

Abstract
Epidemiological evidence conclusively demonstrates that calcium burden is a significant predictor of cardiovascular morbidity and mortality; however, the underlying mechanisms remain largely unknown. These observations have challenged the previously held notion that calcification serves to stabilize the atherosclerotic plaque. Recent studies have shown that microcalcifications that form within the fibrous cap of the plaques lead to the accrual of plaque‐destabilizing mechanical stress. Given the association between calcification morphology and cardiovascular outcomes, it is important to understand the mechanisms leading to calcific mineral deposition and growth from the earliest stages. We highlight the open questions in the field of cardiovascular calcification and include a review of the proposed mechanisms involved in extracellular vesicle‐mediated mineral deposition.
Abbreviations
- ACP
amorphous calcium phosphate
- ANK
ankylosis protein
- CT
computed tomography
- ECM
extracellular matrix
- EV
extracellular vesicle
- FetA
fetuin A
- HU
Hounsfield unit
- MGP
matrix Gla protein
- MV
matrix vesicle
- NC
nucleational core
- NPP
nucleoside pyrophosphohydrolase
- PChol
phosphocholine
- PEA
phosphoethanolamine
- PET
positron emission tomography
- PHOSPHO1
phosphatase orphan 1
- Pi
phosphate
- PPi
pyrophosphate
- TNAP
tissue non‐specific alkaline phosphatase
- VSMC
vascular smooth muscle cell
Vascular calcification predicts cardiovascular morbidity and mortality
Rupture of vulnerable atherosclerotic plaques is the leading cause of heart attacks and strokes (Lloyd‐Jones et al. 2010). The root cause of this acute rupture event is a failure of the collagen‐rich fibrous cap that lies at the boundary of the plaque and the vessel lumen. Rupture of the fibrous cap exposes the underlying plaque contents to blood flow, leading to platelet‐mediated thrombin generation and fibrin deposition. The resulting thrombus occludes the vessel, reducing blood flow and oxygen supply to downstream tissues. Despite the once prevailing clinical view that calcification of the vessel wall is inconsequential to plaque rupture, and possibly plaque stabilizing, recent data indicate that calcium score is a better predictor of acute cardiovascular events than lipid score (Vliegenthart et al. 2005; Martin et al. 2014). Inclusion of calcium score in risk assessments significantly improves the prediction of coronary heart disease and cardiovascular events in at‐risk patients (Elkeles et al. 2008; Gepner et al. 2015; Matsushita et al. 2015). Traditionally, computed tomography (CT) identifies arterial calcium as high attenuating regions (> 130 Hounsfield units (HU)) within the relatively low attenuating vascular tissue (Fig. 1) (Rumberger et al. 1999; Kelly‐Arnold et al. 2013; Han et al. 2015).
Figure 1. High resolution micro‐computed tomography (CT) of explanted human coronary artery .
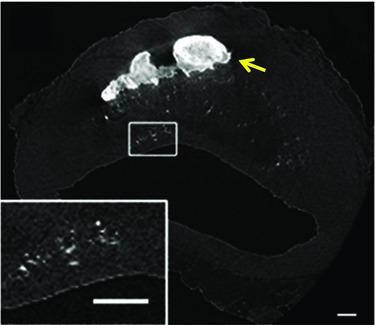
The yellow arrow indicates a large calcification, and the box highlights spotty microcalcifications. (Adapted from Kelly‐Arnold et al. 2013.)
In a study of 1795 asymptomatic participants, compared to participants with an Agatston score lower than 100, the relative risk of coronary heart disease increased to 3.1 for scores of 101–400 HU, to 4.6 for scores of 401–1000 HU, and to 8.3 for scores higher than 1000 HU (Vliegenthart et al. 2005). Further, a retrospective study of 23,057 patients found a significant relationship between the number of coronary arteries (right, left main, left anterior and left circumflex) with calcification in each patient and all‐cause mortality within 7 years of the initial measurement of calcium burden (Tota‐Maharaj et al. 2015). The compiled data from these studies suggest a strong link between cardiovascular calcification and morbidity/mortality that has challenged previously held notions that calcification stabilizes atherosclerotic plaques. Indeed, biomechanical models predict that large calcifications that form within the lipid pool/necrotic core of atherosclerotic plaques increase plaque stability by reducing the deformation of the fibrous cap during systolic pressure (Imoto et al. 2005; Wong et al. 2012; Holzapfel et al. 2014; Ruiz et al. 2015) (Fig. 2). Studies into this apparent contradiction have revealed that calcification morphology and location within the plaque determine the impact of the calcification on plaque stability.
Figure 2. Schematic illustration of the mechanism of atherosclerotic plaque stabilization by a large calcification .

The lipid pool/necrotic core of a non‐calcified plaque is deformable, which allows for high tissue strain in the fibrous cap during systole. Large, dense calcifications help counteract the resulting stress by limiting the degree of fibrous cap deformation that occurs under systolic pressure. (From Ruiz et al. 2015.)
Microcalcification and rupture of the atherosclerotic fibrous cap
The first indication that calcification morphology influences plaque stability came from the observation that ‘spotty’ calcification identified by CT (Ferencik et al. 2012) and ultrasound (Ehara et al. 2004) correlates with cardiovascular events. Further, prospective clinical data from the Multi‐Ethnic Study of Atherosclerosis trial found calcium density to be inversely related to the incidence of cardiovascular events (Criqui et al. 2014). The calcium density score was determined by dividing the Agatston score by the total area score in each patient. Including the density score in the prediction models significantly improved risk prediction for both coronary heart disease and cardiovascular disease events. These data support the predictions that larger, denser calcifications may indeed stabilize atherosclerotic plaques (Imoto et al. 2005; Lin et al. 2006; Wong et al. 2012; Holzapfel et al. 2014), whereas smaller, disperse calcifications seem to contribute to plaque destabilization (Ehara et al. 2004). Thus the morphology of the calcification directly determines the associated cardiovascular risk.
Ultrasound and CT are currently able to image only calcifications larger than 30 μm, whereas clinically relevant microcalcifications that lead to plaque rupture are largely 5–15 μm and visible either in histology or high resolution microCT (Kelly‐Arnold et al. 2013; Maldonado et al. 2013). However, it is possible that a cloud of numerous closely spaced, smaller microcalcifications could appear as a single larger microcalcification in CT. This same observation was made when what was thought to be a single microcalcification at 6.7 μm resolution microCT was shown to be a collection of numerous smaller microcalcifications at 2.1 resolution microCT (Kelly‐Arnold et al. 2013). Future work is needed to connect the observation of ‘spotty’ calcification to the presence of clinically unidentifiable microcalcifications that may provide the link between calcification and biomechanical failure of the atherosclerotic plaque.
Vengrenyuk et al. (2006) provided the first explanation of the biomechanical mechanisms through which subcellular ‘microcalcifications’ within the fibrous cap contribute to the rupture of atherosclerotic plaques. In silico modelling of the mechanical stress distribution within the fibrous cap by finite element analysis shows increased stress surrounding microcalcifications (Fig. 3). The stiff microcalcifications present impurities within the relatively soft vascular tissues (Vengrenyuk et al. 2006). Modelling the stresses under different scenarios indicates that calcification size, morphology, spacing and collagen content combine to determine the likelihood of plaque rupture (Kelly‐Arnold et al. 2013; Maldonado et al. 2013). The largest increase in tissue stress occurs when two closely spaced microcalcifications aligned along the tensile axis of the fibrous cap are pulled apart. The tissue in the intervening gap between the particles can undergo an amplification of stress that is more than fivefold depending on their spacing (Kelly‐Arnold et al. 2013; Maldonado et al. 2013). Large calcifications beneath the cap are stabilizing because they attach to the boundaries of the lipid pool/necrotic core and support the cap from below (Maldonado et al. 2015). Therefore, more work is needed to determine how different calcification morphologies arise and the factors that determine calcification location – especially regarding the development of fibrous cap microcalcifiations.
Figure 3. Microcalcifications within the fibrous cap .
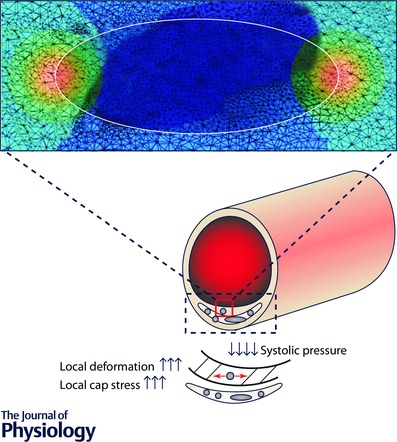
Microcalcifications serve as foci for high levels of local stress within the fibrous cap. Finite element analysis of the local stress levels surrounding microcalcifications within a fibrous cap. The gradient from blue to red indicates a transition from low to high mechanical stress. (Adapted from Vengrenyuk et al. 2006; Ruiz et al. 2015.)
Recently, nano‐analytical electron microscopy techniques revealed structural detail of calcific mineral within cardiovascular tissues that included large regions of calcification and small microcalcifications (Bertazzo et al. 2013). The formation of large calcifications and microcalcifications may involve cell‐derived extracellular vesicles (EVs) that serve as nucleating foci for mineralization (Anderson, 2003; Kapustin et al. 2011; New et al. 2013). Work from our laboratory showed that collagen within the atherosclerotic plaques acts as a scaffold, directing the aggregation of calcifying EVs and thus influencing calcification morphology (Hutcheson et al. 2016). Our laboratory used structured illumination microscopy to directly visualize the aggregation of individual EVs, which appeared to nucleate calcium phosphate mineral in a spherical morphology (Hutcheson et al. 2016), and both nanoparticle tracking analyses and spectroscopic analyses revealed mineral formation and maturation concomitant with EV aggregation. Further, optical and electron micrographs of calcific human arterial plaques showed calcifications composed of aggregated EVs. It remains unclear whether calcification builds solely by aggregation and fusion of EVs or if it is possible for mineral to grow directly from a single EV under certain conditions. MicroCT and histopathological analyses of excised human plaques revealed large macrocalcifications in areas corresponding to the lipid pool/necrotic core of the plaque (Kelly‐Arnold et al. 2013; Maldonado et al. 2015). Detailed histopathological analyses of atherosclerotic plaques showed that lipid pools often contain microcalcifications (Otsuka et al. 2015). Lipid pools of atherosclerotic plaques often transition into necrotic cores over time, though many factors regulating this transition remain poorly understood (Sakakura et al. 2013). The necrotic core does contain both microcalcifications and larger calcifications. Further, small microcalcifications in regions of calcification development preferentially form and align at the borders of the lipid pool/necrotic core (Maldonado et al. 2015). Oxidized forms of cholesterol have shown the propensity to accelerate calcification by osteogenic reprogramming of vascular smooth muscle cells (VSMCs) and interactions with calcifying EVs (Hsu, 2003). Our recent work also showed the formation of microcalcifications in spaces between collagen fibres and in regions of collagen fibre degradation within the fibrous cap of atherosclerotic plaques (Hutcheson et al. 2016). The microcalcifications that form within the fibrous cap preferentially form near the lipid pool/necrotic core (Maldonado et al. 2015). The observation of microcalcifications in the fibrous cap and lipid pool/necrotic core may suggest a role for both VSMCs (Kapustin et al. 2011; Hutcheson et al. 2014) and macrophages (New et al. 2013) in mineral deposition.
Calcification progresses over time in newly formed lesions as microcalcifications appear first in the lipid pool. As the plaque continues to develop the late necrotic core can contain large macrocalcifications with microcalcifications at the periphery. Therefore, within a single patient, as a plaque develops along the lumen, multiple calcification morphologies can be observed as calcifying vesicles diffuse slowly through the lesion to form microcalcifications that build into macrocalcifications (Maldonado et al. 2015). As predicted, lipid pool/necrotic core calcification that occurs beneath a collagen‐rich fibrous cap may serve to stabilize the plaque; however, collagen breakdown at localized regions in vulnerable fibrous caps due to the presence of inflammation and cell migration may lead to the formation of stress‐inducing microcalcifications that cause plaque rupture (Hutcheson et al. 2016). More work is needed to better understand the factors driving EV aggregation and association with extracellular matrix and other plaque components to build calcification. Though these recent studies have provided some explanation of the mechanisms by which EVs may contribute to calcification genesis and growth, the specific processes leading to calcifying EV generation and mineral nucleation remain largely unclear, and traditional clinical imaging modalities cannot readily detect early calcific remodelling to identify at‐risk patients.
Recent advances using positron emission tomography/CT (PET/CT) with 18F‐labelled sodium fluoride (Na18F), an established PET tracer for bone formation and remodelling, may provide new strategies for identifying regions of microcalcification and plaque vulnerability (Chen & Dilsizian, 2013). Coronary uptake of Na18F was found overlaying, adjacent to and distal from regions of CT identified calcifications (Dweck et al. 2012). Additionally, large areas of calcification with no Na18F uptake were observed. This suggests that, as with bone, Na18F uptake in the vasculature is a marker of ongoing calcific remodelling (Dweck et al. 2012). Large, stable calcifications do not exhibit Na18F uptake, whereas active regions of mineralization accumulate Na18F. Future studies are needed to identify cellular and extracellular mechanisms associated with Na18F uptake. The Na18F regions far away from the CT identified calcific regions may represent processes that lead to the development of clinically relevant microcalcifications that cannot be detected by traditional imaging modalities (Dweck et al. 2012). In support of this hypothesis, a prospective clinical trial showed high Na18F accumulation in the culprit coronary plaques in cases of myocardial infarction and in ruptured carotid artery plaques (Joshi et al. 2014). Histological evaluation of these plaques revealed active calcification processes and regions of microcalcifications, indicating that these processes associate with Na18F uptake.
Cell‐mediated processes actively drive mineral nucleation
It is generally believed that many aspects of vascular calcification mirror that of bone formation. Namely, both processes appear to utilize cell‐derived, calcifying vesicles to nucleate and transport mineral to a collagen‐rich matrix. While a competing theory posits that bone mineralization is an acellular process directed by specific zones of the collagen fibril (Glimcher, 1984; Dey et al. 2010; Nudelman et al. 2010), there exists a body of evidence demonstrating the requisite role of cell activity. This hypothesis is exemplified by studies in which the inhibition of cell metabolism or the disruption of cytoskeletal regulation led to a decrease in mineral deposition measured in vitro (Stanford et al. 1995; Drabek et al. 2011). These cellular processes culminate in the generation of vesicles, loaded with enzymatic machinery that concentrates calcium and phosphate ions within the lumen for mineral nucleation.
Similarly, calcifying vesicles have been visualized in and isolated from atherosclerotic vessels (Kim, 1976; Tanimura et al. 1983; Hsu & Camacho, 1999). Inflammation within the atherosclerotic environment may trigger an osteochondrogenic transformation of VSMCs (Tintut et al. 2000; Aikawa et al. 2007). Both osteogenic VSMCs (Tanimura et al. 1986; Kapustin et al. 2011) and macrophages (New et al. 2013) have been implicated in producing calcifying vesicles. The degree of mechanistic overlap between bone and vascular mineralization is unclear. However, literature from the bone mineralization field contains the most extensive exploration of the nucleational properties of bone‐derived vesicles. These findings provide a concrete starting point for unravelling the mechanism of vascular calcification, and as such, we will explore key findings from both fields below. For clarification, moving forward we will refer to cardiovascular extracellular vesicles as EVs and osteochondral matrix vesicles as MVs.
Calcific conditions within an atherosclerotic lesion result in the production of vesicles that are mineralization competent (Fig. 4). Specifically, the pro‐calcific vesicles are proposed to contain lipid, protein and ionic components necessary to form membrane‐anchored structures that are the starting point for calcium phosphate mineral formation, known as the nucleational core (NC) (Wu et al. 1993, 1997 a). The NC is fed with Ca2+/Pi ions from multiple sources. Within the vesicle, phosphatase orphan 1 (PHOSPHO1) cleaves Pi ions primarily from two phospholipids enriched in the EV membrane: phosphoethanolamine (PEA) and phosphocholine (PChol) (Mebarek et al. 2013). Enzymes bound to the external EV membrane, particularly tissue non‐specific alkaline phosphatase (TNAP), nucleoside pyrophosphohydrolase (NPP) 1 and NPP3, liberate Pi from ATP, ADP and PPi in the extravesicular space (Ciancaglini et al. 2010). Pit1 and Pit2 transporters pump these Pi ions into the EV (Anderson, 2003, 2005). Calcium is supplied to the NC by annexins A2, A5, and A6, as well as other calcium transporting enzymes (Kirsch et al. 2000 b). As extensive intraluminal Ca2+/Pi mineral forms, the EV membrane undergoes a loss of structural integrity, and mineral crystals are propagated out onto the collagen‐enriched matrix, guided by proteins that attach the EV membrane to collagen (Wu et al. 2002; Mebarek et al. 2013). The continued extra‐vesicular propagation of mineral is regulated by the ratio of pyrophosphate (PPi), an inhibitor of crystal growth (Francis, 1969), and its derivative, Pi.
Figure 4. Proposed mechanism of vesicle‐mediated mineral formation .
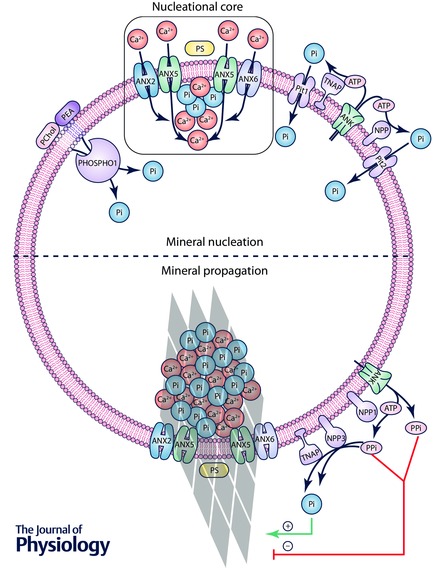
Top, cell‐derived vesicles are preloaded with a nucleational core (NC), composed of annexins A2, A5 and A6, as well as a complex of PS, Ca2+ and Pi. Calcium and phosphate ions are accumulated within the vesicle lumen via multiple routes: intravesicular Pi cleavage from PChol and PEA via PHOSPHO1; Pi cleavage from ATP in the extravesicular space via TNAP and NPP1/3, and import via Pit1/2; and calcium import via annexins A2, A5 and A6. These ions are patterned as calcium phosphate mineral at the NC. Bottom, over time, mineral crystals propagate through the vesicle membrane, where their continued growth is regulated by the ratio of PPi, an inhibitor of crystal growth, to its derivative Pi. Note that the hypothesis that annexins form transmembrane ion channels, as depicted in this figure, is controversial (given that annexins are believed to primarily function as peripheral membrane proteins), thus meriting further investigation.
Membrane‐bound nucleational core is the primary unit of vesicle mineralization
Pivotal studies conducted two decades ago subjected chondrocyte‐derived MVs to a series of chemical and detergent extraction steps to reduce MVs to their most basic unit that retained the ability to nucleate mineral formation (Wu et al. 1993, 1997 a). This and subsequent studies by Wuthier and colleagues demonstrated that this NC is composed of three key components (Genge et al. 2007): amorphous calcium phosphate (ACP); CPLX, a membrane‐associated complex of PS–Ca2+–Pi; and annexin A5 (Wu et al. 1993, 1997 a). While ACP comprises 91.8% of the NC (Wu et al. 1997 a) experiments with in vitro synthesized NCs demonstrated that ACP mediates only 20% of mineral formation (Wu et al. 2008), concluding that CPLX predominately exerts the nucleating power of the NC. Furthermore, annexin A5, and to some degree annexin A6, catalysed the onset and increased the extent of CPLX mineral nucleation by 10‐ to 20‐fold (Genge et al. 2007). Ultimately, Wutheir and colleagues hypothesize that the atomic arrangement of the CPLX–annexin A5 complex provides a molecular template that guides the arrangement of Ca2+/Pi ions into a crystalline pattern (Genge et al. 2007). These hypotheses have been supported by electron microscopy images that demonstrate calcium phosphate deposits localized to the inner MV membrane (Anderson et al. 2005).
Furthermore, it has been proposed that MV budding and NC formation occur simultaneously within the mineralizing chondrocyte. These cells demonstrate an increase in intracellular phosphate (Wuthier, 1977) and a marked increase in intracellular Ca2+ levels near the plasma membrane right before MV budding (Wu et al. 1997 b). The elevated peripheral calcium may drive simultaneous clustering of PS within the cell membrane and binding of annexin A5 to these PS‐rich regions, leading to NC formation that is timed with MV budding (Wu et al. 1993). Indeed, when comparing MVs from mineralizing vs. non‐mineralizing chondrocytes, only those from mineralizing cells contained annexins, demonstrating the specialized loading of mineral‐nucleating MVs with pro‐calcific components (Kirsch et al. 2000 a). EVs elaborated by VSMCs are similarly enriched in annexins A2, A5 and A6, hinting at a shared mechanism of EV loading and mineral nucleation by these two cell types (Chen et al. 2008; Kapustin et al. 2011). However, the presence of an NC has not been identified within vascular EVs, requiring more extensive investigation.
Concerted effort of vesicle machinery concentrates ions for mineral nucleation
Annexins mediate Ca2+ influx via direct and indirect activities
Annexins are hypothesized to contribute to two other bone‐derived MV functions: collagen binding and Ca2+ influx. Annexin A2, A5 and A6 are all known components of MVs, and have all individually been demonstrated to mediate Ca2+ influx into PS‐rich liposomes and non‐calcifying MVs that were supplemented with exogenous annexin A2, A5 and A6 (Kirsch et al. 2000 b). This hypothesis is supported by structural analyses, which revealed that the hydrophilic pore located within the structure of the annexin core is biochemically similar to other Ca2+ channels (Gerke, 2002). However, the postulated role of annexins as Ca2+ channels has yet to be demonstrated in living cells (Gerke, 2002). Furthermore, the annexin‐specific Ca2+ channel blocker K‐201 did not suppress EV mineralization (Kapustin et al. 2011), calling to question the function of annexins as mediators of EV Ca2+ influx. Indeed, annexins are primarily thought of as peripheral membrane‐binding proteins. A dynamic interplay of several factors has been proposed to mediate potential annexin membrane insertion, including lipid bilayer composition, annexin concentration, Ca2+ concentration, a hyperpolarization of transmembrane voltage, membrane destabilization and intracellular/intravesicular acidification (Gerke, 2002). Further studies are necessary to definitively determine if annexins have the capacity to function as cellular and/or vesicular Ca2+ channels.
Only annexin A5 binds types II and X collagen, anchoring MVs to the extracellular matrix (ECM). This collagen binding enhances Ca2+ influx via annexin A5 (Kirsch et al. 2000 a), demonstrating an active interplay between the ECM and MV calcification. Interestingly, EVs isolated from bovine vascular smooth muscle cells (BVSMCs) were only capable of inducing mineral formation along type I collagen fibrils, not type II collagen (Chen et al. 2008). Given that type I collagen is up‐regulated in the setting of atherosclerosis and medial calcification (Rekhter et al. 1993), these findings indicate that EVs are produced with an affinity to promote calcification specific to the ECM environment into which they are released (Chen et al. 2008).
Additionally, there is evidence that annexin A2 may act to induce TNAP activity in MVs. Annexin A2 and TNAP have been shown to colocalize at the periphery of calcifying SaOSLM2 osteoblastic cells. Membrane fractionation has demonstrated that both are more specifically localized to cholesterol‐stabilized lipid rafts. An overexpression of annexin A2 led to an increase in TNAP activity in the lipid‐raft membrane fraction (Gillette & Nielsen‐Preiss, 2004). Similarly, VSMC‐EVs treated with the annexin Ca2+ channel inhibitor K201 have demonstrated a decrease in TNAP activity (Chen et al. 2008).
Intraluminal PHOSPHO1 generates phosphate ions that initiate mineral nucleation
The phosphate ions (PO4 3− or Pi) that combine with Ca2+ to build mineral at the NC are sequestered in larger molecular structures to prevent spontaneous, indiscriminate calcium phosphate precipitation. Enzymes positioned on the inner and outer MV membrane liberate Pi for mineral nucleation. Mice deficient in these enzymes do not undergo skeletal mineralization, even in the presence of abundant serum Pi (Millan, 2013), highlighting the requirement for active, ‘peri‐vesicular’ generation of Pi.
PHOSPHO1, a Mg2+‐dependent hydrolase (Stewart et al. 2003), is the only enzyme thus far identified to generate Pi within MVs and EVs. This enzyme primarily cleaves Pi from PEA and PChol (Mebarek et al. 2013), poorly hydrolysing more classical Pi‐providing substrates associated with bone mineralization, including ATP, ADP and PPi (Ciancaglini et al. 2010). Indeed, recent studies have used MVs from autopsy samples from TNAP deficient patients and from TNAP knockout mice to demonstrate that TNAP is not essential to achieve intraluminal mineralization (Anderson et al. 1997, 2004). Furthermore, calcifying VSMCs have demonstrated an up‐regulation of Phospho1 expression and activity (Kiffer‐Moreira et al. 2013). Both PHOSPHO1 knockout (Phospho1 −/−) VSMCs and VSMCs under pharmacological PHOSPHO1 inhibition undergo a significant reduction in their ability to calcify (Kiffer‐Moreira et al. 2013). Because PHOSPHO1 drives the initial, intravesicular stage of mineralization, it has been proposed that targeting PHOSPHO1 may be more effective than inhibiting TNAP at preventing vascular calcification (Kiffer‐Moreira et al. 2013).
Reduced skeletal mineralization seen in Phospho1 −/− mice is not corrected by a superimposed overexpression of TNAP, indicating that these two enzymes fulfil distinct roles in the process of vesicle‐mediated mineralization (Millan, 2013). However, Phospho1 −/− EVs exhibit a 70% reduction in TNAP activity (Ciancaglini et al. 2010), and PHOSPHO1 inhibition leads to a reduction of TNAP mRNA (Yadav et al. 2011). Thus, just as with the interplay between annexin A2 and TNAP, the complex relationship between PHOSPHO1 and TNAP activity merits further investigation.
Extravesicular Pi is regulated by a host of membrane‐bound enzymes and pumps
PHOSPHO1‐driven Pi generation takes place in conjunction with the coordinated activity of several enzymes and pumps interacting with the extravesicular space. Within osteochondrogenic mineralization, ankylosis protein (ANK) has been proposed to pump ATP (Villa‐Bellosta et al. 2011) and PPi (Zhao et al. 2012) out of MV‐releasing cells, and perhaps MVs themselves. ATP is the principal source of Pi channelled back into MVs for mineral nucleation, with additional contributions from ADP and PPi (Ciancaglini et al. 2010; Millan, 2013). Pi liberation from these molecules is predominantly driven by TNAP, which is expressed in osteoblasts and chondrocytes (Ciancaglini et al. 2010), as well as calcifying VSMCs (Villa‐Bellosta et al. 2011; Haarhaus et al. 2013; Kiffer‐Moreira et al. 2013). However, MV‐mediated ATP hydrolysis persists in the absence of TNAP and PHOSPHO1 (Ciancaglini et al. 2010), and mice with a combined deficiency in PHOSPHO1 protein and TNAP function (Phospho1 −/− Akp2 −/−) still underwent a small degree of skeletal mineralization (Yadav et al. 2011). Thus a third Pi‐generating mechanism likely contributes to mineralization.
Recent studies have demonstrated that the NPP family may also play a role in osteochondrogenic and vascular mineralization. NPP has demonstrated ATPase, ADPase and PPiase activity in MVs, but only when TNAP is absent or inhibited (Ciancaglini et al. 2010). TNAP outcompetes NPP1 to bind ATP, ADP and PPi (Ciancaglini et al. 2010), limiting the degree to which NPP1 can contribute to Pi generation. Applying inhibitors of TNAP, PHOSPHO1 and NPP to wild‐type MVs led to a complete inhibition of ATPase activity (Ciancaglini et al. 2010). Further, not only do rat and mouse aorta express NPP1–3 (Villa‐Bellosta et al. 2011), but NPP1 knockout (Enpp1 −/−) VSMCs cultured with PHOSPHO1 and TNAP inhibitors demonstrate a 93% decrease in mineralization, versus only 65% in wild‐type cells cultured with both inhibitors (Kiffer‐Moreira et al. 2013).
While Ciancaglini et al. argued that the observed compensatory phosphatase activity in TNAP‐deficient bone‐derived MVs was mediated by NPP1 (Ciancaglini et al. 2010), the possible contribution of NPP3 in addition to NPP1 was not addressed. Indeed, while Ciancaglini et al. found that purified NPP1 mediated PPi hydrolysis, Villa‐Bellosta and colleagues demonstrated that PPi hydrolysis was carried out only by NPP3‐transfected HEK cells, observing no phosphatase activity in NPP1 cells (Villa‐Bellosta et al. 2011). Furthermore, explanted aortas from Enpp1 −/− mice did not demonstrate altered PPi hydrolysis (Villa‐Bellosta et al. 2011). Further studies are needed to tease apart the exact roles of NPP1 and NPP3 in vascular calcification.
Nevertheless, these studies have demonstrated that the vast majority of extravesicular Pi generation is driven by TNAP and NPPs (Millan, 2013). Additional MV enzymes that act to liberate Pi in the extravesicular space include NTPDases, which convert ATP to AMP and 2 Pi (Villa‐Bellosta et al. 2011), and ecto‐5′‐nucleotidase, which cleaves AMP to adenosine and Pi (Fish et al. 2013). Liberated Pi may subsequently be pumped into the vesicle via type III sodium phosphate cotransporters Pit1 and Pit2 (Anderson, 2003; Anderson et al. 2005). Several reviews describe the bone forming activity of these phosphate‐handling enzymes in greater detail (Anderson, 2003; Golub, 2011; Wuthier & Lipscomb, 2011; Fish et al. 2013; Millan, 2013).
To become nucleation competent, forming EVs may also require a reduction in the concentration of mineralization inhibitors that they contain, most notably including fetuin‐A (FetA) and matrix Gla protein (MGP). FetA is a circulating glycoprotein produced predominantly by the liver (Dziegielewska et al. 1987). It inhibits MV and EV mineralization by binding to calcium phosphate crystals and preventing further crystal growth (Reynolds et al. 2005). Importantly, several studies have demonstrated that there exists a negative correlation between serum concentration of FetA and severity of cardiovascular calcification (Kapustin et al. 2015). Kapustin et al. recently observed the internalization of FetA by VSMCs via a receptor‐independent mechanism (Kapustin et al. 2015). The FetA was observed in both the late endosomal and lysosomal compartments, and some of it was ultimately packed into EVs, which the authors identified as exosomes. Further, an earlier study from the same group demonstrated that EVs containing FetA were less likely to contain calcium phosphate mineral (Reynolds et al. 2005).
Similarly, MGP is a (vitamin K‐dependent) mineralization inhibitor that regulates MV and EV calcification. Produced directly by VSMCs, MGP is loaded into forming EVs (Reynolds et al. 2004), where it is proposed to bind growing calcium phosphate crystals and prevent further crystal growth (Wuthier & Lipscomb, 2011). The stresses that promote an osteochondrogenic conversion of VSMCs have been hypothesized to lead to a decrease in the amount of FetA and MGP loaded into EVs. Further, calcifying VSMCs tend to produce undercarboxylated MGP, which is non‐functional (Kapustin & Shanahan, 2012). The loss of inhibitors packed into EVs is permissive to EV mineralization and microcalcification.
Restructuring of MV lipid membrane permits crystal escape beyond the intraluminal space
As bone‐derived MVs undergo intraluminal mineral nucleation, the membrane lipid composition undergoes significant turnover to become more permissive for the outgrowth of calcium phosphate crystals (Wu et al. 2002; Mebarek et al. 2013). MVs are loaded with phospholipases, including phospholipase A and C (PLA and PLC), which degrade membrane phospholipids to lysophospholipids (LPLs) that alter membrane curvature and compromise membrane integrity (Wu et al. 2002; Mebarek et al. 2013). The exact means by which calcium phosphate crystals emerge from the MV membrane is poorly understood. Kapustin et al. demonstrated that annexin A6 and PS can be isolated from the inner and outer membranes of VSMC‐derived EVs. Furthermore, several studies used TEM to visualize mineral nucleation that occurred on both the inner (Fig. 5 A) and outer membranes (Fig. 5 B) of EVs (Aikawa et al. 2007; Kapustin et al. 2011; New et al. 2013). Thus, active NCs may be present on both sides of the EV membrane. Phospholipase activity may externalize membrane‐associated NCs, which would allow NC‐guided crystal growth to progress in the extravesicular space (Wuthier & Lipscomb, 2011). Subsequently, annexin would mediate binding of the NC to collagen, which would then further guide crystal outgrowth (Wuthier & Lipscomb, 2011). Similarly, TNAP binds collagen via its crown domain, an interaction that is critical for the activity of the enzyme (Mornet et al. 2001). Calcium phosphate crystals may similarly emerge onto the ECM via this TNAP connection. Ultimately, these calcium phosphate crystals form clusters centred upon the nucleating MV, progressing outward within and along the surrounding collagen fibrils (Wu et al. 2002; Millan, 2013). The collagen meshwork contains hole zones that are aligned to form channels sized appropriately for the entry of calcium phosphate nanocrystals (Golub, 2011). These crystals then propagate within the collagen to fill all available intrafibrillar spaces (Golub, 2011). Therefore, our limited insight into how mineral crystals emerge from nucleating vesicles comes exclusively from the field of bone mineralization. EV‐specific studies are needed to determine if these proposed osteochondrogenic mechanisms are also at play in vascular calcification.
Figure 5. Electron micrographs of mineral formation on EV inner membrane (A) and outer membrane (B) .
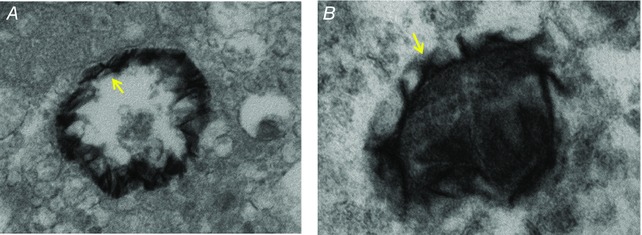
(Adapted from Aikawa et al. 2007.)
Extravesicular mineral propagation regulated by the PPi/Pi ratio
The propagation of any calcium phosphate mineral crystal once it has emerged from the vesicle lumen is dictated by the ratio of PPi to Pi in the extravesicular environment. PPi inhibits mineral propagation by directly binding to the crystal surface and preventing the further addition of Ca2+ and Pi. PPi is supplied to this environment via two mechanisms: ANK, which releases intracellular PPi; and NPP1 and ‐3, which generate PPi chiefly from ATP (Prosdocimo et al. 2009). Studies using explanted aortas in an organ culture model have demonstrated the role of NPP in vascular calcification. Aortic tissue exposed to an NPP inhibitor or isolated from an Enpp1 −/− mouse failed to synthesize PPi from ATP (Villa‐Bellosta et al. 2011). Accordingly, children born with the inability to produce NPP1 suffer from pervasive, fatal arterial calcification (Rutsch et al. 2001). Mouse models that carry a homotypic ANK mutation (ank/ank) that restrict the PPi channelling function of the protein also demonstrate extensive soft tissue calcification (Harmey et al. 2004).
Certain tissue environments are permissive to calcification due to the expression and activity of TNAP, which has been found to conduct 50% of PPi hydrolysis (Villa‐Bellosta et al. 2011). The remaining 50% of PPi hydrolysis is accounted for in party by NPP3. HEK cells transfected with NPP3, but not those with NPP1, demonstrated an increase in PPi hydrolysis. Further, aortic explants from Enpp1 −/− mice hydrolysed PPi to the same degree as wild‐type tissues. Thus, NPP3 appears to be unique in its ability to both generate and hydrolyse PPi. Deficiencies in PPi hydrolysis result in the failure of mineralization to progress past the EV membrane, due to the suppression of extravesicular crystal expansion via PPi binding (Yadav et al. 2011).
New therapeutic targets may be hidden in the process of EV formation
Our increasing knowledge of the mechanisms driving ectopic calcification reveals multiple potential points of intervention. One approach is to target the array of EV membrane‐associated pumps and enzymes that fuel mineral nucleation. Direct targeting of mineral propagation via physicochemical means, which has previously been attempted with bisphosphonates (Lomashvili et al. 2009), is another possible approach. These attempted therapies have failed so far, however, because they compromised bone mineralization. Therefore, more specific therapeutic targets will come to light only after we develop a refined understanding of the differences that delineate calcification driven by bone‐derived MVs versus that by soft tissue‐derived EV.
A third potential target is at the level of vesicle formation, but this is arguably the least understood step in the process of tissue mineralization. Within the context of endochondral ossification, it has been demonstrated that vesicles can bud directly from microvilli of chondrocytes (Wuthier & Lipscomb, 2011). However, electron microscopic studies of osteoblasts have demonstrated the presence of intracellular vesicles loaded with calcium phosphate mineral (Rohde & Mayer, 2007; Boonrungsiman et al. 2012; Nollet et al. 2014). In some cases, intracellular mineral aggregates appeared to form freely within the cytoplasm before being engulfed by a vesicle membrane. Others have observed calcium phosphate mitochondrial granules that may subsequently be loaded into intracellular vesicles. Still others have demonstrated a potential involvement of autophagy (Nollet et al. 2014), cytoskeletal rearrangement (Drabek et al. 2011) and intracellular vesicle trafficking mediated by the ER (Stenbeck & Coxon, 2014) and golgi (Stanford et al. 1995; Rohde & Mayer, 2007). Many of these investigators have observed the exocytosis or secretion of what appear to be non‐membrane‐bound apatite crystals into the ECM (Stanford et al. 1995; Rohde & Mayer, 2007; Nollet et al. 2014). Finally, other studies conducted with osteoblasts and VSMCs provide evidence that calcifying vesicles are exosomes derived from the generation and release of multivesicular bodies (Xiao et al. 2009; Kapustin et al. 2015). Given the complex interplay between many of these systems of intracellular trafficking, more targeted studies are required to determine the origin of calcifying vesicles specifically within vascular tissues before we can design solutions to inhibit their production or disease‐causing activity.
Additional information
Competing interests
We have no conflicts of interest to declare.
Author contributions
J.L.R. and J.D.H. wrote and compiled the manuscript text. S.W. provided critical review of the text. E.A. provided critical review of the text and recommendations on manuscript figures. All authors edited the final text. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
E.A. is supported by grants from the National Institutes of Health (R01HL114805; R01HL109506). J.R. is a Howard Hughes Medical Institute Medical Research Fellow.
Biographies
Jessica L. Ruiz is an MD candidate in the Harvard‐MIT Health Sciences and Technology Program at Harvard Medical School. She joined the laboratory of Elena Aikawa in 2014 to study nucleating mechanisms of vesicle‐mediated calcification in the vasculature and is continuing this work as a 2015–2016 Howard Hughes Medical Institute Medical Research Fellow.

Sheldon Weinbaum, PhD is a CUNY Distinguished Professor Emeritus of Biomedical Engineering at The City College of New York. He is widely recognized for novel biomechanical models that have changed existing views in such areas as transport aspects of arterial disease, vulnerable plaque rupture, mechano‐transduction in bone and the renal tubule, microvascular exchange and bioheat transfer.
Elena Aikawa, MD, PhD is an Associate Professor of Medicine at Harvard Medical School, Principle Investigator at the Center for Excellence in Vascular Biology and Director of the Vascular Biology Program at the Center for Interdisciplinary Cardiovascular Sciences, Brigham and Women's Hospital, Boston, USA. Her current research focuses on the mechanisms of vascular calcification and calcific aortic valve disease.
Joshua D. Hutcheson, PhD joined the Center for Interdisciplinary Cardiovascular Sciences at Harvard Medical School and Brigham and Women's Hospital in September 2012 after completing his PhD in the Department of Biomedical Engineering at Vanderbilt University. His dissertation research focused on understanding the mechanisms of aortic valve disease and the identification of potential therapeutic targets. He has developed new techniques to visualize the earliest events associated with cardiovascular calcification.
This review was presented at the symposium “Extracellular vesicles, exosomes and microparticles in cardiovascular disease”, which took place at Physiology 2015, Cardiff, UK between 6–8 July 2015.
References
- Aikawa E, Nahrendorf M, Figueiredo JL, Swirski FK, Shtatland T, Kohler RH, Jaffer FA, Aikawa M & Weissleder R (2007). Osteogenesis associates with inflammation in early‐stage atherosclerosis evaluated by molecular imaging in vivo. Circulation 116, 2841–2850. [DOI] [PubMed] [Google Scholar]
- Anderson HC (2003). Matrix vesicles and calcification. Curr Rheumatol Rep 5, 222–226. [DOI] [PubMed] [Google Scholar]
- Anderson HC, Garimella R & Tague SE (2005). The role of matrix vesicles in growth plate development and biomineralization. Front Biosci 10, 822–837. [DOI] [PubMed] [Google Scholar]
- Anderson HC, Hsu HH, Morris DC, Fedde KN & Whyte MP (1997). Matrix vesicles in osteomalacic hypophosphatasia bone contain apatite‐like mineral crystals. Am J Pathol 151, 1555–1561. [PMC free article] [PubMed] [Google Scholar]
- Anderson HC, Sipe JB, Hessle L, Dhanyamraju R, Atti E, Camacho NP & Millan JL (2004). Impaired calcification around matrix vesicles of growth plate and bone in alkaline phosphatase‐deficient mice. Am J Pathol 164, 841–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertazzo S, Gentleman E, Cloyd KL, Chester AH, Yacoub MH & Stevens MM (2013). Nano‐analytical electron microscopy reveals fundamental insights into human cardiovascular tissue calcification. Nat Mater 12, 576–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boonrungsiman S, Gentleman E, Carzaniga R, Evans ND, McComb DW, Porter AE & Stevens MM (2012). The role of intracellular calcium phosphate in osteoblast‐mediated bone apatite formation. Proc Natl Acad Sci USA 109, 14170–14175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen NX, O'Neill KD, Chen X & Moe SM (2008). Annexin‐mediated matrix vesicle calcification in vascular smooth muscle cells. J Bone Miner Res 23, 1798–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W & Dilsizian V (2013). Targeted PET/CT imaging of vulnerable atherosclerotic plaques: microcalcification with sodium fluoride and inflammation with fluorodeoxyglucose. Curr Cardiol Rep 15, 364. [DOI] [PubMed] [Google Scholar]
- Ciancaglini P, Yadav MC, Simao AM, Narisawa S, Pizauro JM, Farquharson C, Hoylaerts MF & Millan JL (2010). Kinetic analysis of substrate utilization by native and TNAP‐, NPP1‐, or PHOSPHO1‐deficient matrix vesicles. J Bone Miner Res 25, 716–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criqui MH, Denenberg JO, Ix JH, McClelland RL, Wassel CL, Rifkin DE, Carr JJ, Budoff MJ & Allison MA (2014). Calcium density of coronary artery plaque and risk of incident cardiovascular events. JAMA 311, 271–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey A, Bomans PH, Muller FA, Will J, Frederik PM, de With G & Sommerdijk NA (2010). The role of prenucleation clusters in surface‐induced calcium phosphate crystallization. Nat Mater 9, 1010–1014. [DOI] [PubMed] [Google Scholar]
- Drabek K, van de Peppel J, Eijken M & van Leeuwen JP (2011). GPM6B regulates osteoblast function and induction of mineralization by controlling cytoskeleton and matrix vesicle release. J Bone Miner Res 26, 2045–2051. [DOI] [PubMed] [Google Scholar]
- Dweck MR, Chow MW, Joshi NV, Williams MC, Jones C, Fletcher AM, Richardson H, White A, McKillop G, van Beek EJ, Boon NA, Rudd JH & Newby DE (2012). Coronary arterial 18F‐sodium fluoride uptake: a novel marker of plaque biology. J Am Coll Cardiol 59, 1539–1548. [DOI] [PubMed] [Google Scholar]
- Dziegielewska KM, Mollgard K, Reynolds ML & Saunders NR (1987). A fetuin‐related glycoprotein (alpha 2HS) in human embryonic and fetal development. Cell Tissue Res 248, 33–41. [DOI] [PubMed] [Google Scholar]
- Ehara S, Kobayashi Y, Yoshiyama M, Shimada K, Shimada Y, Fukuda D, Nakamura Y, Yamashita H, Yamagishi H, Takeuchi K, Naruko T, Haze K, Becker AE, Yoshikawa J & Ueda M (2004). Spotty calcification typifies the culprit plaque in patients with acute myocardial infarction: an intravascular ultrasound study. Circulation 110, 3424–3429. [DOI] [PubMed] [Google Scholar]
- Elkeles RS, Godsland IF, Feher MD, Rubens MB, Roughton M, Nugara F, Humphries SE, Richmond W, Flather MD & Group PS (2008). Coronary calcium measurement improves prediction of cardiovascular events in asymptomatic patients with type 2 diabetes: the PREDICT study. Eur Heart J 29, 2244–2251. [DOI] [PubMed] [Google Scholar]
- Ferencik M, Schlett CL, Ghoshhajra BB, Kriegel MF, Joshi SB, Maurovich‐Horvat P, Rogers IS, Banerji D, Bamberg F, Truong QA, Brady TJ, Nagurney JT & Hoffmann U (2012). A computed tomography‐based coronary lesion score to predict acute coronary syndrome among patients with acute chest pain and significant coronary stenosis on coronary computed tomographic angiogram. Am J Cardiol 110, 183–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fish RS, Klootwijk E, Tam FW, Kleta R, Wheeler DC, Unwin RJ & Norman J (2013). ATP and arterial calcification. Eur J Clin Invest 43, 405–412. [DOI] [PubMed] [Google Scholar]
- Francis MD (1969). The inhibition of calcium hydroxypatite crystal growth by polyphosphonates and polyphosphates. Calcif Tissue Res 3, 151–162. [DOI] [PubMed] [Google Scholar]
- Genge BR, Wu LN & Wuthier RE (2007). In vitro modeling of matrix vesicle nucleation: synergistic stimulation of mineral formation by annexin A5 and phosphatidylserine. J Biol Chem 282, 26035–26045. [DOI] [PubMed] [Google Scholar]
- Gepner AD, Young R, Delaney JA, Tattersall MC, Blaha MJ, Post WS, Gottesman RF, Kronmal R, Budoff MJ, Burke GL, Folsom AR, Liu K, Kaufman J & Stein JH (2015). Comparison of coronary artery calcium presence, carotid plaque presence, and carotid intima‐media thickness for cardiovascular disease prediction in the Multi‐Ethnic Study of Atherosclerosis. Circ Cardiovasc Imaging 8, e002262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerke V (2002). Annexins and membrane organisation in the endocytic pathway. Cell Mol Biol Lett 6, 204. [PubMed] [Google Scholar]
- Gillette JM & Nielsen‐Preiss SM (2004). The role of annexin 2 in osteoblastic mineralization. J Cell Sci 117, 441–449. [DOI] [PubMed] [Google Scholar]
- Glimcher MJ (1984). Recent studies of the mineral phase in bone and its possible linkage to the organic matrix by protein‐bound phosphate bonds. Philos Trans R Soc Lond B Biol Sci 304, 479–508. [DOI] [PubMed] [Google Scholar]
- Golub EE (2011). Biomineralization and matrix vesicles in biology and pathology. Semin Immunopathol 33, 409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haarhaus M, Arnqvist HJ & Magnusson P (2013). Calcifying human aortic smooth muscle cells express different bone alkaline phosphatase isoforms, including the novel B1x isoform. J Vasc Res 50, 167–174. [DOI] [PubMed] [Google Scholar]
- Han D, Lee JH, Hartaigh BO & Min JK (2015). Role of computed tomography screening for detection of coronary artery disease. Clin Imaging 40, 307–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmey D, Hessle L, Narisawa S, Johnson KA, Terkeltaub R & Millan JL (2004). Concerted regulation of inorganic pyrophosphate and osteopontin by Akp2, Enpp1, and Ank: an integrated model of the pathogenesis of mineralization disorders. Am J Pathol 164, 1199–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzapfel GA, Mulvihill JJ, Cunnane EM & Walsh MT (2014). Computational approaches for analyzing the mechanics of atherosclerotic plaques: a review. J Biomech 47, 859–869. [DOI] [PubMed] [Google Scholar]
- Hsu HH (2003). In vitro effect of cholesterol on calcifying activity of vesicles isolated from rabbit aortas. Biochim Biophys Acta 1638, 235–240. [DOI] [PubMed] [Google Scholar]
- Hsu HH & Camacho NP (1999). Isolation of calcifiable vesicles from human atherosclerotic aortas. Atherosclerosis 143, 353–362. [DOI] [PubMed] [Google Scholar]
- Hutcheson JD, Goettsch C, Bertazzo S, Maldonado N, Ruiz JL, Goh W, Yabusaki K, Faits T, Bouten C, Franck G, Quillard T, Libby P, Aikawa M, Weinbaum S & Aikawa E (2016). Genesis and growth of extracellular‐vesicle‐derived microcalcification in atherosclerotic plaques. Nat Mater 15, 335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutcheson JD, Goettsch C, Pham T, Iwashita M, Aikawa M, Singh SA & Aikawa E (2014). Enrichment of calcifying extracellular vesicles using density‐based ultracentrifugation protocol. J Extracell Vesicles 3, DOI:10.3402/jev.v3.25129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imoto K, Hiro T, Fujii T, Murashige A, Fukumoto Y, Hashimoto G, Okamura T, Yamada J, Mori K & Matsuzaki M (2005). Longitudinal structural determinants of atherosclerotic plaque vulnerability: a computational analysis of stress distribution using vessel models and three‐dimensional intravascular ultrasound imaging. J Am Coll Cardiol 46, 1507–1515. [DOI] [PubMed] [Google Scholar]
- Joshi NV, Vesey AT, Williams MC, Shah AS, Calvert PA, Craighead FH, Yeoh SE, Wallace W, Salter D, Fletcher AM, van Beek EJ, Flapan AD, Uren NG, Behan MW, Cruden NL, Mills NL, Fox KA, Rudd JH, Dweck MR & Newby DE (2014). 18F‐fluoride positron emission tomography for identification of ruptured and high‐risk coronary atherosclerotic plaques: a prospective clinical trial. Lancet 383, 705–713. [DOI] [PubMed] [Google Scholar]
- Kapustin AN, Chatrou ML, Drozdov I, Zheng Y, Davidson SM, Soong D, Furmanik M, Sanchis P, De Rosales RT, Alvarez‐Hernandez D, Shroff R, Yin X, Muller K, Skepper JN, Mayr M, Reutelingsperger CP, Chester A, Bertazzo S, Schurgers LJ & Shanahan CM (2015). Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ Res 116, 1312–1323. [DOI] [PubMed] [Google Scholar]
- Kapustin AN, Davies JD, Reynolds JL, McNair R, Jones GT, Sidibe A, Schurgers LJ, Skepper JN, Proudfoot D, Mayr M & Shanahan CM (2011). Calcium regulates key components of vascular smooth muscle cell‐derived matrix vesicles to enhance mineralization. Circ Res 109, e1–e12. [DOI] [PubMed] [Google Scholar]
- Kapustin AN & Shanahan CM (2012). Calcium regulation of vascular smooth muscle cell‐derived matrix vesicles. Trends Cardiovasc Med 22, 133–137. [DOI] [PubMed] [Google Scholar]
- Kelly‐Arnold A, Maldonado N, Laudier D, Aikawa E, Cardoso L & Weinbaum S (2013). Revised microcalcification hypothesis for fibrous cap rupture in human coronary arteries. Proc Natl Acad Sci USA 110, 10741–10746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiffer‐Moreira T, Yadav MC, Zhu D, Narisawa S, Sheen C, Stec B, Cosford ND, Dahl R, Farquharson C, Hoylaerts MF, Macrae VE & Millan JL (2013). Pharmacological inhibition of PHOSPHO1 suppresses vascular smooth muscle cell calcification. J Bone Miner Res 28, 81–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KM (1976). Calcification of matrix vesicles in human aortic valve and aortic media. Fed Proc 35, 156–162. [PubMed] [Google Scholar]
- Kirsch T, Harrison G, Golub EE & Nah HD (2000. a). The roles of annexins and types II and X collagen in matrix vesicle‐mediated mineralization of growth plate cartilage. J Biol Chem 275, 35577–35583. [DOI] [PubMed] [Google Scholar]
- Kirsch T, Swoboda B & Nah H (2000. b). Activation of annexin II and V expression, terminal differentiation, mineralization and apoptosis in human osteoarthritic cartilage. Osteoarthritis Cartilage 8, 294–302. [DOI] [PubMed] [Google Scholar]
- Lin TC, Tintut Y, Lyman A, Mack W, Demer LL & Hsiai TK (2006). Mechanical response of a calcified plaque model to fluid shear force. Ann Biomed Eng 34, 1535–1541. [DOI] [PubMed] [Google Scholar]
- Lloyd‐Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, Ferguson TB, Ford E, Furie K, Gillespie C, Go A, Greenlund K, Haase N, Hailpern S, Ho PM, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott MM, Meigs J, Mozaffarian D, Mussolino M, Nichol G, Roger VL, Rosamond W, Sacco R, Sorlie P, Thom T, Wasserthiel‐Smoller S, Wong ND & Wylie‐Rosett J (2010). Heart disease and stroke statistics–2010 update: a report from the American Heart Association. Circulation 121, e46–e215. [DOI] [PubMed] [Google Scholar]
- Lomashvili KA, Monier‐Faugere MC, Wang X, Malluche HH & O'Neill WC (2009). Effect of bisphosphonates on vascular calcification and bone metabolism in experimental renal failure. Kidney Int 75, 617–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado N, Kelly‐Arnold A, Cardoso L & Weinbaum S (2013). The explosive growth of small voids in vulnerable cap rupture; cavitation and interfacial debonding. J Biomech 46, 396–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado N, Kelly‐Arnold A, Laudier D, Weinbaum S & Cardoso L (2015). Imaging and analysis of microcalcifications and lipid/necrotic core calcification in fibrous cap atheroma. Int J Cardiovasc Imaging 31, 1079–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SS, Blaha MJ, Blankstein R, Agatston A, Rivera JJ, Virani SS, Ouyang P, Jones SR, Blumenthal RS, Budoff MJ & Nasir K (2014). Dyslipidemia, coronary artery calcium, and incident atherosclerotic cardiovascular disease: implications for statin therapy from the multi‐ethnic study of atherosclerosis. Circulation 129, 77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita K, Sang Y, Ballew SH, Shlipak M, Katz R, Rosas SE, Peralta CA, Woodward M, Kramer HJ, Jacobs DR, Sarnak MJ & Coresh J (2015). Subclinical atherosclerosis measures for cardiovascular prediction in CKD. J Am Soc Nephrol 26, 439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mebarek S, Abousalham A, Magne D, Do LD, Bandorowicz‐Pikula J, Pikula S & Buchet R (2013). Phospholipases of mineralization competent cells and matrix vesicles: roles in physiological and pathological mineralizations. Int J Mol Sci 14, 5036–5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millan JL (2013). The role of phosphatases in the initiation of skeletal mineralization. Calcif Tissue Int 93, 299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mornet E, Stura E, Lia‐Baldini AS, Stigbrand T, Menez A & Le Du MH (2001). Structural evidence for a functional role of human tissue nonspecific alkaline phosphatase in bone mineralization. J Biol Chem 276, 31171–31178. [DOI] [PubMed] [Google Scholar]
- New SE, Goettsch C, Aikawa M, Marchini JF, Shibasaki M, Yabusaki K, Libby P, Shanahan CM, Croce K & Aikawa E (2013). Macrophage‐derived matrix vesicles: an alternative novel mechanism for microcalcification in atherosclerotic plaques. Circ Res 113, 72–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nollet M, Santucci‐Darmanin S, Breuil V, Al‐Sahlanee R, Cros C, Topi M, Momier D, Samson M, Pagnotta S, Cailleteau L, Battaglia S, Farlay D, Dacquin R, Barois N, Jurdic P, Boivin G, Heymann D, Lafont F, Lu SS, Dempster DW, Carle GF & Pierrefite‐Carle V (2014). Autophagy in osteoblasts is involved in mineralization and bone homeostasis. Autophagy 10, 1965–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nudelman F, Pieterse K, George A, Bomans PH, Friedrich H, Brylka LJ, Hilbers PA, de With G & Sommerdijk NA (2010). The role of collagen in bone apatite formation in the presence of hydroxyapatite nucleation inhibitors. Nat Mater 9, 1004–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsuka F, Kramer MC, Woudstra P, Yahagi K, Ladich E, Finn AV, de Winter RJ, Kolodgie FD, Wight TN, Davis HR, Joner M & Virmani R (2015). Natural progression of atherosclerosis from pathologic intimal thickening to late fibroatheroma in human coronary arteries: A pathology study. Atherosclerosis 241, 772–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosdocimo DA, Douglas DC, Romani AM, O'Neill WC & Dubyak GR (2009). Autocrine ATP release coupled to extracellular pyrophosphate accumulation in vascular smooth muscle cells. Am J Physiol Cell Physiol 296, C828–C839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rekhter MD, Zhang K, Narayanan AS, Phan S, Schork MA & Gordon D (1993). Type I collagen gene expression in human atherosclerosis. Localization to specific plaque regions. Am J Pathol 143, 1634–1648. [PMC free article] [PubMed] [Google Scholar]
- Reynolds JL, Joannides AJ, Skepper JN, McNair R, Schurgers LJ, Proudfoot D, Jahnen‐Dechent W, Weissberg PL & Shanahan CM (2004). Human vascular smooth muscle cells undergo vesicle‐mediated calcification in response to changes in extracellular calcium and phosphate concentrations: a potential mechanism for accelerated vascular calcification in ESRD. J Am Soc Nephrol 15, 2857–2867. [DOI] [PubMed] [Google Scholar]
- Reynolds JL, Skepper JN, McNair R, Kasama T, Gupta K, Weissberg PL, Jahnen‐Dechent W & Shanahan CM (2005). Multifunctional roles for serum protein fetuin‐a in inhibition of human vascular smooth muscle cell calcification. J Am Soc Nephrol 16, 2920–2930. [DOI] [PubMed] [Google Scholar]
- Rohde M & Mayer H (2007). Exocytotic process as a novel model for mineralization by osteoblasts in vitro and in vivo determined by electron microscopic analysis. Calcif Tissue Int 80, 323–336. [DOI] [PubMed] [Google Scholar]
- Ruiz JL, Hutcheson JD & Aikawa E (2015). Cardiovascular calcification: current controversies and novel concepts. Cardiovasc Pathol 24, 207–212. [DOI] [PubMed] [Google Scholar]
- Rumberger JA, Brundage BH, Rader DJ & Kondos G (1999). Electron beam computed tomographic coronary calcium scanning: a review and guidelines for use in asymptomatic persons. Mayo Clin Proc 74, 243–252. [DOI] [PubMed] [Google Scholar]
- Rutsch F, Vaingankar S, Johnson K, Goldfine I, Maddux B, Schauerte P, Kalhoff H, Sano K, Boisvert WA, Superti‐Furga A & Terkeltaub R (2001). PC‐1 nucleoside triphosphate pyrophosphohydrolase deficiency in idiopathic infantile arterial calcification. Am J Pathol 158, 543–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakakura K, Nakano M, Otsuka F, Ladich E, Kolodgie FD & Virmani R (2013). Pathophysiology of atherosclerosis plaque progression. Heart Lung Circ 22, 399–411. [DOI] [PubMed] [Google Scholar]
- Stanford CM, Jacobson PA, Eanes ED, Lembke LA & Midura RJ (1995). Rapidly forming apatitic mineral in an osteoblastic cell line (UMR 106‐01 BSP). J Biol Chem 270, 9420–9428. [DOI] [PubMed] [Google Scholar]
- Stenbeck G & Coxon FP (2014). Role of vesicular trafficking in skeletal dynamics. Curr Opin Pharmacol 16, 7–14. [DOI] [PubMed] [Google Scholar]
- Stewart AJ, Schmid R, Blindauer CA, Paisey SJ & Farquharson C (2003). Comparative modelling of human PHOSPHO1 reveals a new group of phosphatases within the haloacid dehalogenase superfamily. Protein Eng 16, 889–895. [DOI] [PubMed] [Google Scholar]
- Tanimura A, McGregor DH & Anderson HC (1983). Matrix vesicles in atherosclerotic calcification. Proc Soc Exp Biol Med 172, 173–177. [DOI] [PubMed] [Google Scholar]
- Tanimura A, McGregor DH & Anderson HC (1986). Calcification in atherosclerosis. I. Human studies. J Exp Pathol 2, 261–273. [PubMed] [Google Scholar]
- Tintut Y, Patel J, Parhami F & Demer LL (2000). Tumor necrosis factor‐alpha promotes in vitro calcification of vascular cells via the cAMP pathway. Circulation 102, 2636–2642. [DOI] [PubMed] [Google Scholar]
- Tota‐Maharaj R, Joshi PH, Budoff MJ, Whelton S, Zeb I, Rumberger J, Al‐Mallah M, Blumenthal RS, Nasir K & Blaha MJ (2015). Usefulness of regional distribution of coronary artery calcium to improve the prediction of all‐cause mortality. Am J Cardiol 115, 1229–1234. [DOI] [PubMed] [Google Scholar]
- Vengrenyuk Y, Carlier S, Xanthos S, Cardoso L, Ganatos P, Virmani R, Einav S, Gilchrist L & Weinbaum S (2006). A hypothesis for vulnerable plaque rupture due to stress‐induced debonding around cellular microcalcifications in thin fibrous caps. Proc Natl Acad Sci USA 103, 14678–14683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa‐Bellosta R, Wang X, Millan JL, Dubyak GR & O'Neill WC (2011). Extracellular pyrophosphate metabolism and calcification in vascular smooth muscle. Am J Physiol Heart Circ Physiol 301, H61–H68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vliegenthart R, Oudkerk M, Hofman A, Oei HH, van Dijck W, van Rooij FJ & Witteman JC (2005). Coronary calcification improves cardiovascular risk prediction in the elderly. Circulation 112, 572–577. [DOI] [PubMed] [Google Scholar]
- Wong KK, Thavornpattanapong P, Cheung SC, Sun Z & Tu J (2012). Effect of calcification on the mechanical stability of plaque based on a three‐dimensional carotid bifurcation model. BMC Cardiovasc Disord 12, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LN, Genge BR, Dunkelberger DG, LeGeros RZ, Concannon B & Wuthier RE (1997. a). Physicochemical characterization of the nucleational core of matrix vesicles. J Biol Chem 272, 4404–4411. [DOI] [PubMed] [Google Scholar]
- Wu LN, Genge BR, Kang MW, Arsenault AL & Wuthier RE (2002). Changes in phospholipid extractability and composition accompany mineralization of chicken growth plate cartilage matrix vesicles. J Biol Chem 277, 5126–5133. [DOI] [PubMed] [Google Scholar]
- Wu LN, Genge BR & Wuthier RE (2008). Analysis and molecular modeling of the formation, structure, and activity of the phosphatidylserine‐calcium‐phosphate complex associated with biomineralization. J Biol Chem 283, 3827–3838. [DOI] [PubMed] [Google Scholar]
- Wu LN, Wuthier MG, Genge BR & Wuthier RE (1997. b). In situ levels of intracellular Ca2+ and pH in avian growth plate cartilage. Clin Orthop Relat Res 335, 310–324. [PubMed] [Google Scholar]
- Wu LN, Yoshimori T, Genge BR, Sauer GR, Kirsch T, Ishikawa Y & Wuthier RE (1993). Characterization of the nucleational core complex responsible for mineral induction by growth plate cartilage matrix vesicles. J Biol Chem 268, 25084–25094. [PubMed] [Google Scholar]
- Wuthier RE (1977). Electrolytes of isolated epiphyseal chondrocytes, matrix vesicles, and extracellular fluid. Calcif Tissue Res 23, 125–133. [DOI] [PubMed] [Google Scholar]
- Wuthier RE & Lipscomb GF (2011). Matrix vesicles: structure, composition, formation and function in calcification. Front Biosci 16, 2812–2902. [DOI] [PubMed] [Google Scholar]
- Xiao Z, Blonder J, Zhou M & Veenstra TD (2009). Proteomic analysis of extracellular matrix and vesicles. J Proteomics 72, 34–45. [DOI] [PubMed] [Google Scholar]
- Yadav MC, Simao AM, Narisawa S, Huesa C, McKee MD, Farquharson C & Millan JL (2011). Loss of skeletal mineralization by the simultaneous ablation of PHOSPHO1 and alkaline phosphatase function: a unified model of the mechanisms of initiation of skeletal calcification. J Bone Miner Res 26, 286–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G, Xu MJ, Zhao MM, Dai XY, Kong W, Wilson GM, Guan Y, Wang CY & Wang X (2012). Activation of nuclear factor‐kappa B accelerates vascular calcification by inhibiting ankylosis protein homolog expression. Kidney Int 82, 34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]