore was not a result of a change in the QICa (Supplementary Data 2). Thus, as with dynasore, CBD and calmidazolium may inhibit the RRP replenishment by inhibiting endocytosis (see also Supplementary Data 2).
Figure 7.

CBD, calmidazolium and dynasore slow down the RRP replenishment. (a) Sampled Cm induced by ten pulses of 20-ms depolarization at 10 Hz in control conditions (0.1% DMSO), in the presence of 500 μM CBD, 10 μM calmidazolium or 100 μM dynasore. The Cm jump induced by the first 20-ms depolarization was normalized. (b) The capacitance jump (ΔCm) and the accumulated capacitance jump (ΣΔCm) induced by each 20-ms depolarization during 10 such pulses at 10 Hz in the conditions described in a (with same color code as in a). Data were normalized to the capacitance jump induced by the first 20-ms depolarization (control (0.1% DMSO), n = 12; CBD, n = 11; Calm, n = 8; dynasore, n = 5).
We observed an inhibition of RRP replenishment 100 ms after the first pulse; that is, at the second pulse during the ten pulse train. The capacitance jumps induced by the second pulse were 24.6 ± 3.4% (n = 8), 28.4 ± 2.1% (n = 11) and 30.2 ± 2.4% (n = 5) in the presence of calmidazolium, CBD and dynasore, respectively, all of which were significantly lower than that of the control (45.3 ± 3.4%, n = 12, P < 0.01; Fig. 7a,b). Vesicle depletion arising from the block in endocytosis could not account for this difference, as endocytosis retrieves only 0.6% of newly exocytosed vesicles within 100 ms in control conditions (calculated from the measured mean endocytosis τ of ~15.6 s after a 20-ms depolarization). Thus, inhibition of the RRP replenishment was a result of a block of an endocytic mechanism after fusion but before vesicle fission (see Discussion).
Endocytosis overshoot retrieves fused vesicle membrane
Vesicles stranded in low calcium conditions (Figs. 1d and 2) may provide the substrate for endocytosis overshoot. This possibility is difficult to test directly, as low calcium conditions were achieved by including higher concentrations of calcium buffers that precluded the endocytosis overshoot (Fig. 3a,d). To overcome this difficulty, we accumulated vesicles at the plasma membrane by blocking endocytosis with GDPβS (0.3 mM in the pipette instead of GTP), which inhibits dynamin function28. A 20-ms depolarization applied every 20 s for 10–20 repetitions increased the membrane capacitance by 3,278 ± 612 fF (n = 5), after which depolarization-induced endocytosis recovered as a result of activation of GTP-independent endocytosis (Fig. 8a, see ref. 28 for details). When applied after this treatment, ten pulses of 50-ms depolarization at 10 Hz induced an overshoot (2,492 ± 650 fF, n = 5 calyces; Fig. 8a) that was much larger than that induced in controls (254 ± 61 fF, n = 32, P < 0.03; Fig. 3b) but was similar to the membrane accumulation (3,278 ± 612 fF, n = 5) observed before the ten pulse stimulus.
Figure 8.

Endocytosis overshoot retrieves vesicles stranded at the plasma membrane. (a) The capacitance responses to a 20-ms depolarization repeated 11 times every 20 s, followed by a ten pulse train (ten pulses of 50-ms depolarization at 10 Hz, arrows) repeated every 1–2 min three times in a calyx dialyzed with 0.3 mM GDPβS. (b) The capacitance response to a ten pulse train (ten pulses of 50-ms depolarization at 10 Hz, arrows) repeated every 1–2 min three times in a control calyx (0.3 mM GTP).
The overshoot amplitude decreased to nearly 0 as the ten pulse train was repeated 2–4 times (n = 3; Fig. 8a), indicating that the membrane pool retrievable by the overshoot was limited. A similar decrease in the overshoot was observed when the ten pulse train was repeated in calyces containing no GDPβS but showing an overshoot in response to the first ten pulse train (n = 12; Fig. 8b). In the presence of GDPβS, there was no overshoot when the ten pulse train was applied without conditioning pulses, as a result of the initial endocytic block by GDPβS (n = 6, data not shown)28. These results suggest that endocytosis overshoot retrieves a limited pool of vesicles stranded at the plasma membrane.
DISCUSSION
High calcium concentration transients initiate endocytosis
Two pieces of evidence suggest that the [Ca2+]i that initiates efficient endocytosis is high, probably a few or tens of micromolar. First, at a [Ca2+]i of ≤0.75 μM, endocytosis was very slow, with a τ of ~600 s (Fig. 2). Second, a fast (BAPTA), but not a slow (EGTA), calcium chelator abolished endocytosis (Fig. 1c,d), suggesting that a high calcium concentration transient at the release site during depolarization34 initiates endocytosis. EGTA probably reduced this calcium transient, as it partially inhibited release (Fig. 1c)6,37, which explains why EGTA partially inhibited endocytosis (Figs. 1c and 3c–e). Our results suggest that the calcium threshold for initiating endocytosis is higher than for exocytosis, as much less endocytosis than exocytosis occurred in the presence of 10 mM BAPTA or a [Ca2+]i of 0.5–0.75 μM (Figs. 1d and 2). Strong calcium influx increased the rateendo by several thousand-fold over the lowest rate (Figs. 1d and 3b,f), resulting in a rateendo of up to ~32 vesicles per s per active zone immediately after stimulation (Fig. 3a,b,e). Thus, the same calcium influx must trigger both exocytosis and endocytosis. Considering that endocytosis is composed of two steps, membrane invagination and fission1, the control of rateendo (Figs. 1–3) and the fission pore closure (Fig. 4) by calcium suggests that calcium influx initiates endocytosis and speeds up both membrane invagination and fission.
Our data suggest that calcium binding with calmodulin initiates endocytosis. Calmodulin forms a physical complex with voltage-gated calcium channels38 and its N and C lobes may sense different calcium concentrations38, allowing calmodulin to mediate multiple forms of endocytosis. Because no endocytosis occurs when exocytosis is abolished by botulinum neurotoxins6,27, we conclude that fused vesicle membrane is the substrate used by calcium/calmodulin to initiate endocytosis.
Calcium influx initiates all forms of vesicle endocytosis
Our results are consistent with those of earlier studies supporting regulation of endocytosis by extracellular calcium in extremely intense or nonphysiological conditions, such as prolonged high potassium application in synaptosomes or α-latrotoxin application (see also Supplementary Discussion)19–24. Whether this finding can be extrapolated to regulation of endocytosis by calcium influx in physiological conditions is unclear. Furthermore, in these studies, endocytosis was measured indirectly using dye uptake and/or electron microscopy with a single time point, which may not distinguish between endocytosis and recycling, between different endocytosis forms or between partial and full inhibition, and thus between regulation and initiation of endocytosis. Our results resolve these issues at calyces.
We found that endocytosis was mostly abolished at a [Ca2+]o of 0.25 mM, but not 0.5 mM (Fig. 1a,b), which may explain why rateendo is only moderately decreased when the [Ca2+]o is reduced to ~0.5–0.75 mM at hippocampal synapses3,25. Our finding that high [Ca2+]i initiates endocytosis could explain the lack of correlation between a submicromolar residual calcium increase and the rateendo at neuromuscular junctions7.
Our observation that calcium/calmodulin initiated bulk endocytosis during more intense stimulation (Fig. 4, see also ref. 32) may explain why endosome-like structures are observed after more intense stimulation at many synapses (for example, refs. 11,12). At ribbon-type synapses, calcium influx, which is probably above tens of micromolar18, facilitates the occurrence of rapid endocytosis4 (but see ref. 2). At calyces, rapid endocytosis occurs on an increase of calcium influx6. Here, we found that calcium influx speeded up endocytosis by increasing the contribution of rapid endocytosis and decreasing the τ of rapid and slow endocytosis (Fig. 3b,f). Contrary to the general assumption, rapid endocytosis did not have a fixed τ or speed. Notably, the lowest detectable rateendo for rapid endocytosis approached the fastest rateendo for slow endocytosis (Fig. 3b,f). Although rapid and slow endocytosis are suggested to be clathrin independent and clathrin dependent, respectively8,10, both are initiated by calcium/calmodulin (Figs. 1–5) and require GTP hydrolysis and dynamin10,27,28. Thus, rapid and slow endocytosis may share at least some of the same endocytic machinery.
Endocytosis overshoot has been observed in endocrine cells13,16 and calyces14. Although calcium influx is considered to be a candidate for mediating overshoot, it has not been tested thoroughly with calcium buffers. Furthermore, it is unclear whether the overshoot retrieves vesicle membrane. We found that large calcium influx during intense stimulation, including high-frequency action potential–like trains (Fig. 3e,f) that may occur in vivo26 triggered endocytosis overshoot, which retrieved vesicles stranded at the plasma membrane (Fig. 8).
Although very slow endocytosis in near resting conditions may result in stranded vesicles at the plasma membrane, the rate of fusion and retrieval may reach a balance in a duration of ~2–3-fold longer than the time constant of very slow endocytosis (Fig. 2f, see also Supplementary Data 1). Thus, very slow endocytosis may recycle vesicles and participate in maintaining the nerve terminal morphology. Stranded vesicles in near resting conditions (Fig. 2f) might explain why vesicle proteins, such as synaptobrevin and synaptophysin, are present at the plasma membrane of hippocampal boutons3,9,15. Retrieval of stranded vesicles by endocytosis overshoot may prevent nerve terminal expansion and increase the vesicle cycling capacity, which is needed to maintain exocytosis during intense firing. These functions are physiologically relevant, as some neurons, including calyces, may fire from 0 to hundreds of hertz spontaneously and during stimulation39.
Vesicle recycling in resting conditions has been observed at hippocampal synapses40–42. However, observation requires prolonged incubation (~15 min to 10 h) of fluorescent materials for vesicle uptake40 and endocytosis has been estimated at a vesicle per bouton per 100 s41,42. These results are consistent with a very slow rate of endocytosis, as we observed here, and/or a low rate of exocytosis, which was not estimated previously40–42.
Taken together, we found that calcium influx initiated all known forms of endocytosis at calyces, including slow, clathrin-dependent endocytosis, bulk endocytosis, rapid endocytosis and endocytosis overshoot, each of which had a correspondingly higher calcium threshold. Increasing calcium influx increased rateendo by increasing the amplitude of rapid endocytosis (Figs. 1 and 3) and the rate of fission pore closure (Fig. 4) and by decreasing the slow and rapid endocytosis time constants. These findings may explain various forms, rates and amounts of endocytosis reported under various conditions at many synapses and non-neuronal secretory cells over the last three decades. We suggest that calcium influx is the unifying signal that initiates all forms of endocytosis in secretory cells, including nerve terminals.
Vesicle uptake of FM dyes has been observed after sucrose application, which presumably induces calcium-independent exocytosis43,44, implying that calcium-independent endocytosis follows calcium-independent exocytosis. This observation does not counter our conclusion that calcium-initiated endocytosis follows calcium-triggered exocytosis, as the existence of sucrose-induced calcium-independent exocytosis does not overthrow the principle that calcium triggers exocytosis during depolarization.
Calmodulin is the calcium sensor of endocytosis
The results of our pharmacological experiments (Fig. 5) suggest that calmodulin is the calcium sensor for calcium-triggered endocytosis. This finding may apply beyond calyces because, as discussed above, calcium influx may initiate endocytosis at many synapses. In support of this possibility, calmodulin blockers inhibit rapid endocytosis at chromaffin cells33 (but see ref. 45) and calcineurin, a phosphatase activated by calcium/calmodulin, may regulate bulk endocytosis in synaptosomes and cerebellar cultures22,23,46.
The incomplete block of endocytosis by calmodulin blockers (Fig. 5) probably reflects the inefficiency of these inhibitors in vivo, although additional calcium sensors cannot be excluded. Calmodulin might initiate endocytosis by activating the phosphatase calcineurin, which dephosphorylates endocytic proteins22,23,46. This possibility, although supported by our preliminary results, is beyond the scope of the present work. Placing calcium/calmodulin at the initial step opens the possibility of putting together the sequence of protein interactions mediating endocytosis.
Our observation that Syt2 deletion did not affect endocytosis after prolonged (20 ms) depolarization is somewhat surprising to us, as Syt1 deletion slows endocytosis after action potential trains at hippocampal synapses and Drosophila neuromuscular junctions5,47. The difference in the stimulation protocol might explain this discrepancy. In Syt1 and Syt2 knockout animals, synchronous release is abolished, but action potential–evoked release still occurs asynchronously at a few to tens of milliseconds after calcium influx35,48. Thus, asynchronously released vesicles may be exposed to much lower calcium levels during fusion than synchronously released vesicles. This would result in a slowed endocytosis, as the calcium influx controls the rateendo (Figs. 1–4). In our study, most asynchronous release may have occurred during prolonged 20-ms depolarization, during which calcium influx initiates efficient endocytosis.
Endocytosis before fission facilitates RRP replenishment
Calcium/calmodulin facilitates the RRP replenishment34, which is critical in maintaining release during repetitive firing26. Our results suggest that calcium/calmodulin facilitates the RRP replenishment via an endocytic mechanism before fission (Fig. 7).
We propose that vesicle fusion disrupts the release site structure, interfering with subsequent vesicle priming and/or fusion. Calcium/calmodulin may facilitate the structural recovery, and thus the RRP replenishment, by rapid transfer of vesicle membrane and proteins from the release site to the endocytic zone.
ONLINE METHODS
Slice preparation, capacitance and mEPSC recordings and solutions
All experiments were conducted in accordance with the guidelines established by the National Institute of Neurological Disorders and Stroke Intramural Animal Care and Use Committee. Parasagittal brainstem slices (200 μm thick) containing the medial nucleus of the trapezoid body were prepared from Wistar rats or mice using a vibratome36. Unless mentioned otherwise, the animals were 7–10 d old, experiments were carried out in rats at 22–24 °C, the [Ca2+]o was 2 mM, the presynaptic pipette contained 50 μM BAPTA and t tests were used to determine statistical significance. Means are presented as ± standard errors.
Whole-cell capacitance measurements were made with the EPC-9 amplifier and the software lock-in amplifier (PULSE, HEKA) to implement the Lindau-Neher technique (for details, see ref. 29). The frequency of the sinusoidal stimulus was 1,000 Hz, and the peak-to-peak voltage of the sine wave was ≤60 mV. The mEPSCs were recorded with an Axopatch 200B amplifier (Axon Instruments). Data were low-pass filtered at 5 kHz and sampled at 20 kHz (for details, see ref. 29). The holding potential for both pre- and postsynaptic recordings was −80 mV. To reduce the noise, we did not compensate for the postsynaptic series resistance (<15 MΩ) in mEPSC recordings.
For capacitance recordings, we pharmacologically isolated presynaptic Ca2+ currents and postsynaptic AMPA receptor–mediated EPSCs with a bath solution containing 105 mM NaCl, 20 mM TEA-Cl, 2.5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 25 mM NaHCO3, 1.25 mM NaH2PO4, 25 mM dextrose, 0.4 mM ascorbic acid, 3 mM myo-inositol, 2 mM sodium pyruvate, 0.001 mM tetrodotoxin, 0.1 mM 3,4-diaminopyridine and 0.05 mM D(–)-2-amino-5-phosphonovaleric acid(D(-)-AP-5), at pH 7.4 when bubbled with 95% O2 and 5% CO2. The presynaptic pipette contained 125 mM cesium gluconate, 20 mM CsCl, 4 mM MgATP, 10 mM sodium phosphocreatine, 0.3 mM GTP, 10 mM HEPES and 0.05 mM BAPTA, pH 7.2, adjusted with CsOH. When a high concentration of EGTA (10–70 mM) or BAPTA (10 mM) was added to the pipette, the cesium gluconate was reduced to keep the same osmolarity (310–320 mOsm).
For simultaneous recordings of the capacitance and the AMPA receptor–mediated mEPSC (Fig. 2), the bath solution contained 105 mM NaCl, 20 mM TEA-Cl, 2.5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 25 mM NaHCO3, 1.25 mM NaH2PO4, 25 mM dextrose, 0.4 mM ascorbic acid, 3 mM myo-inositol, 2 mM sodium pyruvate, 0.001 mM tetrodotoxin, 0.1 mM 3,4-diaminopyridine, 0.05 mM D(-)-AP-5, 0.01 mM bicuculline and 0.01 mM strychnine, at pH 7.4 when bubbled with 95% O2 and 5% CO2. The presynaptic pipette (2.5–4.5 MΩ) solution contained 105 mM cesium gluconate, 20 mM CsCl, 4 mM MgATP, 10 mM sodium phosphocreatine, 0.3 mM GTP and 10 mM HEPES, pH 7.2, adjusted with CsOH. To this solution, we added 10 mM EGTA and 7.50 mM CaCl2 to clamp the free calcium concentration at 0.5 μM or 10 mM EGTA and 8.55 mM CaCl2 to clamp the free calcium concentration at 0.75 μM. The osmolarity was 310–320 Osm. The free calcium concentration was calculated by assuming a calcium dissociation constant of 0.15 μM (for the binding between EGTA and calcium)49. This was confirmed by the intracellular calcium measurement29. The measured value was 0.48 ± 0.03 μM (n = 3) when free calcium was clamped at 0.5 μM29.
The myosin light chain kinase peptide (MLCK), the MLCK control peptide (MLCK-c, a point mutation) and the CBD were obtained from EMD Chemicals. The scrambled CBD peptide with a sequence of KLRLARLKATKNTFKMLGIA, used as a control, was purchased from 21st Century Biochemicals. DNF and DPF were also obtained from 21st Century Biochemicals. Calmidazolium and the wild-type calmodulin protein were purchased from Sigma.
Measurements of the rate and amount of endocytosis
The rateendo was measured as the capacitance jump divided by the endocytosis time constant, which was similar to the rate of the capacitance decay in the first 2–3 s after stimulation. Similarly, the rateendo for the rapid and slow component of endocytosis (Figs. 3 and 5) was measured as the amplitude of the rapid and the slow component divided by their corresponding endocytosis time constant. When endocytosis was slow or blocked (Figs. 1b,d and 4, and Supplementary Fig. 1), the capacitance decay was approximately linear in the first 10–60 s after stimulation. In these conditions, the rateendo was measured as the mean capacitance decay within 10–60 s of stimulation.
We used rateendo instead of τ to evaluate the rate of endocytosis50 because many manipulations substantially inhibited endocytosis, making it difficult to measure τ. Furthermore, τ may not necessarily reflect the rate of endocytosis. Saturation of the endocytic machinery could result in an increase of τ with a constant rateendo50 (see also Supplementary Data 1). Thus, rateendo is a more accurate measurement of the speed of endocytosis25,50.
The endocytosis overshoot was measured as the difference between the baseline and the capacitance value at ~40–60 s after stimulation. When comparing two groups of data (for example, Figs. 1e, 5a,b and 6), we often normalized our rateendo data in the experimental condition to the respective control group. We did this because the control values for each experiment were somewhat different, as the data for the various manipulations were obtained by several experimenters. However, for each manipulation (such as dialysis of the CBD peptide), the control and experimental data were obtained by one experimenter. By normalizing the data, we avoided meaningless comparisons of the slight differences in control values obtained by different experimenters.
Measurements of bulk endocytosis
The method for measuring large DCSs, which reflect bulk endocytosis, the fission pore conductance change during DCSs and the fission pore diameter change during DCSs were described previously32. To detect DCSs, we low-pass filtered capacitance traces at 30 Hz and differentiated. The differentiation was calculated as the difference between capacitance values of two neighboring samples with an interval of 1 ms. A DCS was identified when the rate of the capacitance decay was more than 50 fF per 100 ms in the differentiated capacitance trace, the size of a DCS was more than 20 fF in the filtered capacitance trace, and the measured series conductance and membrane conductance did not change in parallel with the DCS. A DCS was often superimposed on the smooth capacitance decay. To correct the baseline decay, we fit the baseline decay in 100–500 ms before a DCS with a linear regression line and subtracted from the measured capacitance trace. With a baseline-corrected DCS, we calculated the fission pore conductance and diameter as described previously32.
Synaptotagmin 2 knockout mice
Mice lacking synaptotagmin 2 (Syt2) have been described previously35. In short, they were generated by replacing part of exon 2 through exon 7 of the Syt2 gene with a lacZ cDNA construct using homologous recombination. Mice for these experiments were obtained by heterozygous breeding using standard mouse husbandry procedures. Genotyping was performed by PCR, which was previously confirmed to correlate with Syt2 expression by immunoblotting. In some experiments, we confirmed by fluorescence imaging that Syt2−/− mice did not show any immunoreactivity to antibodies raised against Syt2 (data not shown, but see ref. 35).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institute of Neurological Disorders and Stroke Intramural Research Program of the US National Institutes of Health.
Footnotes
Note: Supplementary information is available on the Nature Neuroscience website.
AUTHOR CONTRIBUTIONS
X.-S.W. conducted the double-patch experiments for Figure 2 and many of the capacitance recordings for other figures. B.D.M. initiated the project, designed and conducted capacitance experiments and helped write the paper. J.X. and J.F. conducted capacitance experiments. L.X. performed the simulations. E.M. and R.A. provided the Syt2−/− mice. L.B. maintained the animal colonies and L.-G.W. supervised the project, designed experiments and wrote the paper.
References
- 1.Wu LG, Ryan TA, Lagnado L. Modes of vesicle retrieval at ribbon synapses, calyx-type synapses and small central synapses. J. Neurosci. 2007;27:11793–11802. doi: 10.1523/JNEUROSCI.3471-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.von Gersdorff H, Matthews G. Inhibition of endocytosis by elevated internal calcium in a synaptic terminal. Nature. 1994;370:652–655. doi: 10.1038/370652a0. [DOI] [PubMed] [Google Scholar]
- 3.Sankaranarayanan S, Ryan TA. Calcium accelerates endocytosis of vSNAREs at hippocampal synapses. Nat. Neurosci. 2001;4:129–136. doi: 10.1038/83949. [DOI] [PubMed] [Google Scholar]
- 4.Neves G, Gomis A, Lagnado L. Calcium influx selects the fast mode of endocytosis in the synaptic terminal of retinal bipolar cells. Proc. Natl. Acad. Sci. USA. 2001;98:15282–15287. doi: 10.1073/pnas.261311698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Poskanzer KE, Fetter RD, Davis GW. Discrete residues in the c(2)b domain of synaptotagmin I independently specify endocytic rate and synaptic vesicle size. Neuron. 2006;50:49–62. doi: 10.1016/j.neuron.2006.02.021. [DOI] [PubMed] [Google Scholar]
- 6.Wu W, Xu J, Wu XS, Wu LG. Activity-dependent acceleration of endocytosis at a central synapse. J. Neurosci. 2005;25:11676–11683. doi: 10.1523/JNEUROSCI.2972-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu LG, Betz WJ. Nerve activity, but not intracellular calcium, determines the time course of endocytosis at the frog neuromuscular junction. Neuron. 1996;17:769–779. doi: 10.1016/s0896-6273(00)80208-1. [DOI] [PubMed] [Google Scholar]
- 8.Artalejo CR, Henley JR, McNiven MA, Palfrey HC. Rapic endocytosis coupled to exocytosis in adrenal chromaffin cells involves Ca2+, GTP, and dynamin, but not clathrin. Proc. Natl. Acad. Sci. USA. 1995;92:8328–8332. doi: 10.1073/pnas.92.18.8328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Granseth B, Odermatt B, Royle SJ, Lagnado L. Clathrin-mediated endocytosis is the dominant mechanism of vesicle retrieval at hippocampal synapses. Neuron. 2006;51:773–786. doi: 10.1016/j.neuron.2006.08.029. [DOI] [PubMed] [Google Scholar]
- 10.Jockusch WJ, Praefcke GJ, McMahon HT, Lagnado L. Clathrin-dependent and clathrin-independent retrieval of synaptic vesicles in retinal bipolar cells. Neuron. 2005;46:869–878. doi: 10.1016/j.neuron.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 11.Richards DA, Guatimosim C, Betz WJ. Two endocytic recycling routes selectively fill two vesicle pools in frog motor nerve terminals. Neuron. 2000;27:551–559. doi: 10.1016/s0896-6273(00)00065-9. [DOI] [PubMed] [Google Scholar]
- 12.Holt M, Cooke A, Wu MM, Lagnado L. Bulk membrane retrieval in the synaptic terminal of retinal bipolar cells. J. Neurosci. 2003;23:1329–1339. doi: 10.1523/JNEUROSCI.23-04-01329.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomas P, Lee AK, Wong JG, Almers W. A triggered mechanism retrieves membrane in seconds after Ca2+-stimulated exocytosis in single pituitary cells. J. Cell Biol. 1994;124:667–675. doi: 10.1083/jcb.124.5.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Renden R, von Gersdorff H. Synaptic vesicle endocytosis at a CNS nerve terminal: faster kinetics at physiological temperatures and increased endocytotic capacity during maturation. J. Neurophysiol. 2007;98:3349–3359. doi: 10.1152/jn.00898.2007. [DOI] [PubMed] [Google Scholar]
- 15.Gandhi SP, Stevens CF. Three modes of synaptic vesicular recycling revealed by single-vesicle imaging. Nature. 2003;423:607–613. doi: 10.1038/nature01677. [DOI] [PubMed] [Google Scholar]
- 16.Smith C, Neher E. Multiple forms of endocytosis in bovine adrenal chromaffin cells. J. Cell Biol. 1997;139:885–894. doi: 10.1083/jcb.139.4.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Balaji J, Ryan TA. Single-vesicle imaging reveals that synaptic vesicle exocytosis and endocytosis are coupled by a single stochastic mode. Proc. Natl. Acad. Sci. USA. 2007;104:20576–20581. doi: 10.1073/pnas.0707574105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beutner D, Voets T, Neher E, Moser T. Calcium dependence of exocytosis and endocytosis at the cochlear inner hair cell afferent synapse. Neuron. 2001;29:681–690. doi: 10.1016/s0896-6273(01)00243-4. [DOI] [PubMed] [Google Scholar]
- 19.Ceccarelli B, Hurlbut WP. Ca2+-dependent recycling of synaptic vesicles at the frog neuromuscular junction. J. Cell Biol. 1980;87:297–303. doi: 10.1083/jcb.87.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Henkel AW, Betz WJ. Monitoring of black widow spider venom (BWSV) induced exo- and endocytosis in living frog motor nerve terminals with FM1–43. Neuropharmacology. 1995;34:1397–1406. doi: 10.1016/0028-3908(95)00126-q. [DOI] [PubMed] [Google Scholar]
- 21.Ramaswami M, Krishnan KS, Kelly RB. Intermediates in synaptic vesicle recycling revealed by optical imaging of Drosophila neuromuscular junctions. Neuron. 1994;13:363–375. doi: 10.1016/0896-6273(94)90353-0. [DOI] [PubMed] [Google Scholar]
- 22.Marks B, McMahon HT. Calcium triggers calcineurin-dependent synaptic vesicle recycling in mammalian nerve terminals. Curr. Biol. 1998;8:740–749. doi: 10.1016/s0960-9822(98)70297-0. [DOI] [PubMed] [Google Scholar]
- 23.Cousin MA, Robinson PJ. Ba2+ does not support synaptic vesicle retrieval in rat cerebrocortical synaptosomes. Neurosci. Lett. 1998;253:1–4. doi: 10.1016/s0304-3940(98)00610-7. [DOI] [PubMed] [Google Scholar]
- 24.Gad H, Low P, Zotova E, Brodin L, Shupliakov O. Dissociation between Ca2+-triggered synaptic vesicle exocytosis and clathrin-mediated endocytosis at a central synapse. Neuron. 1998;21:607–616. doi: 10.1016/s0896-6273(00)80570-x. [DOI] [PubMed] [Google Scholar]
- 25.Balaji J, Armbruster M, Ryan TA. Calcium control of endocytic capacity at a CNS synapse. J. Neurosci. 2008;28:6742–6749. doi: 10.1523/JNEUROSCI.1082-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.von Gersdorff H, Borst JG. Short-term plasticity at the calyx of held. Nat. Rev. Neurosci. 2002;3:53–64. doi: 10.1038/nrn705. [DOI] [PubMed] [Google Scholar]
- 27.Yamashita T, Hige T, Takahashi T. Vesicle endocytosis requires dynamin-dependent GTP hydrolysis at a fast CNS synapse. Science. 2005;307:124–127. doi: 10.1126/science.1103631. [DOI] [PubMed] [Google Scholar]
- 28.Xu J, et al. GTP-independent rapid and slow endocytosis at a central synapse. Nat. Neurosci. 2008;11:45–53. doi: 10.1038/nn2021. [DOI] [PubMed] [Google Scholar]
- 29.Wu XS, et al. The origin of quantal size variation: vesicular glutamate concentration plays a significant role. J. Neurosci. 2007;27:3046–3056. doi: 10.1523/JNEUROSCI.4415-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sätzler K, et al. Three-dimensional reconstruction of a calyx of Held and its postsynaptic principal neuron in the medial nucleus of the trapezoid body. J. Neurosci. 2002;22:10567–10579. doi: 10.1523/JNEUROSCI.22-24-10567.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kushmerick C, Renden R, von Gersdorff H. Physiological temperatures reduce the rate of vesicle pool depletion and short-term depression via an acceleration of vesicle recruitment. J. Neurosci. 2006;26:1366–1377. doi: 10.1523/JNEUROSCI.3889-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu W, Wu LG. Rapid bulk endocytosis and its kinetics of fission pore closure at a central synapse. Proc. Natl. Acad. Sci. USA. 2007;104:10234–10239. doi: 10.1073/pnas.0611512104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Artalejo CR, Elhamdani A, Palfrey HC. Calmodulin is the divalent cation receptor for rapid endocytosis, but not exocytosis, in adrenal chromaffin cells. Neuron. 1996;16:195–205. doi: 10.1016/s0896-6273(00)80036-7. [DOI] [PubMed] [Google Scholar]
- 34.Neher E, Sakaba T. Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron. 2008;59:861–872. doi: 10.1016/j.neuron.2008.08.019. [DOI] [PubMed] [Google Scholar]
- 35.Sun J, et al. A dual-Ca2+-sensor model for neurotransmitter release in a central synapse. Nature. 2007;450:676–682. doi: 10.1038/nature06308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu J, Wu LG. The decrease in the presynaptic calcium current is a major cause of short-term depression at a calyx-type synapse. Neuron. 2005;46:633–645. doi: 10.1016/j.neuron.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 37.Borst JGG, Sakmann B. Calcium influx and transmitter release in a fast CNS synapse. Nature. 1996;383:431–434. doi: 10.1038/383431a0. [DOI] [PubMed] [Google Scholar]
- 38.Tadross MR, Dick IE, Yue DT. Mechanism of local and global Ca2+ sensing by calmodulin in complex with a Ca2+ channel. Cell. 2008;133:1228–1240. doi: 10.1016/j.cell.2008.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kopp-Scheinpflug C, Tolnai S, Malmierca MS, Rubsamen R. The medial nucleus of the trapezoid body: comparative physiology. Neuroscience. 2008;154:160–170. doi: 10.1016/j.neuroscience.2008.01.088. [DOI] [PubMed] [Google Scholar]
- 40.Malgaroli A, et al. Presynaptic component of long-term potentiation visualized at individual hippocampal synapses. Science. 1995;268:1624–1628. doi: 10.1126/science.7777862. [DOI] [PubMed] [Google Scholar]
- 41.Murthy VN, Stevens CF. Reversal of synaptic vesicle docking at central synapses. Nat. Neurosci. 1999;2:503–507. doi: 10.1038/9149. [DOI] [PubMed] [Google Scholar]
- 42.Ryan TA, Reuter H, Smith SJ. Optical detection of a quantal presynaptic membrane turnover. Nature. 1997;388:478–482. doi: 10.1038/41335. [DOI] [PubMed] [Google Scholar]
- 43.Pyle JL, Kavalali ET, Piedras-Renteria ES, Tsien RW. Rapid reuse of readily releasable pool vesicles at hippocampal synapses. Neuron. 2000;28:221–231. doi: 10.1016/s0896-6273(00)00098-2. [DOI] [PubMed] [Google Scholar]
- 44.Rizzoli SO, Betz WJ. The structural organization of the readily releasable pool of synaptic vesicles. Science. 2004;303:2037–2039. doi: 10.1126/science.1094682. [DOI] [PubMed] [Google Scholar]
- 45.Nucifora PG, Fox AP. Barium triggers rapid endocytosis in calf adrenal chromaffin cells. J. Physiol. (Lond.) 1998;508:483–494. doi: 10.1111/j.1469-7793.1998.483bq.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Clayton EL, Evans GJ, Cousin MA. Activity-dependent control of bulk endocytosis by protein dephosphorylation in central nerve terminals. J. Physiol. (Lond.) 2007;585:687–691. doi: 10.1113/jphysiol.2007.137539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nicholson-Tomishima K, Ryan TA. Kinetic efficiency of endocytosis at mammalian CNS synapses requires synaptotagmin I. Proc. Natl. Acad. Sci. USA. 2004;101:16648–16652. doi: 10.1073/pnas.0406968101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu J, Mashimo T, Sudhof TC. Synaptotagmin-1, -2 and -9: Ca2+ sensors for fast release that specify distinct presynaptic properties in subsets of neurons. Neuron. 2007;54:567–581. doi: 10.1016/j.neuron.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 49.Augustine GJ, Neher E. Calcium requirements for secretion in bovine chromaffin cells. J. Physiol. (Lond.) 1992;450:247–271. doi: 10.1113/jphysiol.1992.sp019126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sankaranarayanan S, Ryan TA. Real-time measurements of vesicle-SNARE recycling in synapses of the central nervous system. Nat. Cell Biol. 2000;2:197–204. doi: 10.1038/35008615. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.