

Abstract
Novel imidazo[4,5-c]quinolin-2-ones were synthesized and evaluated in asexual blood stage and late stage gametocyte assays of Plasmodium falciparum, a major causative agent of malaria. The design of these compounds is based on a recently identified lead compound from a high throughput screen. A concise synthesis was developed that allowed for generation of analogues with substitution around both the quinoline and imidazolidinone rings. Through structure-activity relationship studies, a number of potent compounds were identified that possessed excellent antimalarial activity against both the asexual and sexual stages with minimal cytotoxicity in mammalian cells. This is the first report describing SAR and gametocytocidal activity of imidazo[4,5-c]quinolin-2-ones, a new lead series for malaria treatment and prevention.
Keywords: Antimalarial; Plasmodium falciparum; gametocytes; gametocytocidal; imidazo[4,5-c]quinolin-2-one
Graphical Abstract

Malaria remains a major infectious disease affecting populations primarily in underdeveloped countries. Despite the availability of improved drugs, particularly those based on the artemisinins, there were an estimated 198 million cases of malaria worldwide in 2013 with children being most susceptible to death from this disease.1 Eradication of malaria has major challenges that include resistance to current therapies such as chloroquine, and the potential emergence of resistance to the artemisinins, the only class of drugs that can treat multi-drug resistant parasites.2 To date, most drug development efforts, including recent high throughput screens, have focused primarily on the asexual blood stages of Plasmodium falciparum, the malarial parasite responsible for most deaths.
Transmission of malaria requires the intermediacy of the mosquito as a host for male and female gametocytes taken up in a blood meal to undergo fertilization, sporozoite formation, and transmission during a subsequent blood meal. P. falciparum gametocytes develop through five stages (I–V) over their 3 week lifespan in a human host. Since most antimalarials target the asexual parasite, gametocytes survive and are capable of being transmitted even after treatment with antimalarial drugs. Therefore, a therapeutic approach that targets the gametocyte stage is highly desirable, and would be key to any potential malaria eradication strategy by limiting transmission.
Relatively few gametocytocidal drugs have been described in the literature. Currently, the only drug used to effectively clear gametocytes is primaquine, an antimalarial not widely distributed because of its toxicity to individuals with a glucose-6-phosphate dehydrogenase deficiency (Figure 1).3 The compound methylene blue has also been reported to possess potent gametocytocidal activity in vitro4 and in vivo.5,6 Most recently, the compound DDD107498 was reported to target plasmodium elongation factor 2, thereby inhibiting protein synthesis and acting against multiple lifecycle stages including gametocytes (Figure 1).7
Figure 1.

Gametocytocidal antimalarial small molecules.
Gametocytes do not replicate during development; therefore, typical in vitro assays used for monitoring DNA replication and asexual parasite growth are less useful. The development of an alamarBlue assay, recently reported by our groups, takes advantage of a fluorescent oxidation-reduction indicator as a measure of cytosolic metabolic activity and thereby gametocyte viability.8,9 Using this assay, we recently reported the high throughput screen of 5,215 known compounds, and identified 27 gametocytocidal compounds. Among the most potent lead compounds was the mTOR inhibitor Torin 2 with an IC50 of 8 nM against gametocytes and possessing efficacy for blocking malaria transmission in a mouse model.10
Given the structural similarity of imidazo[4,5-c]quinolin-2-ones to Torin 2, these scaffolds were of interest as potential gametocytocidal antimalarial agents. Compounds with this structure have primarily been developed as kinase inhibitors and have become clinical candidates for anticancer indications. For example, NVP-BEZ235 and NVP-BGT226 were identified as dual PI3K and mTOR inhibitors, and are currently in clinical trials for a number of cancers including advanced solid tumors and metastatic breast cancers.11,12,13 A recent publication described the optimization of NVP-BEZ235 for anti-trypanosomal activity and reported promising compounds with improved selectivity over human PI3K and mTOR.14 Given that these agents have safety profiles consistent with clinical use, we reasoned this series merited further study to optimize their antimalarial properties. Described herein is the rational design, synthesis, and establishment of structure-activity relationships that will facilitate the development of imidazo[4,5-c]quinolin-2-ones as gametocytocidal agents.
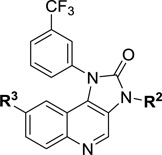
In order to rapidly develop SAR for the imidazo[4,5-c]quinolin-2-one series, a concise synthesis was developed that allows for diversification at three positions around the quinoline and imidazolidinone rings (Scheme 1). The synthesis of analogues commenced with 6-bromo-4-chloro-3-nitroquinoline (5). A nucleophilic aromatic substitution reaction of 5 with various alkyl or aryl amines provided substituted quinolines of the structure 6. Reduction of the nitro group with stannous chloride followed by cyclization upon treatment with diphosgene gave the imidazolidinone ring 8. N-alkylation of this ring was performed with MeI or EtI to afford analogues substituted at position R2 (9). Suzuki–Miyaura coupling of bromides 8 and 9 with desired boronic acids or esters under microwave conditions enables diversification of the quinoline ring and generation of final products. All final analogues were purified by reverse phase HPLC prior to biological evaluation.
Scheme 1.

Reagents and conditions: (i) R1NH2, 1,4-dioxane, 150 °C MW; (ii) SnCl2, EtOH, reflux; (iii) Cl3COCOCl, Et3N, DCM; (iv) MeI or EtI, NaH, DMF, rt; (v) R3-B(OR)2, Pd(PPh3)4, NaHCO3, DMF, H2O, 120 °C MW.
All analogues were tested initially in a blood stage assay with asexual parasites of the chloroquine-sensitive 3D7 strain of P. falciparum.15 The gametocytocidal activity of compounds was determined utilizing the previously described alamarBlue high throughput assay with late stage 3D7 gametocytes (III–V).8–10 In order to measure general cytotoxicity, compounds were tested in the presence of HepG2 cells for cell viability.16 Biological assays were all run in 1536-well format with 11-point dose response curves. All compounds were tested in duplicate, and all assays incorporated Torin 2 (4) as a positive control and DMSO as a negative control.
We initiated SAR studies by synthesizing compounds 11–13 incorporating the aryl substituents of the screening hit Torin 2 (4). The imidazo[4,5-c]quinolin-2-one series allows for relatively easy access to substitution at the –NH of the imidazolidinone ring, providing a handle for further synthetic manipulations. Using this strategy, analogues were synthesized where R1 was kept constant as a 3-trifluoromethylphenyl, and R2 was either unsubstituted or alkyl substituted (Table 1). Compounds 11–13 incorporate the aminopyridine substituent at the R3 position, providing a direct comparison to Torin 2. Of these analogues, compound 12 showed the best antimalarial activity in the asexual assay as well as the gametocyte assay with IC50 values in the low nanomolar range. One notable trend was that methyl substitution provided improved activity relative to either the proton or ethyl variants. Additionally, compound 12 displayed a 4-fold difference in activity between asexual and gametocyte IC50’s, such difference was noticed with most analogues having heterocyclic substituents at R3. While compound 12 was quite potent, it did exhibit HepG2 cytotoxicity in the low micromolar range, which we hoped to diminish through continued SAR.
Table 1.
IC50 values of heteroaromatic substitution at R3.
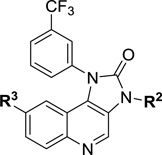 | |||||
|---|---|---|---|---|---|
| Cmpd. | R3 | R2 | Asex. IC50 (µM) |
Gamet. IC50 (µM) |
HepG2 IC50 (µM) |
| 11 | H | 0.091 | 0.097 | 3.11 | |
| 12 | Me | 0.007 | 0.028 | 2.60 | |
| 13 | Et | 0.183 | 0.205 | 3.68 | |
| 14 | H | 2.82 | 11.4 | 2.25 | |
| 15 | Me | 0.095 | 0.236 | 10.3 | |
| 16 | H | 0.313 | 1.14 | 1.50 | |
| 17 | Me | 0.093 | 0.359 | 1.73 | |
| 18 | H | 0.18 | 0.32 | >45.0 | |
| 19 | Me | 0.021 | 0.081 | 14.5 | |
| 20 | H | 0.213 | 0.45 | 2.54 | |
| 21 | Me | 0.017 | 0.052 | 1.33 | |
| 22 | 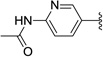 |
H | 0.081 | 0.186 | >45.0 |
| 23 | Me | 0.025 | 0.042 | 3.98 | |
| 24 | H | 0.412 | 4.63 | >45.0 | |
| 25 | Me | 0.158 | 0.294 | 23.5 | |
| 26 |  |
H | 15.4 | 2.35 | >45.0 |
| 27 | Me | 5.78 | 5.27 | 31.6 | |
| 28 |  |
H | 0.214 | 0.347 | 3.24 |
| 29 | Me | 0.065 | 0.157 | 2.63 | |
| 30 | H | 5.03 | 7.46 | >45.0 | |
| 31 | Me | 17.0 | 3.64 | >45.0 | |
Following this initial evaluation based on the structure of Torin 2 (4), analogues were synthesized where the aminopyridine moiety was exchanged for various heterocycles. A few pyridine and substituted pyridine analogues were examined in an effort to understand the relative role of the heterocyclic nitrogen itself in the activity of the aminopyridine substituted compound 12. Unsubstituted 3- and 4-pyridines were examined at the R3 position (14–17). Compounds 15 and 17 possessed similar biological activity, albeit with a 10-fold decrease in both asexual and gametocyte activity compared to 12. When R2 was unsubstituted (14 and 16), there was a significant decrease in activity, as was seen with compound 11. With all of the examined heterocyclic substituents at R3, it was observed that methyl substitution at R2 led to a 2 to 10-fold increase in activity with both the asexual and gametocyte assays.
Substituted pyridine analogues that were examined included methylpyridine (18, 19) acetylaminopyridine (22, 23), and hydroxypyridine (24, 25). Compounds 19 and 23 showed low nanomolar activity in the asexual assay, and good gametocytocidal activity (< 100 nM). The hydroxypyridine compound 25 was slightly less potent than other substituted pyridine analogues. Additionally, compound 21, where R3 is aminopyrimidine, showed low nanomolar activity similar to 12. Notably, the presence of an amino or amide moiety at the para position of heterocycles at R3 showed low micromolar activity in the HepG2 cytotoxicity assay. There were significant reductions in general cytotoxicity with methylpyridine or hydroxypyridine substituents, with HepG2 IC50’s greater than 10 µM. From these data, compound 19 indicates that methylpyridine is a promising substituent at the R3 position because it demonstrates excellent antimalarial activity with low HepG2 cytotoxicity. These results suggest that reductions in mammalian cytotoxicity can be obtained without sacrificing potent antimalarial activity, a critical step in pursuing this series of compounds.
Larger bicyclic heterocycles at the R3 position were also examined. The quinoline ring at R3 (26, 27) led to a significant abrogation of antimalarial activity and was thought to be too bulky at this position. The benzimidazole compound 29, on the other hand, showed good activity in the asexual blood stage assay but was only moderately active in the gametocyte assay. Therefore, larger heterocyclic or aryl substituents were not pursued at R3 for this series of compounds. Additionally, compounds 30 and 31 with no substitution at R3 which were isolated as side products in the Suzuki–Miyaura coupling, showed weak antimalarial activity.
With an understanding of heterocyclic substitutions at the R3 position of the quinoline ring, we turned our attention to phenyl derivatives at this position (Table 2). We started by looking at analogues with an unsubstituted phenyl ring (32, 33) which showed moderate activity for the asexual blood stage assay and little activity in the gametocyte assay. Substitution of the phenyl ring at the 3- or 4-position with primary amines (34–37) was examined to understand the importance of hydrogen bond donors or acceptors at these positions. As seen for analogues containing heterocyclic substituents at R3 (Table 1), phenyl derivatives with methyl substitution at R2 also led to significant improvements in potency for both antimalarial assays (Table 2). Compounds 35 and 37 did show good activity (< 100 nM) in the asexual assay and comparable activity in the gametocyte assay as well. In contrast to heterocyclic substituents at R3, substituted phenyl derivatives generally had less variability between asexual and gametocyte activity, typically showing a 1- to 2-fold difference.
Table 2.
IC50 values of aromatic substitution at R3.
 | |||||
|---|---|---|---|---|---|
| Cmpd. | R3 | R2 | Asex. IC50 (µM) |
Gamet. IC50 (µM) |
HepG2 IC50 (µM) |
| 32 | H | 4.04 | 18.4 | >45.0 | |
| 33 | Me | 0.442 | 4.71 | >45.0 | |
| 34 | H | 0.283 | 3.27 | 7.23 | |
| 35 | Me | 0.086 | 0.087 | 5.53 | |
| 36 | 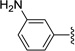 |
H | 0.275 | 0.637 | 6.17 |
| 37 | Me | 0.077 | 0.145 | 13.9 | |
| 38 | H | 0.781 | 1.47 | 4.06 | |
| 39 | Me | 0.114 | 0.229 | 2.33 | |
| 40 | H | 0.109 | 0.294 | >45.0 | |
| 41 | Me | 0.038 | 0.497 | 4.69 | |
| 42 | H | 1.16 | 1.11 | >45.0 | |
| 43 | Me | 0.578 | 0.855 | >45.0 | |
| 44 | 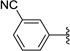 |
H | 0.874 | 2.79 | >45.0 |
| 45 | Me | 0.184 | 2.16 | >45.0 | |
| 46 | H | 2.58 | 6.33 | >45.0 | |
| 47 | Me | 7.33 | 11 | >45.0 | |
| 48 | H | 0.948 | 2.33 | >45.0 | |
| 49 | Me | 0.452 | 0.816 | >45.0 | |
| 50 | H | 1.69 | 3.56 | >45.0 | |
| 51 | Me | 1.75 | 1.86 | >45.0 | |
| 52 | Et | 3.93 | 10.4 | >45.0 | |
| 53 | H | 1.57 | 1.83 | >45.0 | |
| 54 | Me | 3.18 | 5.59 | >45.0 | |
| 55 | H | 5.19 | 7.05 | >45.0 | |
| 56 | Me | 0.733 | 0.524 | >45.0 | |
A number of additional 4-substituted phenyl derivatives were synthesized with both electron withdrawing and donating substituents. The 4-hydroxyphenyl derivative 39 showed slightly lower activity to the 4-aminophenyl 35, suggesting that both hydrogen atoms are beneficial for activity. In comparison, electron withdrawing groups at the 4-position such as nitrile (46, 47), chloro (50–52), and fluoro (53, 54) gave poor activity in the asexual and gametocytocidal assays. The electron donating 4-methylphenyl gave a moderate gametocytocidal IC50 of 524 nM. The 3-benzonitrile 45 derivative demonstrated good asexual activity (184 nM) but poor gametocytocidal activity. We next examined amides, sulfones, and sulfonamides as functional groups incorporating both hydrogen bond acceptors and donors of varying sizes. The primary amide 41 showed excellent asexual activity, but demonstrated a 10-fold lower gametocytocidal activity. The methylsulfone and sulfonamide derivatives 43 and 49, respectively, were similar to each other in activity and had moderate asexual and gametocyte activity. Of note, the HepG2 cytotoxicity of the phenyl derivatives was significantly lower than heterocyclic substitution at R3, with most compounds showing no cytotoxicity at the highest concentration tested (46 µM).
Having considered numerous analogues with changes to R3, we turned our attention to variations at R1 of the imidazolidinone ring (Table 3). Substitution at R1 requires incorporation of the varying anilines from the beginning of the synthesis with the SNAr reaction. In order to directly compare modifications at R1, the aminopyridine group at R3 was held constant, since it was found to be the most potent substituent in Table 1.
Table 3.
IC50 values of substitution at R1.
 | |||||
|---|---|---|---|---|---|
| Cmpd. | R1 | R2 | Asex. IC50 (µM) |
Gamet. IC50 (µM) |
HepG2 IC50 (µM) |
| 57 | H | 0.530 | 0.576 | >45.0 | |
| 58 | Me | 0.104 | 0.237 | >45.0 | |
| 59 | H | 0.386 | 0.482 | >45.0 | |
| 60 | Me | 0.098 | 0.118 | >45.0 | |
| 61 | Me | 0.047 | 0.148 | 2.34 | |
| 62 | 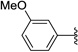 |
Me | 0.088 | 0.126 | 2.53 |
| 63 | Me | 0.069 | 0.219 | 6.54 | |
| 64 | H | 1.35 | 2.87 | 6.50 | |
| 65 | Me | 0.832 | 1.88 | 3.84 | |
| 66 | Me | 1.16 | 0.865 | 11.4 | |
We began by making a relatively simple change from compounds 11 and 12, by incorporating a 4-trifluoromethylphenyl group at R1 (Table 3). One can hypothesize that modulation of R1 may more directly impact the R2 substituent; therefore, we tested the unsubstituted –NH and methyl substitution at position R2. Comparison of compounds 57 and 58 clearly showed that methyl substitution provides a 3- to 5-fold increase in antimalarial activity. The more active compound 58 showed good asexual and gametocytocidal activity; however, these potencies were more than 10-fold lower than the 3-trifluoromethylphenyl derivative (12).
Additional 3- and 4-substituted phenyl derivatives that were examined included variations in electron withdrawing or donating properties. The 4-benzonitrile compound 60 showed good activity in both the asexual and gametocytocidal assays with no measurable HepG2 cytotoxicity. Additionally, substitution with the electron donating methoxy group at either the 4- (61) or 3-position (62) of the phenyl ring demonstrated good asexual and gametocytocidal activity. While cytotoxicity of these compounds was in the low micromolar range, modulation of the R3 position may decrease general cytotoxicity as measured by the HepG2 assays. Interestingly, the unsubstituted phenyl derivative 63 possessed good asexual antimalarial activity and gametocyte activity that was comparable to substituted phenyl derivatives. Alkyl substitution at R1 included both the cyclohexyl derivatives 64 and 65 as well as the cyclopropyl compound 66. All of the alkyl compounds tested to date, have shown a significant decrease in activity compared to the phenyl derivatives. Therefore, our focus at the R1 position has been phenyl or substituted phenyl derivatives. With flexibility regarding the substituents at R1, we anticipate continuing SAR studies with the aim to decrease cytotoxicity, increase solubility, and optimize physical properties of this series of compounds.
In conclusion, by designing a concise synthesis, a number of analogues incorporating changes to both the quinoline ring and imidazolidinone rings of the imidazo[4,5-c]quinolin-2-one series were synthesized. These analogues were tested in previously established P. falciparum asexual blood stage assay and the alamarBlue gametocytocidal assay recently described by our groups. Through these studies, a number of compounds were identified with potent, low nanomolar activity in both the asexual and gametocyte stages. In particular, compound 19 was found to be among the best compounds with excellent antimalarial activity in both assays and low cytotoxicity. We also demonstrated improved cytotoxicity profiles upon incorporation of phenyl derivatives at R3 compared to heterocyclic substitution, and showed that methyl substitution at R2 was key for improved antimalarial activity. Finally, derivatives at position R1, while tolerated, seemed to show little improvement from the 3-trifluoromethylphenyl group (11–12). With these potent compounds in hand, we anticipate more significant efforts aimed at understanding the targets of these phenotypic screens. We have demonstrated key structure-activity relationships around the imidazo[4,5-c]quinolin-2-one series of compounds. Further lead optimization to develop this class of compounds as antimalarials with a focus on gametocytocidal activity aimed at limiting transmission of the malaria parasite is under way in our group, we will report the results in due course.
Supplementary Material
Acknowledgments
The authors would like to thank Paul Shinn, Danielle VanLeer, and Jennifer Bennison for assistance with compound management. This work was supported by the Intramural Research Programs of the National Center for Advancing Translational Sciences and Public Health Service Grant AI101396 from the National Institute of Allergy and Infectious Diseases. TQT is a JSPS Research Fellow in Biomedical and Behavioral Research at the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.World Health Organization; World Malaria Report 2014. (ISBN: 978 92 4 156483 0) [Google Scholar]
- 2.Flannery EL, Chatterjee AK, Winzeler EA. Nature Rev. 2013;11:849. doi: 10.1038/nrmicro3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beutler E. Blood. 1959;14:103. [PubMed] [Google Scholar]
- 4.Peatey CL, Leroy D, Gardiner DL, Trenholme KR. Malaria J. 2012;11:34. doi: 10.1186/1475-2875-11-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coulibaly B, Zoungrana A, Mockenhaupt FP, Schirmer RH, Klose C, Mansmann U, Meissner PE, Muller O. PloS One. 2009;4:5318. doi: 10.1371/journal.pone.0005318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adjalley SH, Johnston GL, Li T, Eastman RT, Ekland EH, Eappen AG, Richman A, Sim BK, Lee MCS, Hoffman SL, Fidock DA. Proc. Nat. Acad. Sci. 2011;108:E1214. doi: 10.1073/pnas.1112037108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baragana B, Hallyburton I, Lee M, Norcross N, Grimaldi R, Otto T, Proto W, Blagborough A, Meister S, Wirjanata G, Ruecker A, Upton L, Abraham T, Almeida M, Pradhan A, Porzelle A, Martinez M, Bolscher J, Woodland A, Norval S, Zuccotto F, Thomas J, Simeons F, Stojanovski L, Osuna-Cabello M, Brock P, Churcher T, Sala K, Zakutansky S, Jimenez-Diaz M, Sanz L, Riley J, Basak R, Campbell M, Avery V, Sauerwein R, Dechering K, Noviyanti R, Campo B, Frearson J, Angulo-Barturen I, Ferrer-Bazaga S, Gamo F, Wyatt P, Leroy D, Siegl P, Delves M, Kyle D, Wittlin S, Marfurt J, Price R, Sinden R, Winzeler E, Charman S, Bebrevska L, Gray D, Campbell S, Fairlamb A, Willis P, Rayner J, Fidock D, Read K, Gilbert I. Nature. 2015;522:315. doi: 10.1038/nature14451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tanaka TQ, Williamson KC. Mol. Biochem. Parasitol. 2011;177:160. doi: 10.1016/j.molbiopara.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tanaka TQ, Dehdashti SJ, Nguyen D, McKew JC, Zheng W, Williamson KC. Mol. Biochem. Parasitol. 2013;188:20. doi: 10.1016/j.molbiopara.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun W, Tanaka TQ, Magle CT, Huang W, Southall N, Huang R, Dehdashti SJ, McKew JC, Williamson KC, Zheng W. Sci. Reports. 2014;4:3743. doi: 10.1038/srep03743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, Brachmann S, Chene P, De Pover A, Schoemaker K, Fabbro D, Gabriel D, Simonen M, Murphy L, Finan P, Sellers W, Garcia-Echeverria C. Mol. Cancer Ther. 2008;7:1851. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- 12.Markman B, Tabernero J, Krop I, Shapiro GI, Siu L, Chen LC, Mita M, Melendez Cuero M, Stutovoet S, Birle D, Anak O, Hackl W, Baselga J. Ann. Oncol. 2012;23:2399. doi: 10.1093/annonc/mds011. [DOI] [PubMed] [Google Scholar]
- 13. www.clinicaltrials.gov.
- 14.Seixas JD, Luengo-Arratta SA, Diaz R, Saldivia M, Rojas-Barros DI, Manzano P, Gonzalez S, Berlanga M, Smith TK, Navarro M, Pollastri MP. J. Med. Chem. 2014;57:4834. doi: 10.1021/jm500361r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trager W, Jensen JB. J. Parasitol. 2005;91:484. doi: 10.1645/0022-3395(2005)091[0484:HMPICC]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 16.Miret S, De Groene EM, Klaffke W. J. Biomol. Screen. 2006;11:184. doi: 10.1177/1087057105283787. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.