

Abstract
Ethnopharmacological relevance
The rate of production of reactive oxygen species (ROS) is determined by mitochondrial metabolic rate. In turn, excessive ROS damage mitochondrial function, which is linked to aging and neurodegenerative conditions. One possible path to prevent oxidative stress could be achieved by reducing mitochondrial respiration in favor of less efficient ATP production via glycolysis. Such a shift in energy metabolism is known as the ‘Warburg effect’. Geissoschizine methyl ether (GM) is one of the active components responsible for the psychotropic effects of Yokukansan, an herbal preparation widely used in China and Japan.
Aim of the study
GM protects neurons from glutamate-induced oxidative cytotoxicity through regulating mitochondrial function and suppressing ROS generation. We investigated the protective mechanism of GM against glutamate-induced oxidative stress in neuronal cells.
Materials and methods
The current study was performed on primary neurons and HT22 cells, a hippocampus neuronal cell line. Cell viability was measured by Calcein AM assay. H2DCFDA staining was used for intracellular ROS measurement. GSH level was measured using the GSH-Glo™ luminescence-based assay. Mitochondrial respiration and glycolysis were measured by the Seahorse Bioscience XFe 96 Extracellular Flux Analyzer. Protein levels were analyzed by western blot analysis.
Results
GM prevented glutamate-induced cytotoxicity in an HT-22 neuronal cell line even with a 9-hour exposure delay. GM blocked glutamate-induced intracellular ROS accumulation through suppressing mitochondrial respiration. Further, we found that GM up-regulated glycolysis and the pentose-phosphate pathway, which is involved in the production of intracellular reducing agent, NADPH. In addition, GM protected primary cortical neurons from both glutamate and buthioninesulfoximine toxicity.
Conclusion
GM prevents glutamate-induced oxidative damage through reducing mitochondrial respiration, which further suppresses ROS generation. In addition, GM up-regulates glycolysis which compensate for the energy depletion induced by mitochondrial respiration inhibition. Overall, our study is the first to report that GM protects neurons from oxidative toxicity by shifting energy metabolism from mitochondrial respiration to glycolysis.
Graphical abstract
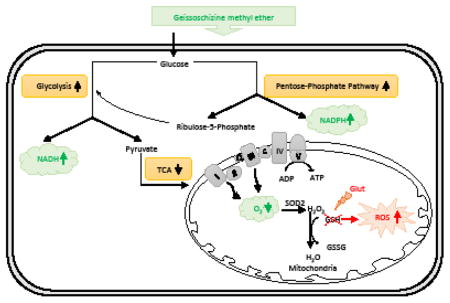
1. Introduction
Mitochondria plays dual roles in cell survival and death. Mitochondrial oxidative phosphorylation is essential for adenosine 5′-triphosphate (ATP) generation (Brookes et al., 2004). During the course of normal oxidative phosphorylation, 0.4–4% of all oxygen consumed is converted into free radicals (Boveris, 1984; Shigenaga et al., 1994). The rate of production of reactive oxygen species (ROS) is essentially determined by the mitochondrial metabolic rate although ROS production can clearly be altered by respiratory chain function and antioxidant concentration (Melov, 2000). Oxidative stress is caused by an imbalance of ROS production and antioxidative capacity. As a vulnerable target of ROS, mitochondrial dysfunction induced by oxidative stress has been linked to aging and neurodegenerative conditions (Lin and Beal, 2006). Therefore, the complexity of mitochondrial ROS metabolism suggests that interventions such as the administration of one or a few antioxidants may be too simplistic. One possible path to suppress oxidative stress could be achieved by reducing mitochondrial oxidative respiration in favor of less efficient ATP production via glucose fermentation. Such a shift in energy metabolism is known as the ‘Warburg effect’, which can reduce the generation of ROS (Bading, 2013).
Glutamate-induced oxidative stress is a well-known pathological factor of neuronal cell death in various neurodegenerative conditions, including ischemia, epileptic seizures, Alzheimer’s diseases, and Parkinson’s diseases (Coyle and Puttfarcken, 1993; Manzanero et al., 2013a; Manzanero et al., 2013b; Ozkul et al., 2007). By blocking cysteine uptake, the synthesis of glutathione (GSH), an important intracellular antioxidant, is inhibited, which further leads to the accumulation of ROS (Choi, 1994; Coyle and Puttfarcken, 1993). The ROS accumulation causes mitochondrial dysfunction and energy failure, which further leads to cell death (Fukui et al., 2009; Landshamer et al., 2008).
Yokukansan (YKS), a traditional herbal preparation, has been used as a remedy in the treatment of neurosis, insomnia, and night crying and irritability in children in China and Japan. It is approved by the Ministry of Health, Labour and Welfare of Japan as a prescription drug (Nishi et al., 2012). It consists of seven crude drugs, Uncariae Ramulus cum Uncis, Bupleuri Radix, Chuanxiong Rhizoma, Angelicae Sinensis Radix, Poria, Atractylodis Rhizoma, and Glycyrrhizae Radix et Rhizoma. Among them, Uncariae Ramulus cum Uncis (Uncaria hook) is botanically from the hooks of Uncaria rhynchophylla (Miq.) Miq. ex Havil. and U. sinensis (Oliv.) Havil. Uncaria hook has been traditionally used for the treatment of hypertension, dizziness, and stroke. Recently, YKS was reported to improve behavioral and psychological symptoms of dementia such as hallucinations, agitation, and aggressiveness in patients with Alzheimer’s disease, Parkinson’s disease and dementia (Hatano et al., 2014; Mizukami et al., 2009; Okahara et al., 2010). It shows protective effects against neuronal cell death induced by glutamate-mediated oxidative stress, and Uncaria hook was found to have the highest potency in the protection (Kawakami et al., 2011). Our previous study reported the neuroprotective effects of 11 oxindole and indole alkaloids, which were isolated from the hooks of Uncaria rhynchophylla (Miq.) Miq. ex Havil, against glutamate-induced oxidative toxicity in neuronal cells. Among them, geissoschizine methyl ether (GM) has the highest potency in the neuroprotection (Qi et al., 2014). Published study reported that GM could reach the brain by crossing the blood-brain barrier in rats following the oral administration of yokukansan. (Imamura et al., 2011). Also, GM was shown to be one of the active components responsible for the psychotropic effects of YKS in several studies (Morita et al., 2014; Nishi et al., 2012; Ueki et al., 2013). However, the underlying mechanisms of GM’s protective role have not been elucidated.
Using a glutamate-induced oxidative toxicity model in a mouse hippocampal neuronal cell line (HT-22 cells) and rat primary cortical neurons, we here investigated the protective mechanism of GM against glutamate-induced oxidative cytotoxicity.
2. Materials and Methods
2.1 Plant material and extraction
GM was isolated from the extract of the hook-bearing branches of U. sinensis (Qi et al., 2014). Its structure (Supplemental Fig 1) was characterized by comparison of its 1H-NMR, 13C -NMR (Supplemental Table and Supplemental Fig 2) and ESI-QTOF-MS/MS data (Supplemental Fig 3). The purity of GM, calculated by HPLC–UV at 245 nm with peak area normalization method, was 98.4% (Supplemental Fig 3). A MeOH extract of the hooks and stems of U. sinensis was prepared by percolation. The n-hexane-, CHCl3-, and n-BuOH-soluble fractions were obtained by partitioning the MeOH extract. According to UPLC analysis, the CHCl3-soluble fraction consisted of such indole alkaloids as GM, hirsuteine, and hirsutine.
2.2 Cell culture
HT-22 cells were the generous gift of Dr. David Schubert (Salk Institute, San Diego, CA). Cells were routinely grown in high glucose Dulbecco’s Modified Eagle Medium (HyClone, South Logan, UT) media supplemented with 10% fetal bovine serum (Altanta Biologicals, Flowery Branch, GA) at 37°C in 5% CO2 humid atmosphere. Passages 6 to 25 were used in the study. As previous reported (Sun et al., 2015), primary cortical neurons were collected from embryonic day 17 Sprague-Dawley rats. Briefly, cells were grown in minimal essential medium (ATCC, Manassas, VA) supplemented with 4.4 g/L glucose and 10% heat-inactivated horse serum (Life Technologies, Carlsbad, CA). After 1 day in vitro culture, cells were exposed to respective treatments.
2.3 Cell viability assay
HT-22 cells were cultured in 96-well plate overnight and then exposed to respective exposures. To perform cell viability assay, media was removed and cells were washed once with phosphate buffered saline (PBS). Cells were incubated with 1 μM Calcein AM (Molecular Probes, Grand Island, NY) in for 15 minutes at 37 °C. The nonfluorescent Calcein AM is converted to a green-fluorescent Calcein after acetoxymethyl ester hydrolysis by intracellular esterases. A Biotek Synergy H1 Hybrid plate reader (excitation 485 emission 530) was used for fluorescence measurement. For the primary cortical neurons, morphology and viability of the cells was imaged using fluorescent microscopy (EVOS® FL Auto Imaging System).
2.4 ROS detection
Intracellular ROS were detected by 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) (Molecular Probes, Grand Island, NY). H2DCFDA, a non-fluorescent dye, can be converted to the fluorescent DCF by ROS. As previous reported (Sun et al., 2015), cells were incubated with 10 μM H2DCFDA for 30 min at 37 °C after respective exposures. Mean fluorescence intensity (MFI) was measured by a Biotek Synergy H1 Hybrid plate reader (excitation 485, emission 530). DCF fluorescence was standardized based on a parallel measurement of cell viability.
2.5 GSH measurement
GSH level was measured using by a GSH-Glo™ GSH Assay (Promega, Madison, WI). A luciferin derivative was converted into luciferin in the presence of glutathione. As described in the manufacturer’s protocol, cells were incubated with Glo-GSHTM reagent for 30 min, followed by incubating with luciferin detection reagent. A Biotek Synergy H1 Hybrid plate reader was used to detect luminescence.
2.6 Mitochondrial respiration measurement
HT-22 cells were plated and cultured overnight in an XFe96 plate. After respective exposures, the media was changed 1 hour before the start of the mitochondrial respiration measurement (Seahorse Bioscience, North Billerica, MA). Oligomycin (1 μM), carbonilcyanide p-triflouromethoxyphenylhydrazone (FCCP, 0.5 μM), antimycin (1 μM) and rotenone (1 μM) (Sigma) were prepared with XFe96 media and loaded into the sensor cartridge. Injections of the components into the wells occurred at the time points specified. Oxygen consumption rate (OCR) was monitored using a Seahorse Bioscience XFe96 Extracellular Flux Analyzer. ATP production was measured using the third OCR measurement prior to addition of oligomycin subtracted from OCR after oligomycin injection. Maximal respiration was calculated using the maximal OCR after FCCP injection subtracted from OCR after rotenone and antimycin injection. To calculate spare capacity, the maximal OCR after FCCP injection was subtracted from the measurement prior to addition of oligomycin. Proton leak was measured using OCR after oligomycin injection subtracted from OCR after rotenone and antimycin injection.
2.7 Glycolysis measurement
HT-22 cells were plated at a density of 15000/well in an XFe96 plate. After respective exposures, glycolysis was measured by Seahorse Bioscience XFe96 Extracellular Flux Analyzer (Seahorse Bioscience, North Billerica, MA). Glucose (10 mM), oligomycin (1 μM) and 2-deoxy-glucose (2-DG, 50 mM) (Sigma) were diluted into XFe96 medium and loaded into the accompanying cartridge. Injections of the components into the wells occurred at the time points specified. Extracellular acidification rate (ECAR) was monitored using a Seahorse Bioscience XFe96 Extracellular Flux Analyzer. Non-glucose-derived ECAR was measured using the minimal measurement after addition of 2-DG. To calculate glycolysis, the maximal ECAR after glucose addition was subtracted from ECAR prior to glucose injection. Glycolytic reserve was measured using the measurement after oligomycin injection subtracted from ECAR after glucose addition. Glycolytic reserve capacity was calculated using the total ECAR for glycolysis and glycolytic reserve dividing by glycolysis measurement.
2.8 Western blot
Cells were lysed in radio-immunoprecipitation assay buffer with cocktail protease inhibitors. Blots were incubated with anti-G6PD antibody (1: 1000 dilution, Santa Cruz) at 4 °C overnight. Membranes were then probed with the fluorescence-conjugated secondary antibody (Li-cor Biotechnology, Lincoln, NE).β-actin was used as loading control (1:1000 dilution, Santa Cruz Biotechnology). Fluorescence was detected using Odyssey CLx imaging system, and UVP software was used for quantification of western blot signals.
2.9 Statistical analysis
The data were shown as means ± SEM. Statistical analyses were performed using one-way ANOVA with Tukey’s post hoc test. GraphPad PRISM 5.0 was used for statistical analyses.
3. Results
3.1 GM protects from GSH-depletion induced toxicity in both HT-22 cells and primary neurons
There was an 80% of HT-22 cells loss after a 24 h exposure of glutamate (5 mM). Simultaneous treatment of GM (20–100 μM) significantly ameliorated glutamate-induced cell death (Figure 1A). In a parallel experiment, tert-Butylhydroquinone, as a positive control, significantly possesses a neuroprotective against glutamate-induced cell death (Supplemental Fig 4) (Sun et al., 2015). We also determined the duration of treatment delay with GM at which protection would be observed. Cells were exposed to glutamate (5 mM) for 24 h. GM was applied at 0–9h after the glutamate exposure. Cell viability was measured at 24 h after glutamate exposure. Up to an 8 h treatment delay, GM prevented more than 50% of the cells from glutamate-induced oxidative toxicity. This protection was reduced when the exposure delay of GM was prolonged to 9 h (Figure 1B). As previous reported (Sun et al., 2015), exposure of primary rat cortical neurons to glutamate (5mM) for 24 h caused the disruption of neural networks and the shrinkage of cell bodies. Co-exposure to GM (50 μM) ameliorated glutamate-induced neuron death (Figure 2A). Glutamate led to a significant decrease in Calcein AM fluorescence, which was preserved by GM (Figure 2B). We also blocked GSH synthesis by using buthioninesulfoximine (BSO), an inhibitor of γ-glutamylcysteine synthetase. BSO exposure (500 μM) induced the morphological changes, which was consistent with Calcein AM data. Simultaneous exposure of GM (50 μM) rescued cells from BSO-induced cell death (Figure 2A and B). We conclude that the protection of GM on oxidative stress-induced cytotoxicity was not specific to a transformed cell line.
Figure 1. GM prevents glutamate-induced oxidative toxicity in HT-22 cells.
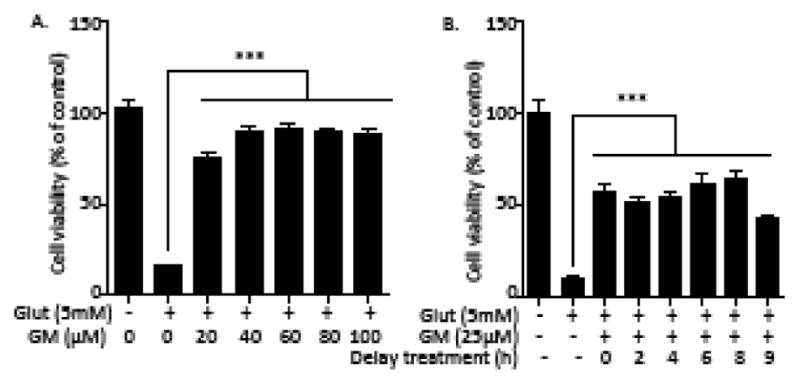
(A) HT-22 cells were exposed to glutamate (5 mM) and GM (20–100 μM) for 24 h. Cell viability was detected by a Calcein AM assay (n=8). (B) Cells were exposed to glutamate (5 mM) for 24 h. GM (25 μM) exposure was applied at 0, 2, 4, 6, 8, 9h after glutamate exposure. Cell viability was detected by aCalcein AM assay (n=8). Representative experiments were independently repeated three times. Results are reported as mean ± SEM. ***P < 0.001 compared with glutamate-treated cells (one-way ANOVA, Tukey’s test).
Figure 2. GM reduces both glutamate- and BSO- induced cell death in immature primary rat cortical neurons.

One-day-old primary cultures prepared from embryonic day 17 rats were exposed to glutamate (5 mM), BSO (500 μM) and GM (25, 50 μM) for 24 h. Phase contrast images and calcein AM staining fluorescence pictures were photographed. DAPI was used for nuclear staining. Representative experiments were independently repeated three times.
3.2 GM prevents glutamate-induced ROS generation
We measured ROS level by H2DCFDA assay. A 3-fold increase of ROS level was induced after a 9h exposure to glutamate. Application of GM at 0 and 6 h after glutamate prevented this ROS accumulation (Figure 3). This indicates that GM inhibits glutamate-induced intracellular ROS accumulation in HT-22 cells.
Figure 3. GM prevents glutamate-induced ROS accumulation in HT-22 cells.
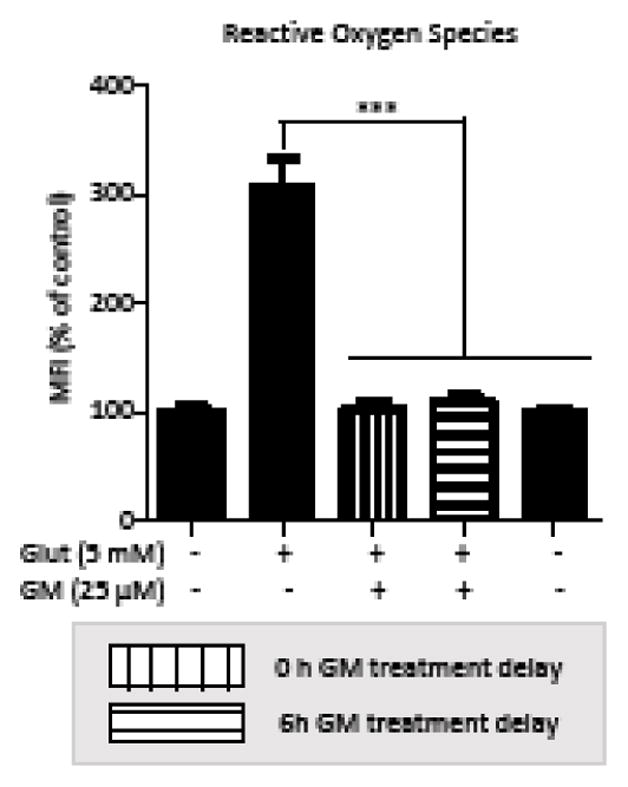
HT-22 cells were exposed to glutamate (5 mM) for 10 h. GM (25 μM) was applied at 0 or 6 h after glutamate exposure. ROS levels were detected by H2DCF and fluorescence was measured by plate reader (n=8). All experiments were repeated three times and the results indicate the mean ± SEM. ***P < 0.001 compared with glutamate-treated cells. (one-way ANOVA, Tukey’s test).
3.3 GM fails to prevent glutamate-induced GSH depletion and does not directly eliminate ROS
Since glutamate directly inhibit the synthesis of GSH, we first investigated if GM protected glutamate-induced cell death through preserving GSH level. Glutamate exposure profoundly reduced GSH level, which was not prevented by GM. Surprisingly, GM alone also induced a decrease of GSH level (Figure 4A). We then determined whether GM is an antioxidant and directly removed intracellular ROS. A 20 min exposure of hydrogen peroxide (H2O2) increased intracellular ROS level; co-exposure with GM failed to reduce ROS level (Figure 4B). These data indicate that GM does not prevent glutamate-induced GSH depletion, and itself does not directly eliminate ROS.
Figure 4. GM fails to block glutamate-induced GHS depletion and does not directly scavenge intracellular ROS.

(A) GSH levels were detected at 9 h after exposure to glutamate (5 mM) and GM (25 μM) (n=5). (B) HT-22 cells were exposed to H2O2 (50 μM) and GM (25 μM) for 20 min. ROS levels were detected by H2DCF and fluorescence was measured by plate reader (n=8). All experiments were repeated three times and the results indicate the mean ± SEM. ***P < 0.001 compared with control (one-way ANOVA, Tukey’s test).
3.4 GM suppresses mitochondrial respiration, and enhances glycolysis and pentose-phosphate pathway activities
Mitochondria are both targets and important sources of ROS (Balaban et al., 2005). Superoxide, a major intracellular ROS, is generated during normal mitochondrial respiration. Reversely, excessive ROS comprises mitochondrial function. Our previous study revealed that glutamate inhibited mitochondrial respiration (Sun et al., 2015). To determine if GM exposure attenuates the exacerbation of mitochondrial respiration under glutamate toxicity, we measured mitochondrial respiration after glutamate and GM exposure. After a 10 h exposure of glutamate (5mM), maximal respiration and spare capacity were decreased by 35 %. Surprisingly, co-exposure of GM induced a significant inhibition of mitochondrial respiration, including ATP production (Figure 5A). Next, we investigated the effect of GM treatment alone on mitochondrial respiration. A 24h exposure of GM (5–50 μM) induced a dose-dependent suppression of mitochondria respiration, as measured by ATP production, maximal respiration, spare capacity and proton leak (Figure 5B).
Figure 5. GM suppresses mitochondrial respiration.

After a 10 h exposure to glutamate (5 mM) and GM (25 μM) (A) or a 24 h exposure to GM (5–50 μM) (B), OCR was recorded by a Seahorse XFe96 flux analyzer (n=8). OCR recording at baseline and subsequent treatment of oligomycin (1μM), FCCP (0.5 μM), and a rotenone and antimycin mixture (1 μM). ATP production, spare capacity, maximum respiration and proton leak were calculated. All experiments were repeated three times and the results indicate the mean ± SEM. *P < 0.05, **P < 0.01, *** P < 0.001 compared with control. #P < 0.05, ### P < 0.001 compared with glutamate-exposed cells. (one-way ANOVA, Tukey’s test).
Previous studies support the notion that the metabolic needs of active neural tissue are met, at least partially, by non-oxidative glucose metabolism (Bélanger et al., 2011). Thus, we evaluated the effect of GM on non-oxidative glucose metabolism. First, we measured glycolytic activity after glutamate and GM exposure. Glycolytic activity was not changed with a 10 h exposure of HT-22 cells to glutamate. GM treatment up-regulated glycolysis accompanied with a reduced glycolytic reserve capacity (Figure 6A). A dose-dependent elevation of glycolysis was observed after a 24 h exposure of GM alone (Figure 6B). With a high concentration of GM exposure (25 and 50 μM), glycolytic reserve capacity was significantly decreased compared to control. This indicates that elevation of glycolytic activity by GM might compensate for the energy depletion caused by mitochondrial suppression.
Figure 6. GM enhances glycolytic activity.

After a 10 h exposure to glutamate (5 mM) and GM (25 μM) (A) or a 24 h exposure to GM (5–50 μM) (B), ECAR was recorded by a Seahorse XFe96 flux analyzer (n=8). ECAR recording at baseline and subsequent exposure toglucose (10 mM), oligomycin(5 μM) and 2-DG (100 mM). Glycolysis, glycolytic reserve, glycolytic reserve capacity and non-glucose-derived ECAR were calculated. All experiments were repeated three times and the results indicate the mean ± SEM. *P < 0.05, **P < 0.01, *** P < 0.001 compared with control. #P < 0.05, ### P < 0.001 compared with glutamate-treated cells. (one-way ANOVA, Tukey’s test).
The pentose-phosphate pathway is a metabolic pathway parallel to glycolysis that generates NADPH, hence contributing to neuronal protection against oxidative damage (Baquer et al., 1988). We evaluated the regulation of GM on the pentose-phosphate pathway by measuring the expression levels of G6PD, the first and rate-controlling enzyme of this pathway. There was a slight increase in G6PD level after a 12 h exposure to GM; a 1.5-fold increase in G6PD expression was observed at 24 hours after GM exposure (Figure 7).
Figure 7. GM up-regulates G6PD expression.
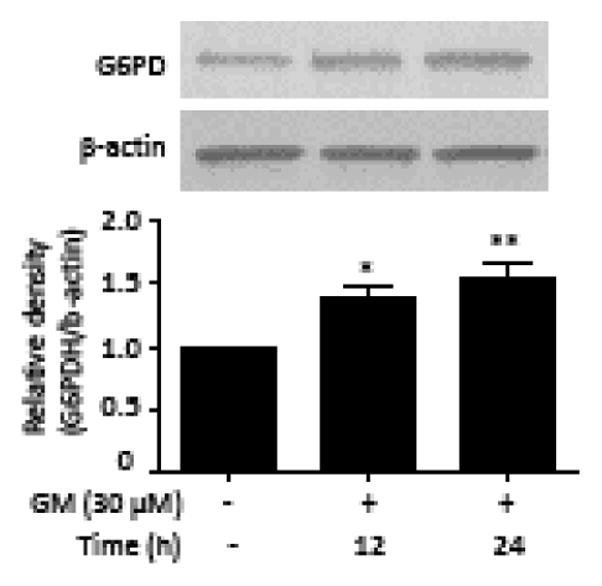
HT-22 cells were exposed to 30 μM GM. Samples were collected at 12, 24 h after GM exposure. Cell extracts were subjected to immunoblot with antibodies specific for G6PD. Quantitation of G6PD was normalized to β-actin. Bars represent normalized relative densities plotted as mean ± SEM calculated from 5 independent blots. *P < 0.05, **P < 0.01 compared with control group (one-way ANOVA, Tukey’s test).
4. Discussion
Free radicals, typically generated from mitochondrial respiration, cause oxidative damage of various intracellular components. In mitochondrial diseases, free radicals may induce modifications of subunits involved in oxidative phosphorylation that result in an increased propensity for the respiratory chain to produce free radicals, and hence, overwhelm the mitochondria’s ability to protect itself from endogenous oxidative stress (Melov, 2000). One potential strategy to reduce oxidative stress is to reduce mitochondrial respiration at the expense of less efficient ATP production via glycolysis. Such a shift in energy metabolism has been observed in cancer cells and is known as the ‘Warburg effect’ (aerobic glycolysis) (Upadhyay et al., 2013). Even though it is illogical for actively metabolic neurons to switch to an inefficient way for energy supplement, such a transition, indeed, is beneficial for preventing ROS generation. As it turns out, some brain areas and cell types can also process glucose in this way. Glucose utilization by the brain is in excess of oxygen consumption by about 10%, implying a non-oxidative use of glucose entering the brain (Magistretti, 2014). We observed the Warburg effect after exposing neuronal cells to GM. A popular hypothesis to explain aerobic glycolysis in cancer cells is that this metabolic phenotype confers a unique advantage for survival and proliferation in the tumor microenvironment (Justus et al., 2015). Interestingly, we monitored the toxicity of GM, and found that GM significantly suppressed cell proliferation without inducing cytotoxicity (Supplemental Fig 4). This indicates that the effect of aerobic glycolysis induced by GM on neurons is different from the impact of aerobic glycolysis on tumorigenesis. Taken together, these data indicate that aerobic glycolysis tightly regulates intracellular redox homeostasis, which is closely linked to resistance against oxidative stimuli. Although GM inhibits mitochondrial ATP production, we should note that the proton leak is also significantly reduced by GM (Figure 5). Oxidative phosphorylation is incompletely coupled, since protons can leak across the inner membrane and relieve mitochondrial membrane potential independently of ATP synthase (Divakaruni et al., 2011). The reduction of proton leak promotes the coupling efficiency for ATP synthesis.
The pentose-phosphate pathway is also involved in glucose utilization. In neurons, the pentose–phosphate pathway is very active, and the rate of glucose oxidized through the pentose–phosphate pathway is about 14% of total glucose metabolism (Rodriguez-Rodriguez et al., 2013). G6PD is the rate-limiting enzyme in the pentose-phosphate pathway (Tian et al., 1999). A significant up-regulation of G6PD expression was observed after a 24h exposure to GM. This indicates that GM enhances pentose-phosphate pathway activity. The pentose-phosphate pathway serves as an entry into glycolysis. Therefore, up-regulation of glycolytic activity after GM exposure might also attribute to the enhanced pentose-phosphate pathway activity. The critical role of the pentose-phosphate pathway in protection against oxidative stress has been demonstrated in neurons and astrocytes (Ben-Yoseph et al., 1996; Bolaños et al., 2008). Besides supplying ribose-5-phosphate for nucleic acid biosynthesis, the pentose-phosphate pathway is a key regulator of cytosolic NADPH regeneration. NADPH is used as a cofactor for the reduction of GSH from its oxidized form (GSSG) (Stanton, 2012). NADPH provides reducing equivalents for biosynthetic reactions and the oxidation-reduction involved in protecting against the toxicity of ROS (Nayernia et al., 2014). Notably, our results showed that GM failed to prevent glutamate-induced GSH depletion. Since glutamate reduces the total amount of intracellular GSH through lowering intracellular cysteine concentration, we speculate that GM has no effect on cysteine uptake and simply an up-regulation of NADPH by GM can not reverse the effect of glutamate on GSH synthesis. Interestingly, GM exposure alone also significantly reduced intracellular GSH level. GM exposure moderates the production of mitochondrial superoxide, which also lowers the demand for GSH consumption for superoxide detoxification. Further, there may be a compensatory reduction of intracellular GSH synthesis in response to the decrease of GSH consumption. This provides a potential explanation of how GM exposure alone decreases GSH level.
A major limitation is that the effect of GM on in vivo neuron-compromising conditions is not elucidated in the current study. Recently published study reported that serotonin receptors are associated with specific binding sites for GM in rat brains (Mizoguchi et al., 2014). It also has shown that GM ameliorated isolation stress-induced aggressive behaviors by stimulating serotonin1A receptors (Nishi et al., 2012). Therefore, whether GM-induced ‘Warburg effect contributes to its neurotrophic effects needs to be addressed in future studies. Notably, a plant contains hundreds of compounds that could contribute to its protective effect. As a preliminary test, the MeOH extract of the hooks and stems of U. sinensis, together with the n-hexane-, and n-BuOH-soluble fractions obtained by partitioning the crude MeOH extract, were examining for protective activity against glutamate-induced cytotoxicity by a Calcein AM cell viability assay.
The MeOH extract, and the n-hexane- and n-BuOH-soluble fractions exhibited little protective activity at a concentration of 10 μg/mL, and cytotoxic effects appeared at higher concentrations. Only the CHCl3-soluble fraction was found to be significantly active, with the IC50 value of the CHCl3-soluble fraction being 5.7 μg/mL, suggesting that the indole alkaloids that were the major ingredients in the CHCl3-soluble fraction possess protective effects against glutamate-induced cytotoxicity. The current study evaluated GM protective effect because it is a major indole alkaloid found in the CHCl3-soluble fraction and has highest potency (Qi et al., 2014). However, it is unknown if Uncaria hook or other alkaloids extracted from Uncaria hook exert neuroprotective effects through the same mechanism, which needs to be addressed in future studies.
Our study demonstrates the protection of GM against oxidative toxicity in both neuronal cell line and primary cortical neurons. In addition, this protection is observed with a 9 h exposure delay of GM through blocking glutamate-induced intracellular ROS accumulation in HT-22 cells. GM suppresses mitochondrial respiration, which contributes to limiting the production of ROS. Further, we found that GM up-regulated glycolysis and the pentose-phosphate pathway, which is involved in the production of NADPH. Overall, this study is the first to demonstrate a novel neuro-protective mechanism of GM against oxidative stress by regulating intracellular metabolism during oxidative challenge.
Acknowledgments
Stephanie Rellick is gratefully acknowledged for proof reading. Saumyendra Sarkar and Sujung Jun are gratefully acknowledged for their assistance with primary cortical culture. Candice Brown is gratefully acknowledged for offering the used of EVOS® FL Auto Imaging System.
Sources of Funding
This work was supported by NIH P01 AG022550, P01 AG027956, P20 GM109098 and U54GM104942 and the National Natural Science Foundation of China (NSFC) (No. 81173544).
List of Abbreviations
- 2-DG
2-deoxy-glucose
- ATP
adenosine 5′-triphosphate
- BSO
buthionine sulfoximine
- DAPI
4′, 6-diamidino-2-phenylindole
- ECAR
extracellular acidification rate
- FCCP
carbonilcyanide p-triflouromethoxyphenylhydrazone
- GM
geissoschizine methyl ether
- GSH
glutathione
- H2DCFDA
2′, 7′-dichlorodihydrofluorescein diacetate
- H2O2
hydrogen peroxide
- MFI
mean fluorescence intensity
- O2·−
superoxide
- OCR
oxygen consumption rate
- PBS
phosphate buffered saline
- ROS
reactive oxygen species
- YKS
Yokukansan
Footnotes
Conflict of interest
None
Authorship Contributions
Participated in research design: Sun, Ren, Yuan, Simpkins
Conducted experiments: Sun, Ren, Qi
Performed data analysis: Sun, Ren, Simpkins
Wrote or contributed to the writing of the manuscript: Sun, Ren, Yuan, Simpkins
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting galley proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bading H. Nuclear calcium signalling in the regulation of brain function. Nature reviews. Neuroscience. 2013;14:593–608. doi: 10.1038/nrn3531. [DOI] [PubMed] [Google Scholar]
- Balaban RS, Nemoto S, Finkel T. Mitochondria, Oxidants, and Aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Baquer NZ, Hothersall JS, McLean P. Function and regulation of the pentose phosphate pathway in brain. Current topics in cellular regulation. 1988;29:265–289. [PubMed] [Google Scholar]
- Bélanger M, Allaman I, Magistretti P. Brain Energy Metabolism: Focus on Astrocyte-Neuron Metabolic Cooperation. Cell Metabolism. 2011;14:724–738. doi: 10.1016/j.cmet.2011.08.016. [DOI] [PubMed] [Google Scholar]
- Ben-Yoseph O, Boxer PA, Ross BD. Assessment of the role of the glutathione and pentose phosphate pathways in the protection of primary cerebrocortical cultures from oxidative stress. Journal of neurochemistry. 1996;66:2329–2337. doi: 10.1046/j.1471-4159.1996.66062329.x. [DOI] [PubMed] [Google Scholar]
- Bolaños JP, Delgado-Esteban M, Herrero-Mendez A, Fernandez-Fernandez S, Almeida A. Regulation of glycolysis and pentose–phosphate pathway by nitric oxide: Impact on neuronal survival. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 2008;1777:789–793. doi: 10.1016/j.bbabio.2008.04.011. [DOI] [PubMed] [Google Scholar]
- Boveris A. Determination of the production of superoxide radicals and hydrogen peroxide in mitochondria. Methods in enzymology. 1984;105:429–435. doi: 10.1016/s0076-6879(84)05060-6. [DOI] [PubMed] [Google Scholar]
- Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. American journal of physiology. Cell physiology. 2004;287:C817–33. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- Choi DW. Chapter 6 Glutamate receptors and the induction of excitotoxic neuronal death. Progress in brain research. 1994;100:47–51. doi: 10.1016/s0079-6123(08)60767-0. [DOI] [PubMed] [Google Scholar]
- Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science (New York, NY) 1993;262:689–695. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- Fukui M, Song J, Choi J, Choi HJ, Zhu BT. Mechanism of glutamate-induced neurotoxicity in HT22 mouse hippocampal cells. European journal of pharmacology. 2009;617:1–11. doi: 10.1016/j.ejphar.2009.06.059. [DOI] [PubMed] [Google Scholar]
- Hatano T, Hattori N, Kawanabe T, Terayama Y, Suzuki N, Iwasaki Y, Fujioka T Yokukansan Parkinson’s Disease Study Group. An exploratory study of the efficacy and safety of yokukansan for neuropsychiatric symptoms in patients with Parkinson’s disease. Journal of neural transmission (Vienna, Austria : 1996) 2014;121:275–281. doi: 10.1007/s00702-013-1105-y. [DOI] [PubMed] [Google Scholar]
- Imamura S, Tabuchi M, Kushida H, Nishi A, Kanno H, Yamaguchi T, Sekiguchi K, Ikarashi Y, Kase Y. The blood-brain barrier permeability of geissoschizine methyl ether in Uncaria hook, a galenical constituent of the traditional Japanese medicine yokukansan. Cellular and molecular neurobiology. 2011;31:787–793. doi: 10.1007/s10571-011-9676-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justus CR, Sanderlin EJ, Yang LV. Molecular Connections between Cancer Cell Metabolism and the Tumor Microenvironment. International journal of molecular sciences. 2015;16:11055–11086. doi: 10.3390/ijms160511055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami Z, Kanno H, Ikarashi Y, Kase Y. Yokukansan, a kampo medicine, protects against glutamate cytotoxicity due to oxidative stress in PC12 cells. Journal of ethnopharmacology. 2011;134:74–81. doi: 10.1016/j.jep.2010.11.063. [DOI] [PubMed] [Google Scholar]
- Landshamer S, Hoehn M, Barth N, Duvezin-Caubet S, Schwake G, Tobaben S, Kazhdan I, Becattini B, Zahler S, Vollmar A, Pellecchia M, Reichert A, Plesnila N, Wagner E, Culmsee C. Bid-induced release of AIF from mitochondria causes immediate neuronal cell death. Cell death and differentiation. 2008;15:1553–1563. doi: 10.1038/cdd.2008.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Magistretti P. Synaptic Plasticity and the Warburg Effect. Cell Metabolism. 2014;19:4–5. doi: 10.1016/j.cmet.2013.12.012. [DOI] [PubMed] [Google Scholar]
- Manzanero S, Santro T, Arumugam TV. Neuronal oxidative stress in acute ischemic stroke: Sources and contribution to cell injury. Neurochemistry international. 2013a;62:712–718. doi: 10.1016/j.neuint.2012.11.009. [DOI] [PubMed] [Google Scholar]
- Manzanero S, Santro T, Arumugam TV. Neuronal oxidative stress in acute ischemic stroke: Sources and contribution to cell injury. Neurochemistry international. 2013b;62:712–718. doi: 10.1016/j.neuint.2012.11.009. [DOI] [PubMed] [Google Scholar]
- Melov S. Mitochondrial oxidative stress. Physiologic consequences and potential for a role in aging. Annals of the New York Academy of Sciences. 2000;908:219–225. doi: 10.1111/j.1749-6632.2000.tb06649.x. [DOI] [PubMed] [Google Scholar]
- Mizoguchi K, Kushida H, Kanno H, Igarashi Y, Nishimura H, Ikarashi Y, Kase Y. Specific binding and characteristics of geissoschizine methyl ether, an indole alkaloid of Uncaria Hook, in the rat brain. Journal of ethnopharmacology. 2014;158(Part A):264–270. doi: 10.1016/j.jep.2014.10.015. [DOI] [PubMed] [Google Scholar]
- Mizukami K, Asada T, Kinoshita T, Tanaka K, Sonohara K, Nakai R, Yamaguchi K, Hanyu H, Kanaya K, Takao T, Okada M, Kudo S, Kotoku H, Iwakiri M, Kurita H, Miyamura T, Kawasaki Y, Omori K, Shiozaki K, Odawara T, Suzuki T, Yamada S, Nakamura Y, Toba K. A randomized cross-over study of a traditional Japanese medicine (kampo), yokukansan, in the treatment of the behavioural and psychological symptoms of dementia. The international journal of neuropsychopharmacology/official scientific journal of the Collegium Internationale Neuropsychopharmacologicum (CINP) 2009;12:191–199. doi: 10.1017/S146114570800970X. [DOI] [PubMed] [Google Scholar]
- Morita S, Tatsumi K, Makinodan M, Okuda H, Kishimoto T, Wanaka A. Geissoschizine methyl ether, an alkaloid from the Uncaria hook, improves remyelination after cuprizone-induced demyelination in medial prefrontal cortex of adult mice. Neurochemical research. 2014;39:59–67. doi: 10.1007/s11064-013-1190-1. [DOI] [PubMed] [Google Scholar]
- Nayernia Z, Jaquet V, Krause KH. New insights on NOX enzymes in the central nervous system. Antioxidants & redox signaling. 2014;20:2815–2837. doi: 10.1089/ars.2013.5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi A, Yamaguchi T, Sekiguchi K, Imamura S, Tabuchi M, Kanno H, Nakai Y, Hashimoto K, Ikarashi Y, Kase Y. Geissoschizine methyl ether, an alkaloid in Uncaria hook, is a potent serotonin1A receptor agonist and candidate for amelioration of aggressiveness and sociality by yokukansan. Neuroscience. 2012;207:124–136. doi: 10.1016/j.neuroscience.2012.01.037. [DOI] [PubMed] [Google Scholar]
- Okahara K, Ishida Y, Hayashi Y, Inoue T, Tsuruta K, Takeuchi K, Yoshimuta H, Kiue K, Ninomiya Y, Kawano J, Yoshida K, Noda S, Tomita S, Fujimoto M, Hosomi J, Mitsuyama Y. Effects of Yokukansan on behavioral and psychological symptoms of dementia in regular treatment for Alzheimer’s disease. Progress in neuro-psychopharmacology & biological psychiatry. 2010;34:532–536. doi: 10.1016/j.pnpbp.2010.02.013. [DOI] [PubMed] [Google Scholar]
- Ozkul A, Akyol A, Yenisey C, Arpaci E, Kiylioglu N, Tataroglu C. Oxidative stress in acute ischemic stroke. Journal of Clinical Neuroscience. 2007;14:1062–1066. doi: 10.1016/j.jocn.2006.11.008. [DOI] [PubMed] [Google Scholar]
- Qi W, Yue SJ, Sun JH, Simpkins JW, Zhang L, Yuan D. Alkaloids from the hook-bearing branch of Uncariarhynchophylla and their neuroprotective effects against glutamate-induced HT22 cell death. Journal of Asian natural products research. 2014;16:876–883. doi: 10.1080/10286020.2014.918109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Rodriguez P, Fernandez E, Bolanos JP. Underestimation of the pentose-phosphate pathway in intact primary neurons as revealed by metabolic flux analysis. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2013;33:1843–1845. doi: 10.1038/jcbfm.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigenaga MK, Hagen TM, Ames BN. Oxidative damage and mitochondrial decay in aging. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:10771–10778. doi: 10.1073/pnas.91.23.10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton RC. Glucose-6-phosphate dehydrogenase, NADPH, and cell survival. IUBMB life. 2012;64:362–369. doi: 10.1002/iub.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Ren X, Simpkins JW. Sequential Upregulation of Superoxide Dismutase 2 and Heme Oxygenase 1 by tert-Butylhydroquinone Protects Mitochondria during Oxidative Stress. Molecular pharmacology. 2015;88:437–449. doi: 10.1124/mol.115.098269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian WN, Braunstein LD, Apse K, Pang J, Rose M, Tian X, Stanton RC. Importance of glucose-6-phosphate dehydrogenase activity in cell death. The American Journal of Physiology. 1999;276:C1121–31. doi: 10.1152/ajpcell.1999.276.5.C1121. [DOI] [PubMed] [Google Scholar]
- Ueki T, Nishi A, Imamura S, Kanno H, Mizoguchi K, Sekiguchi K, Ikarashi Y, Kase Y. Effects of geissoschizine methyl ether, an indole alkaloid in Uncaria hook, a constituent of yokukansan, on human recombinant serotonin 7 receptor. Cellular and molecular neurobiology. 2013;33:129–135. doi: 10.1007/s10571-012-9878-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhyay M, Samal J, Kandpal M, Singh OV, Vivekanandan P. The Warburg effect: Insights from the past decade. Pharmacology & therapeutics. 2013;137:318–330. doi: 10.1016/j.pharmthera.2012.11.003. [DOI] [PubMed] [Google Scholar]