

Abstract
Autophagy is an essential degradative pathway that maintains neuronal homeostasis and prevents axon degeneration. Initial observations suggest that autophagy is spatially regulated in neurons, but how autophagy is regulated in distinct neuronal compartments is unclear. Using live-cell imaging in mouse hippocampal neurons, we establish the compartment-specific mechanisms of constitutive autophagy under basal conditions, as well as in response to stress induced by nutrient deprivation. We find that at steady state, the cell soma contains populations of autophagosomes derived from distinct neuronal compartments and defined by differences in maturation state and dynamics. Axonal autophagosomes enter the soma and remain confined within the somatodendritic domain. This compartmentalization likely facilitates cargo degradation by enabling fusion with proteolytically active lysosomes enriched in the soma. In contrast, autophagosomes generated within the soma are less mobile and tend to cluster. Surprisingly, starvation did not induce autophagy in either the axonal or somatodendritic compartment. While starvation robustly decreased mTORC1 signaling in neurons, this decrease was not sufficient to activate autophagy. Furthermore, pharmacological inhibition of mammalian target of rapamycin with Torin1 also was not sufficient to markedly upregulate neuronal autophagy. These observations suggest that the primary physiological function of autophagy in neurons may not be to mobilize amino acids and other biosynthetic building blocks in response to starvation, in contrast to findings in other cell types. Rather, constitutive autophagy in neurons may function to maintain cellular homeostasis by balancing synthesis and degradation, especially within distal axonal processes far removed from the soma.
SIGNIFICANCE STATEMENT Autophagy is an essential homeostatic process in neurons, but neuron-specific mechanisms are poorly understood. Here, we compare autophagosome dynamics within neuronal compartments. Axonal autophagy is a vectorial process that delivers cargo from the distal axon to the soma. The soma, however, contains autophagosomes at different maturation states, including input received from the axon combined with locally generated autophagosomes. Once in the soma, autophagosomes are confined to the somatodendritic domain, facilitating cargo degradation and recycling of biosynthetic building blocks to primary sites of protein synthesis. Neuronal autophagy is not robustly upregulated in response to starvation or mammalian target of rapamycin inhibition, suggesting that constitutive autophagy in neurons maintains homeostasis by playing an integral role in regulating the quality of the neuronal proteome.
Keywords: autophagy, axonal transport, hippocampal neuron, LC3, mTOR, soma
Introduction
Most neurons in our brain are born during embryogenesis and need to last an entire lifetime. To support their long-term viability and functionality, neurons must have robust housekeeping pathways to ensure the quality and integrity of proteins and organelles in the cytoplasm. Neurons are postmitotic and so cannot simply dilute out proteotoxins by cell division. Thus, neurons are particularly dependent on quality-control pathways, such as autophagy, to maintain cellular homeostasis.
Autophagy is an evolutionarily conserved degradative pathway that eliminates damaged organelles and protein aggregates from the cytoplasm (Xie and Klionsky, 2007; Mizushima et al., 2008). In neurons, autophagy is essential. CNS-specific and neuron-specific loss of autophagy is sufficient to induce neuronal death (Hara et al., 2006; Komatsu et al., 2006, 2007; Nishiyama et al., 2007). Furthermore, autophagy is misregulated in various neurodegenerative diseases characterized by the accumulation of protein aggregates, including Alzheimer's disease, Parkinson's disease, Huntington's disease, and amyotrophic lateral sclerosis (Yamamoto and Yue, 2014). Despite evidence that autophagy is critical in maintaining neuronal homeostasis, surprisingly little is understood about the fundamental mechanisms driving this process in neurons.
We have established that neurons exhibit robust, constitutive autophagy with a clear enhancement of autophagosome formation in the distal axon (Maday et al., 2012; Maday and Holzbaur, 2014). Following formation, autophagosomes undergo robust retrograde transport toward the soma (Hollenbeck, 1993; Yue, 2007; Lee et al., 2011; Maday et al., 2012; Maday and Holzbaur, 2014). As autophagosomes move toward the soma, they mature into degradative autolysosomes (Lee et al., 2011; Maday et al., 2012; Cheng et al., 2015; Wang et al., 2015). Thus autophagy is spatiotemporally regulated along the axon, exhibiting a high degree of compartmentalization within the neuron.
Recent evidence further underscores specific roles of autophagy within distinct compartments of the neuron. For example, the bulk turnover of mitochondria via mitophagy may occur specifically within the soma (Cai et al., 2012; Devireddy et al., 2015). Autophagy is also enhanced within presynaptic and postsynaptic terminals to regulate neuronal activity and synaptic transmission (Hernandez et al., 2012; Shehata et al., 2012). Together, these data suggest that autophagy may be differentially regulated within the axonal and somatodendritic compartments.
Here, we examine autophagosome dynamics in different neuronal compartments (axonal vs somatodendritic) and determine how this process is regulated in response to metabolic stress induced by nutrient deprivation. We find that the cell soma contains different populations of autophagosomes, which are derived from distinct neuronal compartments and are defined by their maturation state and dynamics. Autophagosome distribution is compartmentalized; axonal autophagosomes entering the soma are then restricted to the somatodendritic domain, while autophagosomes originating from dendrites are infrequently observed under basal conditions. Compartmentalization within the neuron likely facilitates degradation events by enabling fusion with proteolytically active lysosomes; these more active lysosomes are enriched in the soma relative to the axon. Surprisingly, we do not observe an induction of autophagy in response to nutrient deprivation or mammalian target of rapamycin (mTOR) inhibition, indicating that the primary role of constitutive autophagy in neurons may not be to mobilize amino acids in response to starvation but to effectively turnover aging proteins and organelles to maintain homeostasis.
Materials and Methods
Reagents
GFP-LC3 transgenic mice, strain name B6.Cg-Tg(CAG-EGFP/LC3)53Nmi/NmiRbrc (Mizushima et al., 2004) were obtained from the RIKEN BioResource Center in Japan. Constructs include YFP-NAVII-III (Addgene #26056), LAMP1-RFP (Addgene #1817), DsRed2-mito (gift from Dr. Thomas Schwarz), and mCherry-LC3. Primary antibodies for immunofluorescence include mouse anti-p62 (Abcam, ab56416), rat anti-lysosomal-associated membrane protein 1 (LAMP1) (Abcam, ab25245), and chicken anti-GFP (Aves Labs, GFP-1020). Secondary antibodies for immunostain include goat anti-mouse Alexa-594 and goat anti-rat Alexa-594 (Invitrogen), and goat anti-chicken Cy2 (Jackson ImmunoResearch Laboratories). Antibodies for immunoblot analysis include rabbit anti-LC3 (Abcam, ab48394), mouse anti-GAPDH (Abcam, ab9484), rabbit anti-pS6 (Cell Signaling Technology, 2215S), rabbit anti-S6 (Cell Signaling Technology, 2217), mouse anti-α-tubulin (Sigma-Aldrich, T9026), rabbit anti-calnexin (Stressgen, SPA-860), and donkey anti-mouse and rabbit peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories).
Primary hippocampal culture
The hippocampus was dissected from mouse embryos of either sex at day 15.5. Hippocampal neurons were isolated by 0.25% trypsin digestion for 10 min at 37°C, followed by trituration through a small-bore glass Pasteur pipette. We plated 750,000 hippocampal neurons in a 10 cm dish filled with eight 25 mm acid-washed glass coverslips coated with 500 μg/ml poly-l-lysine. To determine the polarity of single axons during live-cell imaging, GFP-LC3 transgenic neurons were diluted 1:20 with nontransgenic neurons isolated from littermates. For live-cell experiments with transfected markers and fixed-cell immunolabeling, GFP-LC3 transgenic neurons were plated undiluted. Axons were identified by morphologic criteria established by Kaech and Banker (2006). Wild-type mouse hippocampal neurons used in Figure 5G–I were obtained from the Neurons R Us core facility at the University of Pennsylvania, prepared from C57BL/6 mouse embryos at day 18. Neurons were grown for 8–10 DIV in maintenance media (Neurobasal medium supplemented with 2% B-27, 37.5 mm NaCl, 33 mm glucose, 2 mm GlutaMAX, and 100 U/ml penicillin and 100 μg/ml streptomycin) at 37°C in a 5% CO2 incubator. Every 3–4 d, 20–30% media was replaced; 1 μm AraC (antimitotic drug) was added to the first feed. For transfection, coverslips (7 DIV) were transferred to a six-well tray and transfected with 1.8 μg of DNA using Lipofectamine 2000 (Invitrogen) and incubated for 16–18 h. All animal protocols were approved by the Institutional Animal Care and Use Committee at the University of Pennsylvania.
Figure 5.
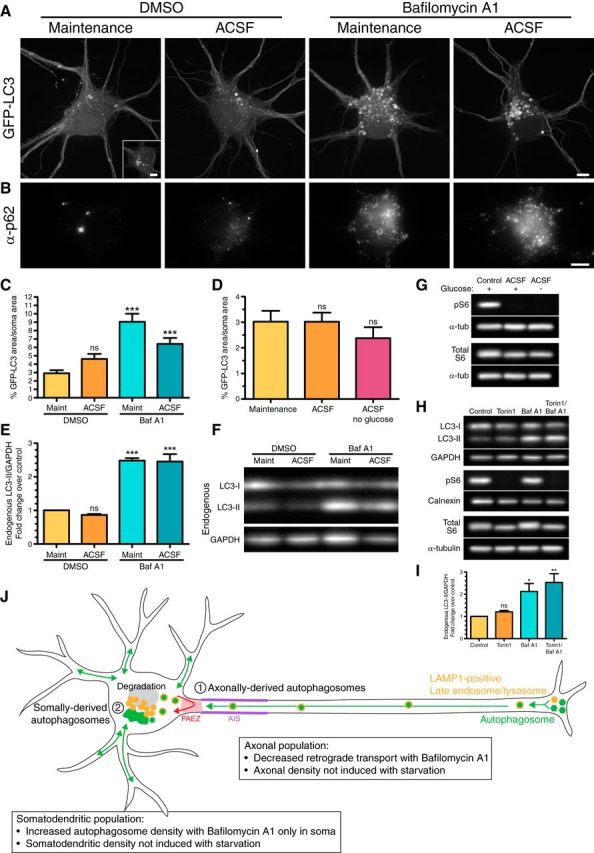
Autophagosomes accumulate in the soma with bafilomycin A1 treatment. A, Maximum projections of z-stacks of the soma of GFP-LC3 transgenic hippocampal neurons (8–10 DIV). Inset, hippocampal neurons also display larger clusters of GFP-LC3. Scale bar, 5 μm. B, GFP-LC3-expressing hippocampal neurons were immunostained for p62. Scale bar, 5 μm. C, Quantitation of GFP-LC3 area normalized to the soma area (mean ± SEM; n = 24–36 neurons from 5–6 experiments; 8–10 DIV). ***p ≤ 0.001 (one-way ANOVA with Dunnett's post hoc test). D, Quantitation of GFP-LC3 area normalized to the soma area (mean ± SEM; n = 30–33 neurons from 3–4 experiments; 8–9 DIV; one-way ANOVA with Dunnett's post hoc test). E, Quantitation of immunoblotting of lysates from cultured hippocampal neurons. LC3-II levels were normalized to GAPDH (mean ± SEM; n = 3 experiments; SDS-PAGE and immunoblotting were performed in duplicate for each experiment, 8–10 DIV). ***p ≤ 0.001 (one-way ANOVA with Dunnett's post hoc test). F, Immunoblot analysis of lysates from cultured hippocampal neurons for endogenous LC3 and GAPDH (loading control). G, Immunoblot analysis of lysates from cultured wild-type hippocampal neurons for mTORC1 activity. Alpha-tubulin loading controls are shown below each corresponding immunoblot. H, I, Immunoblot analysis and corresponding quantitation of LC3-II levels in lysates from cultured wild-type hippocampal neurons treated with Torin1. LC3-II levels were normalized to GAPDH (mean ± SEM; n = 3 experiments; 8–10 DIV). *p ≤ 0.05; **p ≤ 0.01 (one-way ANOVA with Dunnett's post hoc test). Calnexin and α-tubulin serve as loading controls for pS6 and total S6 immunoblots, respectively. J, Model schematic of the compartment-specific regulation of autophagy in neurons. ns, Not significant.
Starvation and Torin1 treatments
For starvation experiments, neurons were washed in either maintenance media or artificial CSF (ACSF; 10 mm HEPES, pH 7.4, 125 mm NaCl, 5 mm KCl, 2 mm CaCl2, 1 mm MgCl2, 10 mm glucose), and incubated in maintenance media with 100 nm bafilomycin A1 (Sigma-Aldrich) or equivalent volume of DMSO (solvent control), or ACSF with 100 nm bafilomycin A1 or equivalent volume of DMSO (solvent control), respectively. For Western blotting and immunostain, neurons were incubated in these conditions for 4 h. For live-cell imaging, neurons were incubated in starvation conditions for 2 h and then transferred to new media for live-cell imaging for 2 h, totaling 4 h of treatment. Neurons in maintenance media were transferred to Hibernate E (BrainBits) supplemented with 2% B-27, 2 mm GlutaMAX, and either 100 nm bafilomycin A1 or DMSO. Samples in ACSF were transferred to new ACSF with 100 nm bafilomycin A1 or DMSO. For the starvation experiment in Figure 5G, wild-type hippocampal neurons were washed in either maintenance media, ACSF with glucose, or ACSF without glucose, followed by a 4 h incubation in maintenance media, ACSF with glucose, or ACSF without glucose, respectively. For the Torin1 treatment in Figure 5H,I, wild-type hippocampal neurons were incubated 4 h in maintenance media supplemented with either 250 nm Torin1 (Calbiochem), 100 nm bafilomycin A1, 250 nm Torin1 and 100 nm bafilomycin A1, or an equivalent volume of DMSO as a solvent control.
Live-cell imaging
For live-cell imaging of neurons under basal conditions (Figs. 1A–D,F–H, 2A,D,E), neurons were imaged in Hibernate E (BrainBits) supplemented with 2% B-27 and 2 mm GlutaMAX. Twenty-five millimeter coverslips with hippocampal neurons were sandwiched within a Chamlide CMB magnetic imaging chamber (BioVision Technologies). Live-cell microscopy was performed on a PerkinElmer UltraVIEW Vox Spinning Disk Confocal with a Nikon Eclipse Ti Inverted Microscope with the Perfect Focus System using an Apochromat 100× 1.49 numerical aperture oil-immersion objective (Nikon) in an environmental chamber at 37°C. Digital images were acquired with a Hamamatsu EMCCD C9100-50 camera using Volocity software (PerkinElmer). Solid-state 488 and 561 nm lasers (PerkinElmer) were used for excitation. Axons were imaged at 1–2 s per frame for 5 min. Z-stacks were obtained of the entire cell body at 0.1 μm sections. Whenever possible, axons and cell bodies were obtained from the same neuron.
Figure 1.
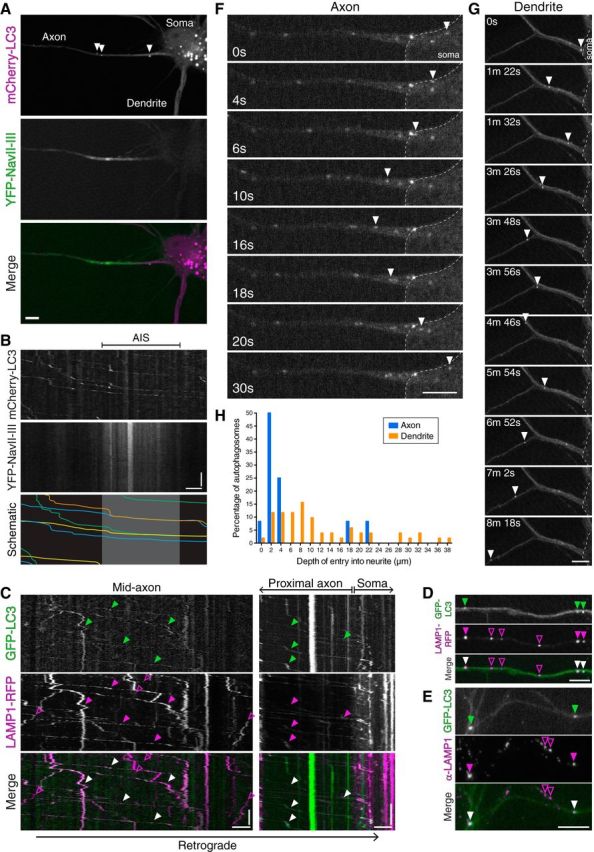
Axonal, LAMP1-positive autophagosomes enter the soma through the AIS and become restricted to the somatodendritic domain. A, Hippocampal neuron, grown 8 DIV, develops axon and dendritic processes. Neurons were transfected with mCherry-LC3 and YFP-NaVII-II, a marker for the AIS. Arrowheads denote autophagic vesicles. B, Kymographs of mCherry-LC3 motility through the AIS. For clarity, the mCherry-LC3 kymograph was traced and shown below; the gray box denotes the AIS region. C, Kymograph analysis of GFP-LC3 and LAMP1-RFP motility in the mid-axon and proximal axon. Closed arrowheads denote retrogradely moving autophagosomes that comigrate along the mid-axon with LAMP1-RFP, and enter the soma positive for LAMP1-RFP. Open arrowheads denote LAMP1-RFP-positive organelles that are negative for GFP-LC3. D, Still images along the mid-axon generated from the movie analyzed in C. Closed arrowheads denote autophagosomes positive for LAMP1-RFP. Open purple arrowheads denote organelles positive for LAMP1-RFP only. E, GFP-LC3 transgenic hippocampal neurons 10 DIV were immunostained for LAMP1. Closed arrowheads denote autophagosomes in the axon that are positive for endogenous LAMP1. Open arrowheads denote endogenous LAMP1-positive organelles that are negative for GFP-LC3. F, Time series of autophagosome motility at the base of an axon in a hippocampal neuron grown 10 DIV. Arrowheads denote a GFP-LC3-positive autophagosome rejected from entry into the axon. G, Time series of autophagosome motility into a dendrite of a hippocampal neuron grown 8 DIV. Arrowheads denote a GFP-LC3-positive autophagosome that robustly enters into the dendritic shaft. H, Frequency distribution of distances that autophagosomes enter from the base of each neurite population (axons, n = 12 events from 6 neurons from 3 experiments; dendrites, n = 51 events from 15 neurons from 5 experiments; 5–10 DIV). Values for dendrites are under-represented since autophagosomes move beyond the field of view. Scale bars: horizontal, 5 μm; vertical, 1 min.
Figure 2.
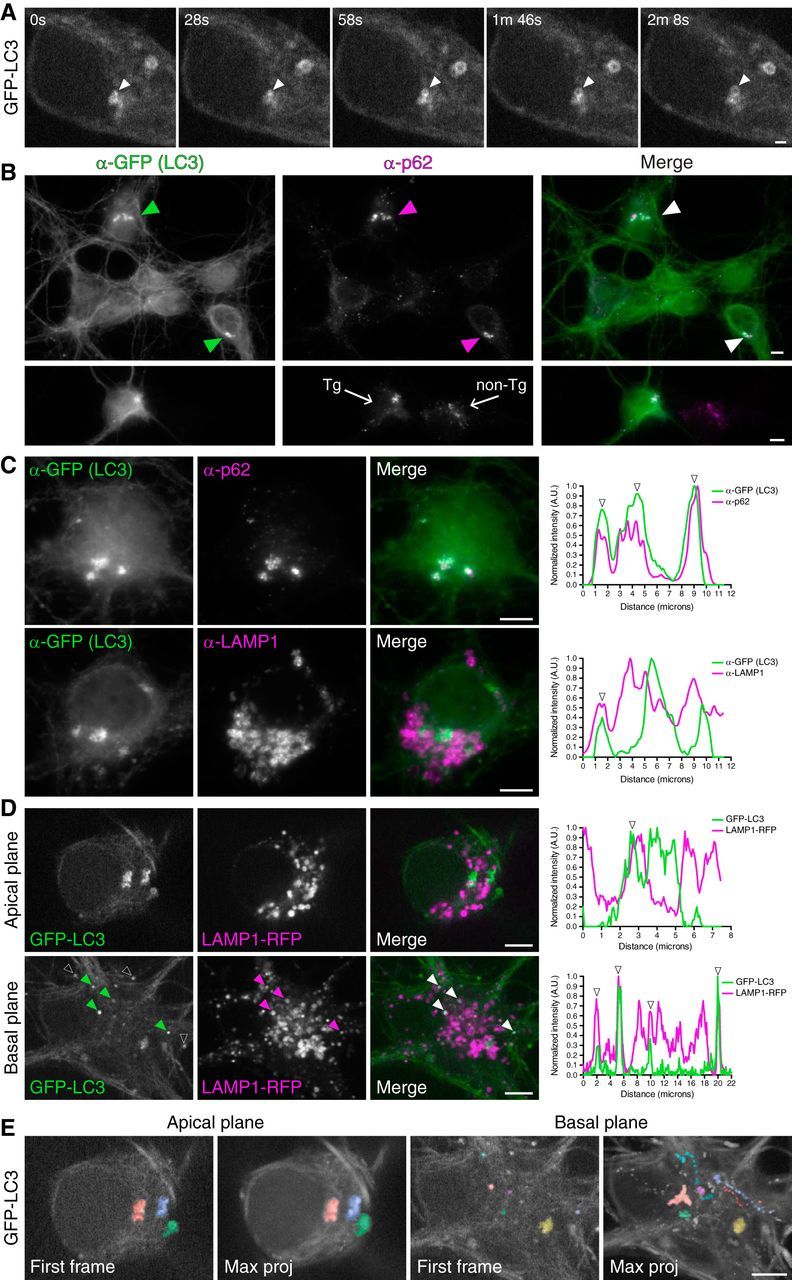
The soma contains autophagosomes from distinct compartments at different stages of maturation. A, Time series of autophagosome biogenesis in the soma of a hippocampal neuron grown 6 DIV. Arrowheads denote a newly forming autophagosome. Scale bar, 1 μm. B, GFP-LC3 transgenic hippocampal neurons grown 10 DIV were fixed and immunostained for GFP and p62. Arrowheads denote cells with GFP-LC3-positive structures that are copositive for p62. Lower panels are GFP-LC3 transgenic neurons cocultured with nontransgenic neurons (10 DIV); the presence of p62-positive clusters is not dependent on expression of GFP-LC3. C, GFP-LC3 transgenic hippocampal neurons grown 10 DIV were immunostained for GFP, p62, and LAMP1. Corresponding line scans are shown on the right; arrowheads denote overlapping peaks of fluorescence intensity. D, Stills from live-cell imaging of hippocampal neurons on 8 DIV expressing GFP-LC3 and LAMP1-RFP. Shown is an apical and basal plane of two individual cells and corresponding line-scan analysis. Closed arrowheads on basal images denote organelles positive for both GFP-LC3 and LAMP1-RFP. Colocalized puncta may appear slightly offset due to the sequential capture of images, green followed by purple. Arrowheads on line scans denote peaks of overlapping fluorescence. Open white arrowheads on GFP-LC3 basal image denote autophagosomes in neighboring axons that are not transfected with LAMP1-RFP. E, First frames and corresponding maximum projections of the entire frames of GFP-LC3 movies acquired in the apical or basal plane of two different hippocampal neurons grown 8 DIV. Individual autophagosomes are color-coded to visualize the entire trajectory in the maximum projection. Scale bars: B–E, 5 μm.
Immunostaining
Hippocampal neurons at 10 DIV were fixed for 10 min in 4% PFA/4% sucrose previously warmed to 37°C. Cells were washed two times in PBS (50 mm NaPO4, 150 mm NaCl, pH 7.4), permeabilized for 5 min in 0.1% TX-100 in PBS, washed two times in PBS, and then blocked for 1 h in 5% goat serum, 1% BSA in PBS. Samples were then incubated in primary antibody diluted in block solution for 1 h, washed three times for 5 min in PBS, incubated in secondary antibody diluted in block solution for 1 h, washed three times for 5 min in PBS, and mounted in ProLong Gold (Life Technologies). Images were acquired on a Leica DMI6000B inverted epifluorescence microscope using an Apochromat 63× 1.4 numerical aperture oil-immersion objective with a 1.6× optical magnification (Leica Microsystems). Digital images were acquired with a Hamamatsu ORCA-R2 charge-coupled device camera using LAS-AF software (Leica Microsystems).
Immunoblotting
We plated 240,000 GFP-LC3 transgenic neurons per 6 cm dish coated with 50 μg/ml poly-l-lysine, grown for 8–10 DIV, and starved according to the conditions described above. Neurons were then washed in PBS (50 mm NaPO4, 150 mm NaCl, pH 7.4) and lysed in RIPA buffer [50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% Triton X-100, 0.5% deoxycholate, 0.1% SDS, and 1× complete protease inhibitor mixture (Roche)] for 30 min on ice. For samples measuring phospho-S6 levels, Halt Protease and Phosphatase inhibitor mixture (ThermoFisher Scientific) along with 5 mm EDTA were substituted for the Roche complete protease inhibitor mixture. Samples were centrifuged at 15,800 × g for 15 min at 4°C. Supernatants were analyzed by SDS-PAGE and transferred onto an Immobilon P PVDF membrane. Membranes were blocked in 5% milk in Tris-buffered saline-Igepal (24.8 mm Tris-HCl, pH 7.4, 2.7 mm KCl, 137 mm NaCl, 0.05% Igepal) for 30 min at room temperature followed by incubation in primary antibody diluted in block solution for overnight at 4°C, rocking. Membranes were washed 3 × 20 min in HRP wash buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 0.1% BSA, 0.05% Igepal), and peroxidase-conjugated secondary antibodies diluted in HRP wash buffer were applied for 45 min. Membranes were washed 3 × 20 min in HRP wash buffer and developed using the SuperSignal West Pico Chemiluminescent Substrate (ThermoScientific).
Image analysis
Entry into neurites.
In Fiji (Schindelin et al., 2012), a line was drawn from the base of the axon or dendrite (defined as where the neurite widens and opens to cell soma) to the farthest distance into the neurite that an autophagosome reached during the entire duration of the movie. The length of this line was recorded and values for entry into axons and dendrites were plotted as a histogram.
Colocalization between LC3 and LAMP1.
Discrete puncta with signals above background that colocalized or shared ∼80% overlap were binned as copositive. With live-cell imaging, puncta that were comigrating were binned as copositive.
Autophagosome transport and density.
Kymographs were generated in Fiji using a line width of three pixels. From the kymographs, autophagosome average velocity through the axon initial segment (AIS) region (defined as 5–30 μm from the soma) was calculated as displacement divided by time. For kymographs generated in the mid-axon (>50 μm from soma), we determined the percentage of autophagosomes moving in the net retrograde direction (displacement of ≥5 μm within 5 min) versus net anterograde direction (displacement of ≥5 μm within 5 min). Autophagosomes that exhibited a confined motility (displacement of <5 μm within the 5 min imaging window) were binned as bidirectional or stationary. Area flux (number of autophagosomes/100 μm/min) was determined from each kymograph as the sum of retrograde, anterograde, and bidirectional/stationary vesicles normalized by kymograph length and time. Autophagosome density was determined from the first frame of the movie by counting the number of autophagosomes normalized by distance of the axon.
Line scans.
Line scans of GFP-LC3, LAMP1, and p62 in the soma were generated using the plot profile tool in Fiji (line width of one pixel). A mean background intensity value was determined for each channel from a box drawn within the cell. Intensity values for a given marker were normalized by subtracting the respective mean background, and dividing the dataset by its maximum.
GFP-LC3 area in the soma.
To measure the total cellular GFP-LC3, maximum projections were generated in Fiji of the z-stacks of the soma. LC3-positive regions were identified and segmented using ilastik. Segmentations from ilastik were imported into Fiji and the total LC3 area was measured using the analyze particles function. The total area of LC3 particles for a given cell body was then divided by the total area of the cell body.
Quantitation of Western blotting.
Bands were quantitated using the gel analyzer tool in Fiji. To control for differences in sample loading between lanes, values for LC3-II were divided by values for GAPDH from the corresponding lanes. Treatments were then normalized relative to the corresponding control sample (maintenance + DMSO) and expressed as a fold difference above control.
Figure preparation and statistical tests.
All image measurements were obtained from the raw data. GraphPad Prism was used to plot graphs and perform statistical tests; statistical tests are denoted within each figure legend. When performing a one-way ANOVA with Dunnett's post hoc test, values were compared with the maintenance control column. In the case of the two-way ANOVA with Bonferroni's post hoc test, row means were compared. Images were prepared in Fiji and Adobe Photoshop, adjusting contrast and brightness equally to all images within a series. Figures were assembled in Adobe Illustrator.
Results
The soma contains autophagosomes at different maturation states originating from distinct neuronal compartments
Previous work has identified a robust pathway for autophagosome biogenesis and maturation in axons (Lee et al., 2011; Maday et al., 2012; Maday and Holzbaur, 2014), suggesting that there may be compartment-specific regulation of autophagy in neurons. To explore this possibility, we examined autophagy in different neuronal compartments under basal versus stress conditions. To track the fate of autophagosomes originating in the axon, we performed live-cell imaging of autophagosomes in the proximal axon as they transit through the AIS to enter the cell soma. Mouse hippocampal neurons were transfected with mCherry-LC3 to label autophagosomes and autolysosomes. YFP-NaVII-III was used to mark the AIS. At 8 DIV, a well defined axon and dendrites are established and the YFP-NaVII-III localizes to the AIS region of the proximal axon (Fig. 1A).
We observed robust and processive movement of autophagic vesicles through the YFP-NaVII-III-positive AIS region as they enter into the soma (Fig. 1B). Previous reports suggest the AIS functions as a filter to sort vesicles into the axon and observed a ∼2-fold decrease in velocities as vesicles move through this region (Song et al., 2009). To measure the average velocity of autophagosomes moving in the retrograde direction through the AIS region (∼5–30 μm from soma), hippocampal neurons were isolated from GFP-LC3 transgenic mice (Mizushima et al., 2004) and cultured for 8–10 DIV. The average velocity of GFP-LC3-positive puncta within the AIS region was 0.55 ± 0.03 μm/s (mean ± SEM; n = 134 vesicles from 13 neurons from 5 experiments), not significantly different from our previous reported value of 0.62 ± 0.06 μm/s measured in the mid-axon (>50 μm from soma) of hippocampal neurons from the same developmental time period (Maday and Holzbaur, 2014). Thus, retrogradely moving autophagosomes do not slow down as they transit through the AIS region.
To determine the maturation state of autophagosomes entering the soma from the axon, we performed dual-color live-cell imaging of GFP-LC3 with the late endosomal/lysosomal marker LAMP1-RFP in hippocampal neurons grown 8–10 DIV. Imaging along the axon reveals retrogradely moving GFP-LC3 puncta that robustly colocalize with LAMP1-RFP in the mid-axon as well as in the proximal axon as they enter into the soma (Fig. 1C,D). Kymographs indicate comigration of LC3 and LAMP1, strongly suggesting that they are the same compartment (Fig. 1C). In the mid-axon, 34 of 37 (∼92%) GFP-LC3-positive puncta were positive for LAMP1-RFP (measured from five neurons from two experiments). Interestingly, one of the autophagosomes that was LAMP1 negative was newly formed, evidenced by an increase in size and fluorescence with time (Maday et al., 2012; Maday and Holzbaur, 2014). This result is consistent with other reports of formation along the mid-axon (Ashrafi et al., 2014), although we find these events to be infrequent under basal conditions. As a confirmatory approach, we assessed axonal GFP-LC3-positive puncta for the presence of endogenous LAMP1 by immunostain, and found that 38 of 54 (∼70%) GFP-LC3-positive puncta are positive for endogenous LAMP1 (from ≥17 axons from two experiments; Fig. 1E). Thus, the vast majority of autophagosomes entering the soma from the axon are positive for the late endosomal/lysosomal marker LAMP1, indicating that these compartments have begun to fuse within the axon before reaching the soma.
To track the destination of autophagosomes that reach the soma, we measured the sorting of autophagosomes between the axon and dendrites. Autophagosomes from the soma sometimes re-enter the base of the proximal axon, but are promptly rejected and return back to the soma (Fig. 1F; n = 12 events). In contrast, autophagosomes entering the dendrite from the soma navigate deeply into the dendritic shaft (Fig. 1G; n = 51 events). Quantitation of the distances autophagosomes enter into each neurite population revealed a ∼3.6-fold higher median distance in dendrites compared with axons, 8.3 μm compared with 2.3 μm, respectively (Fig. 1H). Interestingly, despite their ability to navigate into dendrites, the vast majority of autophagosomes eventually return to the soma. Under our conditions of basal autophagy, autophagosome biogenesis in dendrites was infrequent, indicating that the soma receives autophagosomes forming primarily from the axonal compartment and fewer from dendrites. Thus, autophagosomes represent a primarily unidirectional and retrograde cargo that can exit the axon, but not re-enter the axon. Upon entry into the soma, autophagosomes are restricted to the somatodendritic domain, further supporting the highly compartmentalized nature of the autophagy pathway in neurons.
Next we wanted to determine whether all the autophagosomes in the soma are derived from the axon or whether there is also local formation. To address this possibility, we performed live-cell imaging of GFP-LC3 in the cell soma of hippocampal neurons. In Figure 2A, a GFP-LC3 punctum appears and enlarges to form a ring structure, indicative of autophagosome formation as previously observed in the distal axons of dorsal root ganglion and hippocampal neurons (Maday et al., 2012; Maday and Holzbaur, 2014). Thus, autophagosomes can also be generated locally within the soma.
To more completely define the somal population of autophagosomes, we immunostained for GFP and observed two populations of neurons based on the GFP-LC3 distribution in the soma. Approximately 52% of hippocampal neurons had larger GFP-LC3-positive structures (>2 μm in the longest axis) in the soma (Fig. 2B). We immunostained these larger LC3-positive structures for p62, an adaptor that links ubiquitinated cargo to the LC3 machinery during engulfment (Pankiv et al., 2007). Shown in Figure 2B,C (top), these large GFP-LC3-positive structures are positive for p62. Line-scan analysis indicates that multiple individual p62 puncta overlap with a more continuous LC3 signal, suggesting that p62 puncta may be enclosed within a larger autophagosome structure (Fig. 2C, top). Furthermore, the presence of p62-positive clusters is not dependent on expression of the GFP-LC3 transgene, as p62-positive clusters were also present in nontransgenic neurons (Fig. 2B, bottom). Thus, GFP-LC3-positive structures within the soma contain the cargo adaptor p62, suggesting that these structures represent bona fide autophagosomes.
To assess the maturation state of autophagosomes in the soma, we measured colocalization with LAMP1. In basal regions of the cell, we observed an enrichment of discrete GFP-LC3-positive puncta that were robustly positive for LAMP1-RFP (Fig. 2D). Line-scan analysis shows overlapping peaks of LC3 and LAMP1 fluorescence intensity, indicating colocalization (Fig. 2D). This population of autophagosomes likely represents the LC3/LAMP1 copositive organelles originating from the axon that feeds into more basal regions of the soma. In contrast, the larger GFP-LC3-positive structures were enriched in apical regions of the soma, and exhibited only partial overlap with endogenous LAMP1 (Fig. 2C, bottom) or the live-cell marker LAMP1-RFP (Fig. 2D, apical plane). Thus, we suggest that autophagosomes in the soma can be found in different stages of maturation, with axonally derived autophagosomes more likely to be more uniformly LAMP1 positive than autophagosomes formed within the soma itself.
To further distinguish between these populations, we performed live-cell imaging of GFP-LC3 and LAMP1-RFP in the soma. Analysis of motility patterns reveals that these two populations exhibit unique dynamics. The LAMP1-positive autophagosomes likely derived from the axon are quite motile and are found predominantly within basal regions of the cell (Fig. 2E). In contrast, the larger LAMP1-negative, LC3-positive structures observed within apical regions of the soma are primarily stationary or oscillate within a confined region of a few micrometers (Fig. 2E).
Together, these results support the presence of multiple populations of autophagosomes within the soma defined by different maturation states and motility patterns. One population of autophagosomes likely originates in the axon, is LAMP1 positive, and remains highly motile once reaching the soma. However, the soma also contains LAMP1-negative autophagosomes, likely formed locally within this compartment. These LC3 structures tend to cluster and are predominantly stationary. These results further support the hypothesis that autophagy in primary neurons is highly compartmentalized (Maday and Holzbaur, 2014; Devireddy et al., 2015).
Amino acid deprivation does not effectively upregulate autophagy in the axon
Next, we wanted to ask whether these populations of autophagosomes are differentially regulated in a compartment-specific manner in response to stress. Since autophagy was originally identified in yeast as a response to starvation (Tsukada and Ohsumi, 1993), we applied stress in the form of nutrient deprivation. To starve neurons of amino acids, we incubated hippocampal neurons in ACSF, a buffered salt solution supplemented with glucose. Neurons were incubated in either maintenance media (Neurobasal media supplemented with glucose and B-27) or ACSF for 2 h, and we then performed live-cell imaging for 2 h with continued treatment. Since levels of autophagy are a balance between the antagonistic activities of autophagosome formation and autophagosome degradation, the total effect of any starvation-induced autophagy was measured by treating with 100 nm bafilomycin A1. Bafilomycin A1 inhibits the vacuolar H+-pump and blocks lysosome-mediated degradation and also autophagosome–lysosome fusion (Yamamoto et al., 1998). Thus, with bafilomycin A1, we can measure autophagy induction in the absence of autophagy clearance.
We first measured autophagosome density along the axon by determining the number of autophagosomes per 100 μm. Surprisingly, we observed no change in autophagosome density along the axon in response to starvation (Fig. 3A,B). Treatment with ACSF alone did not increase autophagy and treatment in combination with bafilomycin A1 did not further increase the density of axonal autophagosomes (Fig. 3A,B). We also measured autophagosome flux as defined by the total number of autophagosomes observed within a region of interest along the axon over time. Again, we observed no change in autophagosome area flux in response to starvation with and without bafilomycin A1 (Fig. 3A,C). Together, these data indicate that autophagy in the axon is not effectively upregulated by amino acid deprivation.
Figure 3.
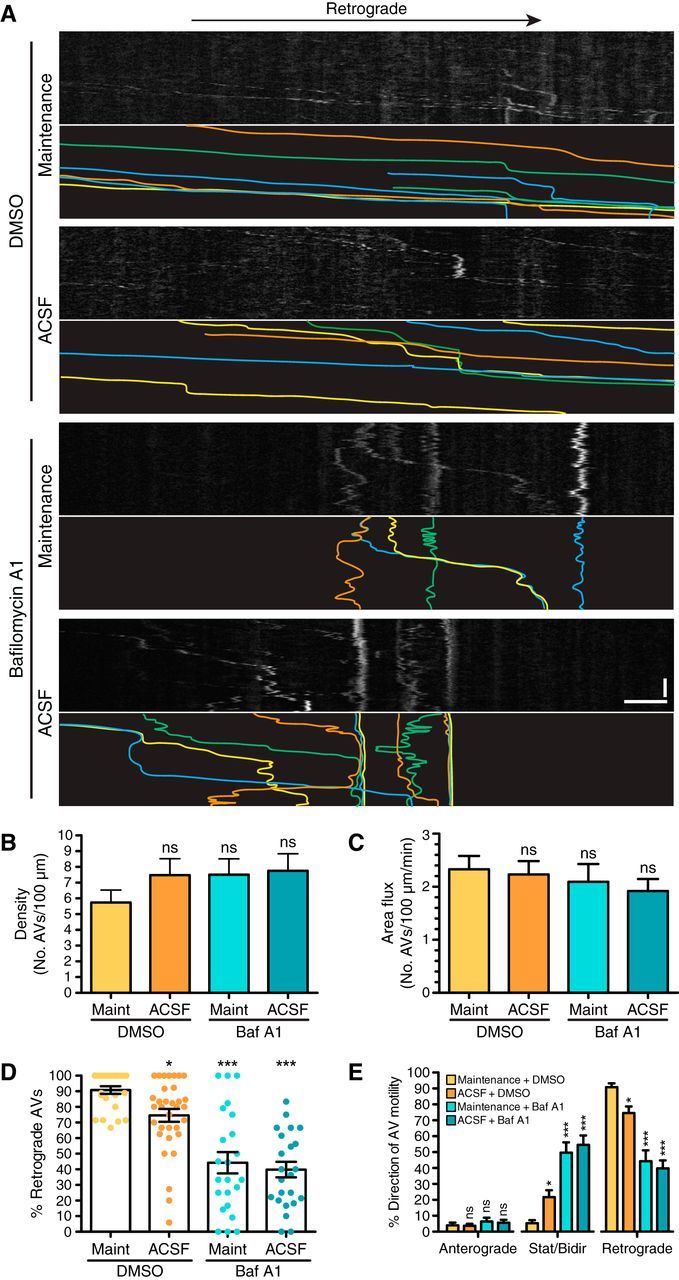
Autophagy is not upregulated along the axon in response to amino acid starvation. A, Kymographs from live-cell imaging of GFP-LC3 transgenic hippocampal neurons (8 DIV). For clarity, GFP-LC3 tracks are traced and shown below each corresponding kymograph. Scale bars: horizontal, 5 μm; vertical, 1 min. B, Quantitation of autophagosome density (number of autophagosomes per 100 μm axon in the first frame of movie) in the mid-axon (mean ± SEM; n = 23–32 neurons from 5–6 experiments; 8–10 DIV; one-way ANOVA with Dunnett's post hoc test). C, Quantitation of autophagosome area flux (number of autophagosomes per 100 μm per minute) in the mid-axon (mean ± SEM; n = 23–33 neurons from 5–6 experiments; 8–10 DIV; one-way ANOVA with Dunnett's post hoc test). D, Quantitation of autophagosomes undergoing retrograde transport in the mid-axon (mean ± SEM; n = 23–33 neurons from 5–6 experiments; 8–10 DIV). *p ≤ 0.05; ***p ≤ 0.001 (one-way ANOVA with Dunnett's post hoc test). E, Quantitation of autophagosome motility along the mid-axon of hippocampal neurons (mean ± SEM; n = 23–33 neurons from 5–6 experiments; 8–10 DIV). *p ≤ 0.05; ***p ≤ 0.001 (one-way ANOVA with Dunnett's post hoc test within each grouping designated by x-axis breaks). ns, Not significant; AVs, autophagic vacuoles.
Interestingly, we did observe a decrease in the percentage of autophagosomes engaged in retrograde transport in neurons treated with bafilomycin A1 (Fig. 3D). Retrograde transport decreased by ∼50% in both maintenance-treated and ACSF-treated conditions supplemented with bafilomycin A1, suggesting that bafilomycin A1 treatment alone reduces motility. This decrease in retrograde transport is accompanied by a corresponding increase in nonprocessive autophagosomes that exhibit motility within a confined region (Fig. 3E). This effect is not due to starvation, since there is only a slight decrease in retrograde transport in neurons treated with ACSF alone. Bafilomycin A1, however, did not arrest autophagosome transport completely, as there is a wide range of responses within the neuronal population. Together, these data suggest that inhibiting autophagosome maturation decreases retrograde transport. However, since this is not a complete arrest in transport, ∼50% of axonal autophagosomes are likely to reach the cell soma in the presence of bafilomycin A1.
To rule out the possibility that reduced autophagosome transport is due to decreased cell viability with drug treatment, we measured mitochondrial transport in maintenance/DMSO and ACSF/bafilomycin A1 conditions. We observed no change in mitochondrial density along the axon, with 15.7 ± 1.4 (mean ± SEM) mitochondria/100 μm in maintenance/DMSO and 16.5 ± 1.4 (mean ± SEM) mitochondria/100 μm in ACSF/bafilomycin A1 (n = 13–17 neurons from 2 experiments). There was also no change in area flux along the axon, with 3.7 ± 0.3 (mean ± SEM) mitochondria/100 μm/min in maintenance/DMSO and 3.7 ± 0.3 (mean ± SEM) mitochondria/100 μm/min in ACSF/bafilomycin A1 (n = 13–17 neurons from 2 experiments). Further, the percentage of motile mitochondria was not altered in either the anterograde or retrograde direction (Fig. 4A,B). Thus, since mitochondria sustain their motility in the presence of bafilomycin A1, the decrease in autophagosome transport is not due to decreased cell viability or alterations to the cellular energy state, but suggests rather a reduction specifically in autophagosome transport.
Figure 4.

Glucose deprivation does not induce autophagosome formation but impairs retrograde transport along the axon. A, Straightened en face images and corresponding kymographs of DsRed2-mito along the mid-axon of hippocampal neurons 8 DIV. The retrograde direction is toward the right. Scale bars: horizontal, 5 μm; vertical, 1 min. B, Quantitation of mitochondrial motility along the axon of hippocampal neurons grown 8 DIV (mean ± SEM; n = 13–17 neurons from 2 experiments; two-way ANOVA with Bonferroni's post hoc test). C, Quantitation of autophagosome density (number of autophagosomes per 100 μm axon in the first frame of movie) in the mid-axon (mean ± SEM; n = 27–32 neurons from 3–4 experiments; 8–9 DIV; one-way ANOVA with Dunnett's post hoc test). D, Quantitation of autophagosome area flux (number of autophagosomes per 100 μm per minute) in the mid-axon (mean ± SEM; n = 27–33 neurons from 3–4 experiments; 8–9 DIV; one-way ANOVA with Dunnett's post hoc test). E, Quantitation of autophagosomes undergoing retrograde transport in the mid-axon (mean ± SEM; n = 12–18 neurons from 3–4 experiments; 8–9 DIV). ***p ≤ 0.001 (two-way ANOVA with Bonferroni's post hoc test). ns, Not significant; AVs, autophagic vacuoles.
Based on these observations, amino acid depletion does not induce autophagy along the axon. We then considered whether this lack of response is due to incomplete starvation since glucose is still present. Thus, we removed glucose from the ACSF treatment and determined the effect on autophagy in the axon. Surprisingly, we did not observe any change in autophagosome density or area flux along the axon in these conditions either (Fig. 4C,D), indicating that complete starvation (no amino acids and glucose) does not induce enhanced autophagy in the axon. Glucose depletion did, however, affect retrograde transport. In the fourth hour of treatment, retrograde transport decreased by ∼50%, suggesting insufficient ATP to fuel axonal transport (Fig. 4E). As we saw no enhancement of autophagy in the axon after a 4 h incubation of hippocampal neurons in minimal media, our findings suggest that axonal autophagy is a constitutive process that is not effectively upregulated in response to starvation.
Autophagosomes accumulate in the soma in response to bafilomycin A1 but not starvation
To further investigate the compartmentalized regulation of autophagy in neurons, we examined the soma under conditions of starvation. We obtained paired data from the axons imaged in Figure 3 and generated z-stacks of the corresponding soma (maximum projections shown in Fig. 5A). With live-cell imaging, we determined the GFP-LC3 distribution in the soma. One population of neurons contained discrete, smaller GFP-LC3 puncta (Fig. 5A), while a second population of neurons contained larger GFP-LC3-positive structures (Fig. 5A, inset), similar to our immunostain results (Fig. 2B). ACSF treatment did not increase the number of autophagosomes in the soma. However, we observed a dramatic increase in GFP-LC3 puncta with bafilomycin A1 treatment in neurons under either maintenance or ACSF conditions (Fig. 5A). This effect was restricted to the soma, as we did not observe an increase in GFP-LC3 in dendrites under ACSF conditions with and without bafilomycin A1 (Fig. 5A). Bafilomycin A1 also dramatically increased the number of p62 puncta in the soma (Fig. 5B). In some neurons, we observed that ACSF treatment appeared to breakdown and disassemble the larger LC3 structures as well as p62-positive clusters. However, this was not a consistent effect across datasets (Fig. 5A,B).
We quantitated the area occupied by GFP-LC3 in the soma using ilastik, a machine learning approach to identifying and segmenting objects of interest. While there was a slight increase in the area of GFP-LC3 fluorescence in neurons treated with ACSF, this effect was not statistically significant (Fig. 5C). Removing glucose from ACSF did not further enhance autophagy levels in the soma (Fig. 5D). The most significant effect was observed in the presence of bafilomycin A1. Bafilomycin A1 dramatically increased GFP-LC3 area ∼3.1-fold for neurons in maintenance media, and ∼2.2-fold for neurons in ACSF, compared with the maintenance/DMSO control (Fig. 5C). Thus, under the conditions tested, neither amino acid nor energy deprivation upregulated autophagy in the somatodendritic or axonal compartment of primary hippocampal neurons. Bafilomycin A1, however, dramatically increases the area labeled with GFP-LC3 in the soma, although not in dendrites or the axon. These results suggest that the final destination and site for the bulk of autophagosome clearance/degradation is in the soma, rather than locally within the axon.
As a complementary approach, we also determined the levels of autophagy during starvation with and without bafilomycin A1 by immunoblot analysis of lysates from cultured hippocampal neurons. Levels of autophagosome-associated lipidated LC3, LC3-II, did not change with ACSF treatment alone, but were ∼2.5-fold higher in bafilomycin A1-treated conditions (in either maintenance or ACSF; Fig. 5E,F). This increase measured in total cell lysates by immunoblotting is strikingly similar to the increase we observed in the soma by live-cell imaging (Fig. 5C). Thus, the accumulation of autophagosomes in the soma accounts for the increase in autophagy upon bafilomycin A1 treatment. Since bafilomycin A1 did not arrest retrograde transport completely, the increase in LC3-II in the soma is a sum of axonal autophagosomes that still reach the soma combined with locally synthesized autophagosomes.
Since starvation alone did not induce autophagy in hippocampal neurons, we wanted to determine whether mTORC1, a negative regulator of autophagy, was effectively inhibited under our starvation conditions. To test this, neurons were treated for 4 h in either maintenance media, ACSF with glucose, or ACSF without glucose, and samples were immunoblotted for phospho-S6, a downstream substrate of mTORC1. Active mTORC1 phosphorylates S6 kinase, which in turn phosphorylates ribosomal S6 (Laplante and Sabatini, 2009). Thus, levels of phosphorylated S6 serve as a readout of mTORC1 activity. We observed a dramatic decrease in pS6 levels under starvation conditions (ACSF with or without glucose) compared with the maintenance control, with minimal variance in total S6 protein (Fig. 5G). These results indicate that mTORC1 activity is effectively inhibited under our starvation paradigm, indicating that neurons are in fact responding to nutrient deprivation. However, under these same conditions we did not see an upregulation of autophagy, suggesting that mTOR may not be an effective regulator of autophagy in primary neurons.
To test this hypothesis directly, we inhibited mTOR function with Torin1, an ATP-competitive inhibitor. Hippocampal neurons were treated with 250 nm Torin1 and analyzed by immunoblot for endogenous LC3-II levels. Treatment with Torin1 alone did not significantly increase LC3-II levels compared with DMSO-treated control cells (Fig. 5H,I). To measure autophagic flux, bafilomycin A1 was added alone or in combination with Torin1. On its own, bafilomycin A1 significantly increased LC3-II levels (Fig. 5H,I) as expected. Treatment with bafilomycin A1 in combination with Torin1 resulted in only a minimal increase in LC3-II compared with bafilomycin A1 treatment alone. To confirm the effectiveness of Torin1, we immunoblotted lysates for pS6 and observed a robust decrease in pS6 levels with minimal alterations in total S6 levels, indicating that Torin1 inactivates mTORC1 function in neurons. Together, these results suggest that inhibition of mTOR does not robustly activate autophagy in cultured hippocampal neurons.
Discussion
Here, we investigate the regulation of autophagy within different compartments of the neuron under basal versus stress conditions. We identify distinct populations of autophagosomes within the soma. These organelles are derived from multiple compartments within the neuron and are defined by the maturation state and motility. Autophagosomes derived from the axon are largely LAMP1 positive and motile (Fig. 5J). The soma also contains LAMP1-negative autophagosomes, likely formed locally within this compartment (Fig. 5J). These LAMP1-negative autophagosomes are generally clustered and relatively immobile. Blocking lysosome function with bafilomycin A1 leads to autophagosome accumulation in the soma only, not in the axon or dendrites, suggesting the soma is the primary site of autophagosome clearance. Thus, robust retrograde transport of autophagosomes along the axon delivers autophagosomes to the soma to facilitate degradation and recycling of amino acids for new biosynthetic reactions.
Axonal autophagosomes pass through the AIS with little resistance and no change in velocity. The majority of axonal autophagosomes are positive for the late endosomal/lysosomal marker LAMP1, suggesting that initial fusion between these compartments occurs in the axon. This fusion event follows completion of autophagosome biogenesis since we noted an example of a LAMP1-negative autophagosome undergoing formation along the mid-axon. These findings in hippocampal neurons are consistent with previous observations in dorsal root ganglion neurons where autophagosomes are preferentially generated in the distal axon and mature into LAMP1-positive organelles during retrograde transport toward the soma (Maday et al., 2012), indicating a conserved mechanism of autophagosome maturation along the axon of various neuronal subtypes.
Once autophagosomes enter the soma, they are restricted to the somatodendritic domain (Fig. 5J) and are unable to re-enter the axon. In contrast, autophagosomes can readily enter dendrites. These dynamics are consistent with a dynein-dominant mechanism of motility. In the axon, dynein drives the retrograde transport of autophagosomes (Maday et al., 2012) along the uniformly plus-end out array of microtubules (Baas et al., 1988). Axonal microtubules are thus ideally oriented to allow exit but not re-entry of dynein-driven cargos. Microtubule tracks in the proximal two-thirds of the dendrite, however, are mixed polarity with ∼40% of microtubules with minus ends directed outward (Baas et al., 1988; Kleele et al., 2014). Thus, microtubules at the base of dendrites are in the proper orientation for dynein-mediated entry. Consistent with this, recruitment of dynein to peroxisomes is sufficient to drive their transport into dendrites along the mixed microtubule array (Kapitein et al., 2010). Although both kinesin-1 and kinesin-2 are bound to autophagosomes (Maday et al., 2012), we suggest dynein remains the dominant motor driving autophagosome motility throughout the lifetime of this organelle.
The observed dynamics support the existence of a one-way filter at the base of the axon that restricts entry but not exit of dynein-driven organelles (Fig. 5J). The AIS was previously proposed to function as an actin-based cytoplasmic filter to restrict the entry of dendritic cargoes (Song et al., 2009). Recent reports have challenged this hypothesis by demonstrating that dendritic cargoes are rejected from the axon before AIS development, emphasizing the contributions of sorting mechanisms at the trans-Golgi network (TGN; Petersen et al., 2014). Furthermore, super-resolution and electron micrograph data have challenged the existence of a dense actin meshwork at the AIS (Xu et al., 2013; Jones et al., 2014; D'Este et al., 2015), although these results do not exclude the existence of layered mechanisms to maintain axon–dendrite identity. Autophagosomes represent a unique organelle population not derived from the TGN, but rather from ER membranes in the distal axon (Maday and Holzbaur, 2014). Therefore, exclusion of autophagosomes from axonal re-entry requires additional mechanisms that may occur at the recently defined preaxonal exclusion zone (PAEZ), proximal to the AIS region (Farías et al., 2015). Penetration through the PAEZ requires appropriately bound kinesin-1 motors (Farias et al., 2015). While kinesin-1 motors are bound to neuronal autophagosomes, these motors are likely maintained in an autoinhibited state via scaffolding proteins, including JIP1 and huntingtin (Fu et al., 2014; Wong and Holzbaur, 2014). Thus, our results are also consistent with a gating function at the base of the axon in the vicinity of the PAEZ that maintains the compartment-specific distribution of organelles and functions within the neuron.
In contrast to the uniform and vectorial pathway defined for axonal autophagosomes, we find that the soma contains autophagosomes at different maturation states. Axonal autophagosomes enter the soma largely LAMP1 positive, but the soma also contains LAMP1-negative autophagosomes likely generated locally within this compartment (Fig. 5J). In fact, we observed events of autophagosome biogenesis evident by the appearance of GFP-LC3-positive puncta that expand into ring-like structures, similar to previous observations in the distal axon (Maday et al., 2012; Maday and Holzbaur, 2014). The number of autophagosomes forming in the soma was difficult to determine because of the depth of the soma and the rapid nature of autophagosome biogenesis (4–6 min). Our previous findings in DRG neurons indicated an enrichment of autophagosome formation in the distal axon compared with the soma (Maday and Holzbaur, 2014). However, this polarization may be more pronounced in developing neurons compared with synaptically connected hippocampal neurons.
Blocking lysosome function with bafilomycin A1 increased autophagosomes in the soma but not in the axon or dendrites, supporting the idea that the soma has a role as the primary site for degradation of autophagic cargo. Consistent with this, impaired retrograde transports result in immature autophagosomes and the accumulation of undigested cargo within autophagosomes (Fu et al., 2014; Wong and Holzbaur, 2014). Previous reports indicate higher proteolytic activity in somal lysosomes compared with axonal lysosomes (Lee et al., 2011; Gowrishankar et al., 2015; Xie et al., 2015), suggesting a gradient of degradative activity. Thus, autophagosomes likely undergo retrograde transport to achieve full maturation into degradative compartments by fusing with proteolytically active lysosomes enriched in the soma.
Bafilomycin A1 treatment attenuated the retrograde transport of autophagosomes, as previously noted (Lee et al., 2011; Wang et al., 2015). Bafilomycin A1 also inhibits the motility of late endosomes and lysosomes (Lee et al., 2011). Together, these results suggest that the acidification state of degradative organelles affects their transport, although the underlying mechanisms are not yet clear. Thus, organelle function may be tightly coupled to transport, and vice versa.
The role of starvation and mTORC1 signaling in regulating neuronal autophagy has been controversial. To study the effects of nutrient deprivation on neuronal tissue in vivo, Mizushima et al. (2004) deprived GFP-LC3 transgenic mice of food for 24 h and examined the effects on various tissues. Nutrient deprivation induced autophagy in many tissues, such as the liver, muscle, kidney, and heart. However, no induction was observed in the brain (Mizushima et al., 2004). Since other tissues, such as the liver and muscle will cannibalize themselves to supply the brain with nutrients, and the brain can survive on ketones for energy (Rabinowitz and White, 2010; Ezaki et al., 2011), neuronal tissue is less likely to experience starvation conditions unless extreme circumstances arise. Additionally, neurons rely on neighboring glial cells for lactate (Saab et al., 2013). Examination of cell-autonomous effects of starvation on neurons in vitro has yielded varying effects. One study observed an induction of autophagy in cultured cortical neurons within 30 min of nutrient removal, although glial contributions cannot be completely ruled out (Young et al., 2009). Another group found an induction of autophagy following a 24 h incubation in salt solution coupled with prolonged treatment with protease inhibitors (Boland et al., 2008). In contrast, the Finkbeiner laboratory reported no induction of autophagy in cultured striatal or cortical neurons following mTOR inhibition with rapamycin or everolimus (Tsvetkov et al., 2010). Mixed results have also been reported from in vivo studies using either rapamycin or everolimus to affect the clearance of protein aggregates via autophagy (Ravikumar et al., 2004; Fox et al., 2010).
In our study, starvation conditions that effectively activate autophagy in other cell types [e.g., hepatocytes and HeLa cells (Köchl et al., 2006; Tsvetkov et al., 2010)] were not sufficient to induce autophagy in hippocampal neurons. We observed consistent densities of autophagosomes along the axon, ∼5–6 autophagic vacuoles per 100 μm, in control versus starvation conditions. Autophagosomes underwent robust retrograde transport in ACSF, indicating sustained cell viability in starvation medium. Glucose deprivation, however, attenuated retrograde transport in the fourth hour of treatment, consistent with localized glycolysis providing ATP to power transport (Zala et al., 2013).
While starvation effectively reduced mTORC1 signaling in hippocampal neurons, this was not sufficient to markedly activate autophagy. Further, inhibition of mTOR directly with Torin1 also did not robustly induce autophagy in neurons, consistent with previous reports that inhibition of mTOR with rapamycin or everolimus was ineffective in enhancing autophagy in neurons (Fox et al., 2010; Tsvetkov et al., 2010). Together, these results indicate that autophagy in cultured neurons is not significantly upregulated by nutrient deprivation or mTOR inhibition under the conditions tested.
We propose that the primary physiological function of autophagy in neurons may not be to mobilize amino acids in response to starvation. Rather, autophagy has a critical role in regulating neuronal development and homeostasis (Fimia et al., 2007; Shen and Ganetzky, 2009; Tang et al., 2014; Yamamoto and Yue, 2014). Autophagy has been linked to synaptic activity (Hernandez et al., 2012; Shehata et al., 2012; Wang et al., 2015). Autophagy also plays a critical role in maintaining homeostasis of the distal axon (Komatsu et al., 2007; Nishiyama et al., 2007), where it regulates the quality of the distal proteome (Maday and Holzbaur, 2014). Importantly, in the context of disease, the limited ability of the neuron to upregulate autophagy may be a point of vulnerability rendering the axon susceptible to stress.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants K99NS082619 and R00NS082619 to S.M. and NIH Grant NS060698 to E.L.F.H. We gratefully acknowledge the technical assistance of Karen Jahn and Mariko Tokito. We thank Swathi Ayloo, Amy Ghiretti, Mara Olenick, and Alison Twelvetrees for helpful discussion. We also thank Jeffrey Nirschl for technical assistance with ilastik. We thank Joseph Baur and Cassie Tran for technical advice and reagents for the mTOR analysis.
The authors declare no competing financial interests.
References
- Ashrafi G, Schlehe JS, LaVoie MJ, Schwarz TL. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J Cell Biol. 2014;206:655–670. doi: 10.1083/jcb.201401070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baas PW, Deitch JS, Black MM, Banker GA. Polarity orientation of microtubules in hippocampal neurons: uniformity in the axon and nonuniformity in the dendrite. Proc Natl Acad Sci U S A. 1988;85:8335–8339. doi: 10.1073/pnas.85.21.8335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, Nixon RA. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer's disease. J Neurosci. 2008;28:6926–6937. doi: 10.1523/JNEUROSCI.0800-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Q, Zakaria HM, Simone A, Sheng ZH. Spatial parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons. Curr Biol. 2012;22:545–552. doi: 10.1016/j.cub.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng XT, Zhou B, Lin MY, Cai Q, Sheng ZH. Axonal autophagosomes recruit dynein for retrograde transport through fusion with late endosomes. J Cell Biol. 2015;209:377–386. doi: 10.1083/jcb.201412046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Este E, Kamin D, Göttfert F, El-Hady A, Hell SW. STED nanoscopy reveals the ubiquity of subcortical cytoskeleton periodicity in living neurons. Cell Rep. 2015;10:1246–1251. doi: 10.1016/j.celrep.2015.02.007. [DOI] [PubMed] [Google Scholar]
- Devireddy S, Liu A, Lampe T, Hollenbeck PJ. The organization of mitochondrial quality control and life cycle in the nervous system in vivo in the absence of PINK1. J Neurosci. 2015;35:9391–9401. doi: 10.1523/JNEUROSCI.1198-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezaki J, Matsumoto N, Takeda-Ezaki M, Komatsu M, Takahashi K, Hiraoka Y, Taka H, Fujimura T, Takehana K, Yoshida M, Iwata J, Tanida I, Furuya N, Zheng DM, Tada N, Tanaka K, Kominami E, Ueno T. Liver autophagy contributes to the maintenance of blood glucose and amino acid levels. Autophagy. 2011;7:727–736. doi: 10.4161/auto.7.7.15371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farías GG, Guardia CM, Britt DJ, Guo X, Bonifacino JS. Sorting of dendritic and axonal vesicles at the pre-axonal exclusion zone. Cell Rep. 2015;13:1221–1232. doi: 10.1016/j.celrep.2015.09.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, Corazzari M, Fuoco C, Ucar A, Schwartz P, Gruss P, Piacentini M, Chowdhury K, Cecconi F. Ambra1 regulates autophagy and development of the nervous system. Nature. 2007;447:1121–1125. doi: 10.1038/nature05925. [DOI] [PubMed] [Google Scholar]
- Fox JH, Connor T, Chopra V, Dorsey K, Kama JA, Bleckmann D, Betschart C, Hoyer D, Frentzel S, Difiglia M, Paganetti P, Hersch SM. The mTOR kinase inhibitor everolimus decreases S6 kinase phosphorylation but fails to reduce mutant huntingtin levels in brain and is not neuroprotective in the R6/2 mouse model of Huntington's disease. Mol Neurodegener. 2010;5:26. doi: 10.1186/1750-1326-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu MM, Nirschl JJ, Holzbaur EL. LC3 binding to the scaffolding protein JIP1 regulates processive dynein-driven transport of autophagosomes. Dev Cell. 2014;29:577–590. doi: 10.1016/j.devcel.2014.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowrishankar S, Yuan P, Wu Y, Schrag M, Paradise S, Grutzendler J, De Camilli P, Ferguson SM. Massive accumulation of luminal protease-deficient axonal lysosomes at Alzheimer's disease amyloid plaques. Proc Natl Acad Sci U S A. 2015;112:E3699–E3708. doi: 10.1073/pnas.1510329112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Hernandez D, Torres CA, Setlik W, Cebrián C, Mosharov EV, Tang G, Cheng HC, Kholodilov N, Yarygina O, Burke RE, Gershon M, Sulzer D. Regulation of presynaptic neurotransmission by macroautophagy. Neuron. 2012;74:277–284. doi: 10.1016/j.neuron.2012.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbeck PJ. Products of endocytosis and autophagy are retrieved from axons by regulated retrograde organelle transport. J Cell Biol. 1993;121:305–315. doi: 10.1083/jcb.121.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SL, Korobova F, Svitkina T. Axon initial segment cytoskeleton comprises a multiprotein submembranous coat containing sparse actin filaments. J Cell Biol. 2014;205:67–81. doi: 10.1083/jcb.201401045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- Kapitein LC, Schlager MA, Kuijpers M, Wulf PS, van Spronsen M, MacKintosh FC, Hoogenraad CC. Mixed microtubules steer dynein-driven cargo transport into dendrites. Curr Biol. 2010;20:290–299. doi: 10.1016/j.cub.2009.12.052. [DOI] [PubMed] [Google Scholar]
- Kleele T, Marinković P, Williams PR, Stern S, Weigand EE, Engerer P, Naumann R, Hartmann J, Karl RM, Bradke F, Bishop D, Herms J, Konnerth A, Kerschensteiner M, Godinho L, Misgeld T. An assay to image neuronal microtubule dynamics in mice. Nat Commun. 2014;5:4827. doi: 10.1038/ncomms5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köchl R, Hu XW, Chan EY, Tooze SA. Microtubules facilitate autophagosome formation and fusion of autophagosomes with endosomes. Traffic. 2006;7:129–145. doi: 10.1111/j.1600-0854.2005.00368.x. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Wang QJ, Holstein GR, Friedrich VL, Jr, Iwata J, Kominami E, Chait BT, Tanaka K, Yue Z. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci U S A. 2007;104:14489–14494. doi: 10.1073/pnas.0701311104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009;122:3589–3594. doi: 10.1242/jcs.051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Sato Y, Nixon RA. Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer's-like axonal dystrophy. J Neurosci. 2011;31:7817–7830. doi: 10.1523/JNEUROSCI.6412-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maday S, Holzbaur EL. Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Dev Cell. 2014;30:71–85. doi: 10.1016/j.devcel.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maday S, Wallace KE, Holzbaur EL. Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J Cell Biol. 2012;196:407–417. doi: 10.1083/jcb.201106120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama J, Miura E, Mizushima N, Watanabe M, Yuzaki M. Aberrant membranes and double-membrane structures accumulate in the axons of Atg5-null Purkinje cells before neuronal death. Autophagy. 2007;3:591–596. doi: 10.4161/auto.4964. [DOI] [PubMed] [Google Scholar]
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- Petersen JD, Kaech S, Banker G. Selective microtubule-based transport of dendritic membrane proteins arises in concert with axon specification. J Neurosci. 2014;34:4135–4147. doi: 10.1523/JNEUROSCI.3779-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowitz JD, White E. Autophagy and metabolism. Science. 2010;330:1344–1348. doi: 10.1126/science.1193497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ, Rubinsztein DC. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- Saab AS, Tzvetanova ID, Nave KA. The role of myelin and oligodendrocytes in axonal energy metabolism. Curr Opin Neurobiol. 2013;23:1065–1072. doi: 10.1016/j.conb.2013.09.008. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shehata M, Matsumura H, Okubo-Suzuki R, Ohkawa N, Inokuchi K. Neuronal stimulation induces autophagy in hippocampal neurons that is involved in AMPA receptor degradation after chemical long-term depression. J Neurosci. 2012;32:10413–10422. doi: 10.1523/JNEUROSCI.4533-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Ganetzky B. Autophagy promotes synapse development in Drosophila. J Cell Biol. 2009;187:71–79. doi: 10.1083/jcb.200907109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song AH, Wang D, Chen G, Li Y, Luo J, Duan S, Poo MM. A selective filter for cytoplasmic transport at the axon initial segment. Cell. 2009;136:1148–1160. doi: 10.1016/j.cell.2009.01.016. [DOI] [PubMed] [Google Scholar]
- Tang G, Gudsnuk K, Kuo SH, Cotrina ML, Rosoklija G, Sosunov A, Sonders MS, Kanter E, Castagna C, Yamamoto A, Yue Z, Arancio O, Peterson BS, Champagne F, Dwork AJ, Goldman J, Sulzer D. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron. 2014;83:1131–1143. doi: 10.1016/j.neuron.2014.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada M, Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993;333:169–174. doi: 10.1016/0014-5793(93)80398-E. [DOI] [PubMed] [Google Scholar]
- Tsvetkov AS, Miller J, Arrasate M, Wong JS, Pleiss MA, Finkbeiner S. A small-molecule scaffold induces autophagy in primary neurons and protects against toxicity in a Huntington disease model. Proc Natl Acad Sci U S A. 2010;107:16982–16987. doi: 10.1073/pnas.1004498107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Martin S, Papadopulos A, Harper CB, Mavlyutov TA, Niranjan D, Glass NR, Cooper-White JJ, Sibarita JB, Choquet D, Davletov B, Meunier FA. Control of autophagosome axonal retrograde flux by presynaptic activity unveiled using botulinum neurotoxin type A. J Neurosci. 2015;35:6179–6194. doi: 10.1523/JNEUROSCI.3757-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YC, Holzbaur EL. The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J Neurosci. 2014;34:1293–1305. doi: 10.1523/JNEUROSCI.1870-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Zhou B, Lin MY, Wang S, Foust KD, Sheng ZH. Endolysosomal deficits augment mitochondria pathology in spinal motor neurons of asymptomatic fALS mice. Neuron. 2015;87:355–370. doi: 10.1016/j.neuron.2015.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9:1102–1109. doi: 10.1038/ncb1007-1102. [DOI] [PubMed] [Google Scholar]
- Xu K, Zhong G, Zhuang X. Actin, spectrin, and associated proteins form a periodic cytoskeletal structure in axons. Science. 2013;339:452–456. doi: 10.1126/science.1232251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A, Yue Z. Autophagy and its normal and pathogenic states in the brain. Annu Rev Neurosci. 2014;37:55–78. doi: 10.1146/annurev-neuro-071013-014149. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct. 1998;23:33–42. doi: 10.1247/csf.23.33. [DOI] [PubMed] [Google Scholar]
- Young JE, Martinez RA, La Spada AR. Nutrient deprivation induces neuronal autophagy and implicates reduced insulin signaling in neuroprotective autophagy activation. J Biol Chem. 2009;284:2363–2373. doi: 10.1074/jbc.M806088200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Z. Regulation of neuronal autophagy in axon: implication of autophagy in axonal function and dysfunction/degeneration. Autophagy. 2007;3:139–141. doi: 10.4161/auto.3602. [DOI] [PubMed] [Google Scholar]
- Zala D, Hinckelmann MV, Yu H, Lyra da Cunha MM, Liot G, Cordelières FP, Marco S, Saudou F. Vesicular glycolysis provides on-board energy for fast axonal transport. Cell. 2013;152:479–491. doi: 10.1016/j.cell.2012.12.029. [DOI] [PubMed] [Google Scholar]