

Abstract
Key points
Mitochondrial respiratory sensitivity to ADP is thought to influence muscle fitness and is partly regulated by cytosolic–mitochondrial diffusion of ADP or phosphate shuttling via creatine/phosphocreatine (Cr/PCr) through mitochondrial creatine kinase (mtCK).
Previous measurements of respiration in vitro with Cr (saturate mtCK) or without (ADP/ATP diffusion) show mixed responses of ADP sensitivity following acute exercise vs. less sensitivity after chronic exercise.
In human muscle, modelling in vivo ‘exercising’ [Cr:PCr] during in vitro assessments revealed novel responses to exercise that differ from detections with or without Cr (±Cr).
Acute exercise increased ADP sensitivity when measured without Cr but had no effect ±Cr or with +Cr:PCr, whereas chronic exercise increased sensitivity ±Cr but lowered sensitivity with +Cr:PCr despite increased markers of mitochondrial oxidative capacity.
Controlling in vivo conditions during in vitro respiratory assessments reveals responses to exercise that differ from typical ±Cr comparisons and challenges our understanding of how exercise improves metabolic control in human muscle.
Abstract
Mitochondrial respiratory control by ADP (K mapp) is viewed as a critical regulator of muscle energy homeostasis. However, acute exercise increases, decreases or has no effect on K mapp in human muscle, whereas chronic exercise surprisingly decreases sensitivity despite greater mitochondrial content. We hypothesized that modelling in vivo mitochondrial creatine kinase (mtCK)‐dependent phosphate‐shuttling conditions in vitro would reveal increased sensitivity (lower K mapp) after acute and chronic exercise. The K mapp was determined in vitro with 20 mm Cr (+Cr), 0 mm Cr (−Cr) or ‘in vivo exercising’ 20 mm Cr/2.4 mm PCr (Cr:PCr) on vastus lateralis biopsies sampled from 11 men before, immediately after and 3 h after exercise on the first, fifth and ninth sessions over 3 weeks. Dynamic responses to acute exercise occurred throughout training, whereby the first session did not change K mapp with in vivo Cr:PCr despite increases in −Cr. The fifth session decreased sensitivity with Cr:PCr or +Cr despite no change in −Cr. Chronic exercise increased sensitivity ±Cr in association with increased electron transport chain content (+33–62% complexes I–V), supporting classic proposals that link increased sensitivity to oxidative capacity. However, in vivo Cr:PCr reveals a perplexing decreased sensitivity, contrasting the increases seen ±Cr. Functional responses occurred without changes in fibre type or proteins regulating mitochondrial–cytosolic energy exchange (mtCK, VDAC and ANT). Despite the dynamic responses seen with ±Cr, modelling in vivo phosphate‐shuttling conditions in vitro reveals that ADP sensitivity is unchanged after high‐intensity exercise and is decreased after training. These findings challenge our understanding of how exercise regulates skeletal muscle energy homeostasis.
Key points
Mitochondrial respiratory sensitivity to ADP is thought to influence muscle fitness and is partly regulated by cytosolic–mitochondrial diffusion of ADP or phosphate shuttling via creatine/phosphocreatine (Cr/PCr) through mitochondrial creatine kinase (mtCK).
Previous measurements of respiration in vitro with Cr (saturate mtCK) or without (ADP/ATP diffusion) show mixed responses of ADP sensitivity following acute exercise vs. less sensitivity after chronic exercise.
In human muscle, modelling in vivo ‘exercising’ [Cr:PCr] during in vitro assessments revealed novel responses to exercise that differ from detections with or without Cr (±Cr).
Acute exercise increased ADP sensitivity when measured without Cr but had no effect ±Cr or with +Cr:PCr, whereas chronic exercise increased sensitivity ±Cr but lowered sensitivity with +Cr:PCr despite increased markers of mitochondrial oxidative capacity.
Controlling in vivo conditions during in vitro respiratory assessments reveals responses to exercise that differ from typical ±Cr comparisons and challenges our understanding of how exercise improves metabolic control in human muscle.
Abbreviations
- ADPf
free ADP
- ANT
adenine nucleotide translocator
- Cr
creatine
- mtCK
mitochondrial creatine kinase
- OXPHOS
oxidative phosphorylation
- PCr
phosphocreatine
- PmFB
permeabilized muscle fibres
- VDAC
voltage‐dependent anion carrier
Introduction
Mitochondrial respiratory control by ADP is believed to be a critical regulator of energy homeostasis during muscle contraction. Both classic and contemporary investigations have proposed that a greater mitochondrial oxidative capacity improves respiratory sensitivity to free ADP (ADPf), such that a smaller rise in ADPf is required to stimulate a given rate of oxidative ATP synthesis, particularly at the onset of exercise, leading to reduced substrate phosphorylation from phosphocreatine (PCr), glycogenolysis and lactate formation (Holloszy, 1967; Gollnick & King, 1969; Henriksson, 1977; Dudley et al. 1982, 1987; Hurley et al. 1986; Kiens et al. 1993; Green et al. 1995; Phillips et al. 1996; LeBlanc et al. 2004; Perry et al. 2008). Indeed, this model is supported by consistent reports of a greater ATP/ADPf during exercise in highly oxidative vs. less oxidative skeletal muscle (Dudley et al. 1982, 1987) and following endurance exercise training in rodents and humans with increased markers of mitochondrial content (Holloszy, 1967; Gollnick & King, 1969; Green et al. 1995). These apparent improvements in mitochondrial sensitivity to ADP are viewed as a fundamental mechanism by which muscles acclimatize to exercise, leading to fatigue resistance and greater endurance.
A corollary of this model is that a given [ADPf] will stimulate a greater rate of mitochondrial respiration if mitochondrial oxidative capacity is increased. However, using in vitro measurements of mitochondrial respiration in permeabilized muscle fibres, respiratory sensitivity to ADP is instead reduced in muscle with a greater proportion of type I fibres and greater mitochondrial content (Kuznetsov et al. 1996; Tonkonogi et al. 1998) and in exercise‐trained/athletic human muscle (Mettauer et al. 2001; Zoll et al. 2002, 2003 b), which also has a greater proportion of type I fibres and greater mitochondrial content (Mettauer et al. 2001; Zoll et al. 2002). While increased sensitivity seen in rats after training (Burelle & Hochachka, 2002) supports a positive relationship between mitochondrial content and respiratory sensitivity to ADP (Holloszy, 1967; Dudley et al. 1987), the decreases in humans have led to a proposal that lower ADP sensitivity after chronic exercise is beneficial by preventing erratic increases in ATP synthesis in response to a rise in [ADPf] (Walsh et al. 2001 a; Zoll et al. 2002, 2003 a,2003 b). This proposal is seemingly inconsistent with the known attenuations in exercising [ADPf] after endurance training (Green et al. 1995; Phillips et al. 1996; LeBlanc et al. 2004; Perry et al. 2008). However, it is nonetheless based on direct measurements of respiration that do not align with the classic model linking greater mitochondrial content to improved ADP sensitivity. To date, no study has reconciled how improved metabolic control after chronic exercise is related to decreased in vitro respiratory responses to ADP in muscle with a greater proportion of type I fibres.
However, previous acute and chronic investigations alike have not modelled in vivo creatine (Cr) and PCr concentrations found during exercise that may regulate respiratory control by ADP. Specifically, in vitro respiration is typically measured in the presence of Cr, to saturate the more efficient mitochondrial creatine kinase (mtCK)‐dependent phosphate shuttling, or in the absence of Cr, to promote the less efficient ADP/ATP diffusion model of mitochondrial–cytosolic energy exchange (Tonkonogi et al. 1998, 1999; Mettauer et al. 2001; Walsh et al. 2001 a; Burelle & Hochachka, 2002; Zoll et al. 2002; Perry et al. 2012). In the phosphate‐shuttling model, mtCK in the inner mitochondrial membrane space transfers a phosphate from ATP to Cr, with the PCr product diffusing to the cytosol approximately sevenfold faster than ATP. The PCr is dephosphorylated by cytosolic creatine kinases at sites of ATP utilization, with the Cr product diffusing back to the mitochondria ∼2000‐fold faster than ADP (Wallimann et al. 2011). Such ‘phosphate shuttling’ is viewed as a predominant method of energy exchange in muscle and incorporates ADP/ATP and Cr/PCr transport through mitochondrial adenine nucleotide translocase (ANT) and the voltage‐dependent anion channel (VDAC; Wyss et al. 1992; Ventura‐Clapier et al. 1998; Kay et al. 2000; Schlattner et al. 2006; Wallimann et al. 2011; Guzun et al. 2012). These systems have previously been depicted by the model shown in Fig. 1 (Tonkonogi et al. 1998; Walsh et al. 2001 b; Zoll et al. 2002). Given the potent influence of Cr:PCr ratios on regulation of ADP‐stimulated respiration (Walsh et al. 2001 b), reconstructing in vivo exercising [Cr:PCr] during in vitro assessments of respiration could be a crucial experimental approach for reconciling the opposing models of how mitochondrial oxidative capacity is related to ADP sensitivity post‐training, as suggested previously (Burelle & Hochachka, 2002).
Figure 1. Schematic representation of mitochondrial–cytosolic energy exchange models .
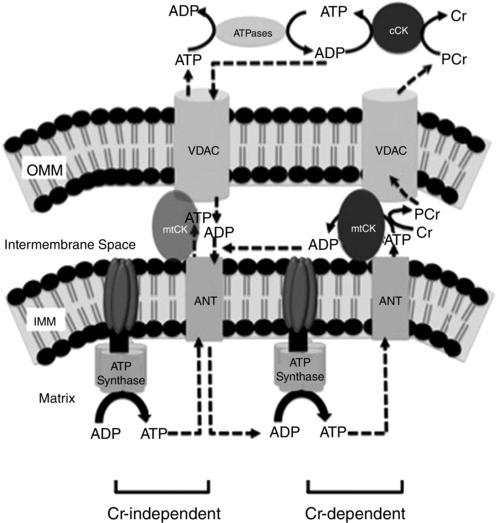
The left side depicts the creatine (Cr)‐independent model of energy exchange, in which ADP/ATP diffusion is the sole method of energy transfer from the mitochondrial matrix to the cytosol. The right side depicts the Cr‐dependent ‘phosphate‐shuttling’ mechanism of energy transfer. In this model, ATP produced through oxidative phosphorylation (OXPHOS) in the mitochondrial matrix diffuses through adenine nucleotide translocator (ANT) into the intermembrane space, where mitochondrial creatine kinase (mtCK) facilitates the transfer of the high‐energy phosphate from ATP to Cr. The PCr product diffuses out of the mitochondria through the voltage‐dependent anion carrier (VDAC) to local cytosolic creatine kinase (cCK)–ATPase complexes. Abbreviations: IMM, inner mitochondrial membrane; and OMM, outer mitochondrial membrane. Figure adapted from Aliev et al. (2011) and Guzun et al. (2012).
Likewise, the acute responses to exercise may be more accurately detected in vitro if respiration is assessed in the presence of ‘exercising’ concentrations of Cr:PCr. Indeed, acute exercise decreases sensitivity with ‘resting’ muscle concentrations of Cr:PCr in vitro that was not evident with Cr alone (Perry et al. 2012). However, to our knowledge, acute exercise has only been reported to lower or have no effect on respiratory sensitivity to ADP after acute exercise when determined with or without Cr (Tonkonogi et al. 1998) or with resting muscle Cr:PCr (Perry et al. 2012) in vitro. The decreased sensitivity post‐training and mixed results after acute exercise are therefore difficult to interpret given that skeletal muscle is never completely devoid of Cr or PCr in vivo.
The purpose of the present study was to determine whether acute and chronic exercise increases human skeletal muscle respiratory sensitivity to ADP when modelling in vivo ‘exercising’ [Cr:PCr], and whether greater sensitivity occurs in conjunction with increased markers of mitochondrial oxidative capacity as classically proposed (Holloszy, 1967). We hypothesized that short‐term exercise training would increase respiratory sensitivity to ADP measured in vitro with in vivo ‘exercising’ [Cr:PCr] previously reported to exist in human muscle sampled during exercise (Perry et al. 2008). Using a short‐term high‐intensity interval exercise protocol, we also hypothesized that this increased ADP sensitivity would occur in the absence of changes in fibre‐type proportions but in conjunction with increased markers of mitochondrial oxidative capacity, as well as increased VDAC, ANT and mtCK. Finally, we hypothesized that increased ADP sensitivity with chronic exercise would follow repeated transient increases in sensitivity during acute exercise.
Methods
Human participants, exercise testing and muscle biopsies
Eleven healthy, recreationally active men were recruited to participate in this investigation. Their mean ± SEM age, height, weight, body mass index and peak oxygen uptake () were 24.8 ± 1.0 years, 180.4 ± 1.8 cm, 75.5 ± 3.4 kg, 23.2 ± 0.8 kg m−2 and 51.9 ± 1.9 ml kg−1 min−1, respectively. All participants were non‐smokers, free of disease and not taking prescription medications or supplements. Participants were given both oral and written information about experimental procedures before providing their informed consent. All experimental procedures with human participants were approved by the Research Ethics Board at York University (Toronto, ON, Canada) and conformed to the Declaration of Helsinki.
Participants initially completed a standardized graded test on a cycle ergometer (Lode, Groningen, The Netherlands). After at least 72 h, participants returned to the laboratory and completed a practice high‐intensity interval exercise session. The session involved 10 × 4 min intervals at 91% maximal heart rate (∼83% ), with 2 min of rest in between each interval. After 1–2 weeks, participants reported to the clinical laboratory for the first experiment. With the participant lying supine on a bed, a single skeletal muscle sample was obtained from the lateral aspect of the vastus lateralis muscle by percutaneous needle biopsy technique using a spring‐loaded 14 gauge Medax Biofeather disposable needle (San Possidonio, MO, Italy) under local subcutaneous anaesthesia (∼2 ml of 2% xylocaine without noradrenaline). A 12 gauge cannula was used to puncture the skin at ∼45 deg to a depth of 2 cm and guide the needle to an additional depth of 2 cm approximately parallel to the longitudinal direction of muscle fibres. Four to five cuts (10–20 mg each) were sampled, with the needle rotated ∼30 deg between cuts over a period of ∼1 min, with slight pressure applied on the skin over the needle prior to each cut. Each cut was removed from the needle with sterile forceps or surgical blades before the subsequent cut was made. The first two samples were used for preparation of fibre bundles, and the remaining tissue was frozen in liquid nitrogen, stored at −86°C and used for Western blotting and other measurements (described in the following subsections).
After applying a bandage to the ∼2‐mm‐diameter biopsy site, participants were moved to the cycle ergometer and performed the exercise session at the predetermined wattage to maintain 91% maximal heart rate. Immediately after this first exercise session (T1), the participant moved back to the bed, and a second biopsy was taken from the opposite leg to the pre‐exercise biopsy. Participants remained at rest in the sitting position for 3 h, after which a third and final biopsy was taken from the initial leg sampled pre‐exercise. A new incision was made immediately before each biopsy at a distance of at least 3 cm from any previous biopsy within the same trial. All subsequent biopsies in exercise sessions 5 (T5) and 9 (T9) were also performed on the leg opposite to the last biopsy. Furthermore, each new participant would receive their first T1 pre‐exercise biopsy on the leg opposite to the previous participant.
Participants completed eight additional exercise sessions over 3 weeks (alternating Mondays, Wednesdays and Fridays), with the same biopsy procedure completed again in exercise sessions T5 and T9. Throughout the nine exercise sessions, cycling workload (in watts) was increased when required to maintain the targeted heart rate as fitness improved. The was reassessed 48–72 h after the ninth session.
Preparation of permeabilized muscle fibres (PmFBs)
This technique is partly adapted from previous methods (Kuznetsov et al. 1996; Tonkonogi et al. 1998) and has been described elsewhere (Anderson et al. 2007; Perry et al. 2011, 2012). Briefly, small portions (∼25 mg wet weight) of muscle were dissected from each biopsy and placed in ice‐cold biopsy preservation solution (BIOPS), containing (mm): 50 Mes hydrate, 7.23 K2EGTA, 2.77 CaK2EGTA, 20 imidazole, 0.5 dithiothreitol, 20 taurine, 5.77 ATP, 15 PCr and 6.56 MgCl2.6H2O (pH 7.1, adjusted using potassium hydroxide (KOH)). The muscle was trimmed of connective tissue and fat and divided into several small muscle bundles (∼2–7 mm, 1.0–2.5 mg wet weight). Each bundle was gently separated along the longitudinal axis with a pair of antimagnetic needle‐tipped forceps under magnification (Zeiss 2000, Germany). Bundles were then treated with 30 μg ml−1 saponin in BIOPS and incubated on a rotor for 30 min at 4°C. Saponin at 30 μg ml−1 has previously been shown to optimize respiration in human skeletal muscle (Kane et al. 2011), albeit in obese females. We have unpublished observations that 30 μg ml−1 provides similar respiratory kinetics to 50 μg ml−1 but decreases the frequency and magnitude of cytochrome c responses (mentioned in the “Mitochondrial respiration in PmFBs” section) in PmFB from human males at 37°C, whereas 50 μg ml−1 is optimal for assays at room temperature. Saponin is a mild, cholesterol‐specific detergent that selectively permeabilizes the sarcolemmal membranes while keeping mitochondrial membranes, which contain little cholesterol, intact (Veksler et al. 1987; Kuznetsov et al. 2008). Following permeabilization, the PmFBs were placed in mitochondrial respiration medium (MiR05) containing (mm): 0.5 EGTA, 10 KH2PO4, 3 MgCl2.6H2O, 60 potassium lactobionate, 20 Hepes, 20 taurine, 110 sucrose and 1 mg ml−1 fatty acid‐free BSA (pH 7.1). The PmFBs were washed at 4°C (<30 min) in MiR05 until the respiratory measurements were initiated.
Mitochondrial respiration in PmFBs
High‐resolution measurements of O2 consumption were made in 2 ml of respiration medium (MiR05) using the Oroboros Oxygraph‐2k (Oroboros Instruments, Corp., Innsbruck, Austria), with stirring at 750 r.p.m. at 37°C. Respiration medium contained either 20 mm Cr (+Cr) or 2.4 mm PCr and 20 mm Cr or neither (−Cr) to modify kinetics of mtCK (Saks et al. 1994, 1995; Walsh et al. 2001 b; Anmann et al. 2006). For ADP‐stimulated respiratory kinetics, 5 mm glutamate and 5 mm malate were added as complex I substrates (via generation of NADH to saturate electron entry into complex I) followed by ADP titrations in stepwise increments. All three conditions (−Cr, 20 mm Cr and 2.4 mm PCr/20 mm Cr) began with a titration of 25, 50, 125, 200, 275 and 500 μm, 1, 2 and 4 mm ADP. Above 4 mm, additions of 4 mm ADP were made until a plateau across two successive ADP additions was reached. In the −Cr conditions, a plateau was generally reached at 20–24 mm ADP. In the 20 mm Cr conditions, a plateau was generally reached at 4–8 mm ADP, and in the PCr/Cr conditions, a plateau was reached at 16–20 mm ADP. All experiments were completed before the oxygraph chamber reached 150 μm [O2]. Permeabilized muscle fibres spontaneously contract in assay medium, a phenomenon which can be prevented by the myosin II‐specific inhibitor blebbistatin (Perry et al. 2011). We have previously demonstrated that microbiopsies yield lower K mapp for ADP than Bergström biopsies, possibly because of greater ADP‐induced contraction and disintegration of PmFBs in vitro (Hughes et al. 2015). Given that blebbistatin normalized respiratory kinetics in microbiopsy PmFBs to the level seen in PmFBs from Bergström samples, we included 25 mm blebbistatin dissolved in DMSO (5 mm stock) to prevent spontaneous contraction in all PmFB experiments. Consistent with our previous work (Perry et al. 2011), we noted that some PmFBs disintegrated during respirometric assessments in the absence of blebbistatin. This appeared to be related to very low respiratory kinetics, which resulted in poor Michaelis–Menten modelling and very low K mapp for ADP in certain participants (see Table 1). Blebbistatin prevented PmFB disintegration, as reported previously (Perry et al. 2011, 2012), and permitted robust detection of respiratory kinetics and accurate modelling of K mapp for ADP (Table 1).
Table 1.
Comparison of K mapp to ADP values in the absence and presence of blebbistatin in the absence of Cr in participants A–C
| T1 | T5 | T9 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Pre‐ | Post‐ | 3 h Post‐ | Pre‐ | Post‐ | 3 h Post‐ | Pre‐ | Post‐ | 3 h Post‐ | |
| Subject | exercise | exercise | exercise | exercise | exercise | exercise | exercise | exercise | exercise |
| No creatine; no blebbistatin | |||||||||
| A | 149 | 55 | 70 | 163 | 47 | 111 | 36 | 17 | 46 |
| B | 94 | 69 | 52 | 105 | 212 | 76 | 66 | 51 | 56 |
| C | 43 | 119 | 37 | 44 | 18 | 12 | 39 | 4 | 1 |
| Mean | 95 | 81 | 53 | 104 | 93 | 66 | 47 | 24 | 34 |
| No creatine; blebbistatin present | |||||||||
| A | 1214 | 857 | 107 | 825 | 778 | 918 | 616 | 432 | 600 |
| B | 1112 | 743 | 851 | 1040 | 1087 | 928 | 729 | 685 | 251 |
| C | 1307 | 1213 | 881 | 549 | 785 | 708 | 446 | 395 | 133 |
| Mean | 1211 | 938 | 613 | 805 | 883 | 851 | 597 | 504 | 328 |
Polarographic oxygen measurements were acquired at 2 s intervals, with the rate of respiration derived from 40 data points, and expressed as picomoles per second. Cytochrome c was added to test for mitochondrial membrane integrity, with all experiments demonstrating <10% increase in respiration.
The K mapp for ADP was determined through the Michaelis–Menten enzyme kinetics, fitting the model [Y = V max × X/(K m + X)], where X is [free ADP] (ADPf) and Y is the O2 flux at [ADPf], using Prism (GraphPad Software, Inc., La Jolla, CA, USA), as published previously (Perry et al. 2011). A representative trace is shown in Fig. 2 as an example of data used to fit with a Michaelis–Menten equation. The mean r 2 for all non‐linear regression (a measure of goodness of fit) was 0.9237 ± 0.0069 (SEM). The K mapp for ADP was higher than in previous studies performed at room temperature and/or in the absence of blebbistatin (Tonkonogi et al. 1998; Mettauer et al. 2001; Walsh et al. 2001 a; Zoll et al. 2002, 2003 b); however, this higher value is consistent with our previous report (Perry et al. 2011) that indicates a positive relationship between K mapp for ADP and temperature as well as the fact that in vitro contraction (in the absence of blebbistatin) lowers K mapp for ADP.
Figure 2. Representative trace of ADP‐stimulated respiration in permeabilized muscle fibre bundles .
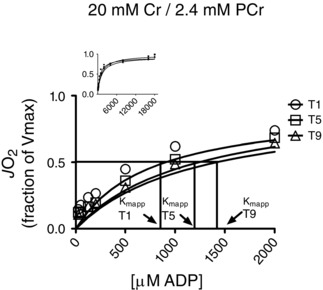
In this trace, respiration was measured on samples taken before the first (T1), fifth (T5) and ninth (T9) exercise training sessions. Data obtained were fitted with a Michaelis–Menten equation. JO2, oxygen flux; Kmapp, apparent KM.
Western blot analyses
An aliquot of frozen muscle (10–30 mg) from each pretraining biopsy was homogenized in a plastic microcentrifuge tube with a tapered teflon pestle in ice‐cold buffer containing (mm): 40 Hepes, 120 NaCl, 1 EDTA, 10 NaHP2O7.10H2O pyrophosphate, 10 β‐glycerophosphate, 10 NaF and 0.3% CHAPS detergent (pH 7.1 adjusted using KOH). Protein concentrations were determined using a BCA assay (Life Technologies, Carlsbad, CA, USA). Fifty micrograms of denatured protein was loaded for Western blotting and resolved by SDS‐PAGE on 6–12% polyacrylamide gels, depending on the molecular weight of the protein, and transferred to a low‐fluorescence polyvinylidene difluoride membrane. Membranes were blocked with LI‐COR Odyssey Blocking Buffer (LI‐COR, Lincoln NE, USA) and immunoblotted overnight (4°C) with antibodies specific for each protein. A commercially available monoclonal antibody was used to detect electron transport chain proteins (human OXPHOS Cocktail, ab110411; Abcam, Cambridge, UK, 1:208 dilution), including V‐ATP5A (54 kDa), III‐UQCRC2 (48 kDa), II‐SDHB (29 kDa), IV‐COX II (22 kDa) and I‐NDUFB8 (18 kDa). Commercially available polyclonal antibodies were used to detect VDAC 1 (AV35122, 33 kDa; Sigma‐Aldrich, St Louis, MO, USA, 1: 500), VDAC 2 (HPA043475, 33 kDa; Sigma‐Aldrich, 1:500), VDAC 3 (HPA026864, 33 kDa; Sigma‐Aldrich, 1:1000), ANT 1 (ab54418, 32 kDa; Abcam, 1:40), ANT 2 (ab118125, 32 kDa; Abcam, 1:1000) and sarcomeric s‐mtCK (generous gift from Dr Uwe Schlattner, Grenoble, France; 42 kDa, 1:1000). The mtCK antibody has been validated previously to confirm specificity (Schlattner et al. 2002).
Common loading controls could not be assessed on the same gel because of size overlap with OXPHOS proteins. Furthermore, detection of other proteins is difficult given that the supplier recommends avoidance of denaturing by heat to optimize detection of certain OXPHOS bands. Considering that our low coefficient of variation in measuring protein densities (6.4%, data not shown) suggests minimal error in loading multiple lanes, we chose to correct for errors in sample preparation by normalizing to controls assessed on a second gel for all prepared samples. Specifically, an average of β‐tubulin (T8328, 52 kDa; Sigma‐Aldrich, 1:1000) and α‐actinin (ab82247, 103 kDa; Abcam, 3.5 μg/mL) was used given that either exhibited variability within some participants that was verified with repeated homogenates prepared from the same samples. This observation is similar to a previous report whereby variability in five proteins were detected across repeated biopsies within a participant, similar to the present investigation (Caron et al. 2011). All other Western blots were normalized in the same manner to ensure consistency.
After overnight incubation in primary antibodies, membranes were washed three times, for 5 min each time, in TBST and incubated for 1 h at room temperature with the corresponding infrared fluorescent secondary antibody (LI‐COR). Immunoreactive proteins were detected by infared imaging (LI‐COR CLx; LI‐COR) and quantified by densitometry (ImageJ, http://imagej.nih.gov/ij/). A double band was detected at 33 kDa for ANT 1 for all subjects and, as such, both bands were quantified.
Fibre type: myosin heavy chain (MyHC) isoforms
The MyHC isoforms were resolved using PAGE adapted from methods previously described (Kohn & Myburgh, 2006; O'Neill et al. 2011). Briefly, using 10 μg of vastus lateralis muscle from the 3 h postexercise biopsies at T1, T5 and T9, proteins were resolved using a 0.75 mm SDS‐PAGE gel consisting of a stacking gel with 4% bis‐acrylamide and a separating gel with 10% bis‐acrylamide [final concentration separating: 30% glycerol, 0.2 m Tris–HCl (pH 8.8), 0.1 m glycine, 0.4% SDS, 0.1% ammonium persulfate and 0.05% tetramethylethylenediamine (TEMED); and stacking: 0.125 m Tris–HCl (pH 6.8), 0.1% SDS, 0.1% ammonium persulfate and 0.05% TEMED]. The electrophoresis buffer consisted of 100 mm Tris, 150 mm glycine, 0.1% SDS and 0.12% β‐mercaptoethanol (Kohn & Myburgh, 2006). The gels were run on a Minigel electrophoresis system (Bio‐Rad Laboratories, Rockford, IL, USA) at 4°C at a constant current of 20 mA until proteins entered the stacking gel and then at 140 mV for a further 20 h. Following electrophoresis, gels were silver stained using a Silver Stain for Mass Spectrometry kit following the manufacturer's recommendations (Pierce, Rockford, IL, USA). Bands were identified as described by Kohn & Myburgh (2006).
Statistics
Results are expressed as means ± SEM. The level of significance was established at P < 0.05 for all statistics. A one‐way ANOVA with repeated measures was used to test for differences in the following: (i) K mapp between each pre‐exercise sample across the training period (T1 pre‐exercise vs. T5 pre‐exercise vs. T9 pre‐exercise); (ii) K mapp within a training session (pre‐exercise, postexercise and 3 h postexercise); (iii) the change in K mapp from pre‐ to postexercise (‘ΔK mapp’ within each session) across T1, T5 and T9; and (iv) densitometry values between the pre‐exercise biopsies across T1, T5 and T9. When a significant F ratio was obtained, post hoc analyses were completed using a least‐squares difference.
Results
Whole‐body responses to training
The mean power output during the training sessions increased by 9% between T1 and T9 (from 240 ± 13 to 260 ± 4 W; P < 0.05, data not shown). Average exercise heart rate throughout 10 intervals did not change with training and averaged 91% across training sessions. The increased by 8.7% following training (51.9 ± 1.9 to 56.4 ± 2.1 ml kg–1 min–1; P < 0.05).
Effects of acute and chronic exercise on mtCK‐independent ADP sensitivity
We first determined the effect of an acute bout of high‐intensity interval exercise on mitochondrial respiratory sensitivity to ADP in PmFBs. When modelling ADP/ATP diffusion in the absence of Cr (mtCK independent), exercise increased respiratory sensitivity to ADP (decreased K mapp) in both postexercise biopsies at T1 (immediately postexercise −30% in K mapp; 3 h postexercise −36% in K mapp; P < 0.05 vs. pre‐exercise; Fig. 3 A). This acute effect of exercise was not observed at T5 or T9 (Fig. 3 A). However, chronic exercise increased Cr‐independent sensitivity in pre‐exercise muscle by 40% at T9 vs. T1 pre‐exercise (P < 0.05; Fig. 3 A).
Figure 3. The effect of acute and chronic exercise on mitochondrial respiratory sensitivity to ADP (Kmapp) in human skeletal muscle .
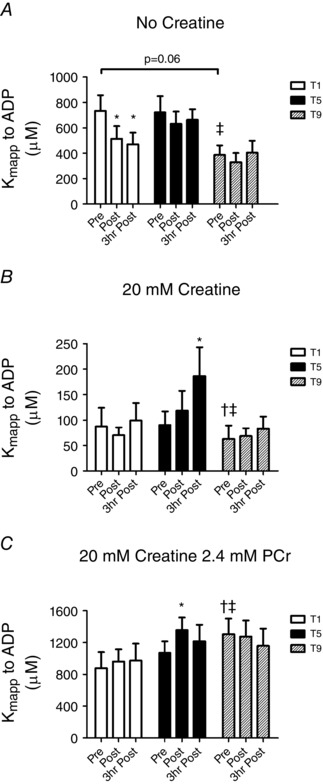
The K mapp to ADP was measured in the absence of creatine (A), in the presence of 20 mm creatine (B) or at in vivo concentrations of 20 mm Cr and 2.4 mm PCr (C) previously reported in human skeletal muscle during exercise (Perry et al. 2008). Results are shown as means + SEM; n = 11; *P < 0.05 compared with pre‐exercise within same trial, † P < 0.05 compared with T1 pre‐exercise and ‡ P < 0.05 compared with T5 pre‐exercise.
Effects of acute and chronic exercise on Cr‐dependent ADP sensitivity
In contrast to the Cr‐independent system, there was no effect of a single bout of exercise in the presence of 20 mm Cr at T1 (Fig. 3 B). However, following T5, this Cr‐dependent condition showed an unexpected decrease in sensitivity (increased K mapp) 3 h postexercise relative to T5 pre‐exercise (immediately postexercise +26% in K mapp; 3 h postexercise +110% in K mapp; P < 0.05; Fig. 3 B). No acute increases in sensitivity were seen after T9 (Fig. 3 B). However, chronic exercise increased Cr‐dependent sensitivity in pre‐exercise muscle 29% by T9 pre‐exercise (P < 0.05; Fig. 3 B).
Effects of acute and chronic exercise on Cr:PCr‐dependent ADP sensitivity
There was no effect of a single bout of exercise on K mapp in the presence of previously reported (Perry et al. 2008) muscle concentrations of Cr and PCr (20 mm and 2.4 mm) found during this specific high‐intensity interval exercise (Fig. 3 C). The Cr:PCr‐dependent conditions also showed a significant decrease in sensitivity following exercise at T5 (postexercise −27% in K mapp; P < 0.05, Fig. 3 C) that was recovered 3 h postexercise. No further changes in sensitivity were seen following T9 (Fig. 3 C). Contrary to the Cr‐independent and Cr‐dependent conditions, chronic exercise decreased sensitivity by T9 (T9 pre‐exercise +22% in K mapp vs. T5 pre‐exercise and +48% in K mapp vs. T1 pre‐exercise; P < 0.05; Fig. 3 C).
Phosphate‐shuttling protein contents
Initially, the contents of the electron transport chain proteins were measured using an OXPHOS cocktail (Fig. 4). Complex I increased 56% by T5 (P < 0.05) and 47% by T9 vs. T1 (P < 0.05), with no difference between T9 and T5. Complex II did not change with training. Complex III at T9 increased 29% relative to T1 (P < 0.05), with no difference between T5 and T1 or T5 and T9. Complex IV increased by 32% at T5 relative to T1 (P < 0.05) and by 36% at T9 relative to T1 (P < 0.05), with no differences between T5 and T9. Complex V protein content was unchanged with exercise (Fig. 4). The average of all five complexes increased 24% from T1 to T5 (P < 0.05) and 30% from T1 to T9 (P < 0.05), with no change between T5 and T9.
Figure 4. Human skeletal muscle protein content of mitochondrial electron transport system proteins (subunits of complexes I–V) throughout nine sessions of high‐intensity interval exercise .
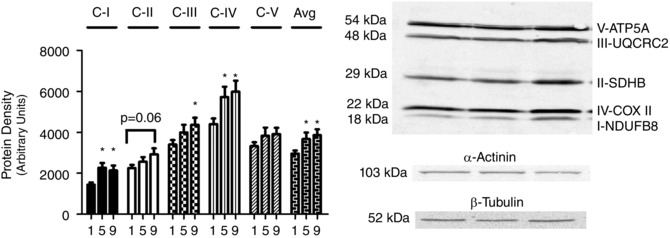
α‐Actinin and β‐tubulin were used as loading controls as described in the Methods section. Results are shown as means + SEM; n = 11; *P < 0.05 compared with T1. Avg is the average of all 5 complex protein densities.
The content of proteins involved in mitochondrial–cytosolic energy exchange were measured prior to exercise at all three time points. Training did not change the contents of VDAC isoforms 1, 2 and 3 (Fig. 5), ANT 1 and ANT 2 (Fig. 6) or mtCK (Fig. 7).
Figure 5. Human skeletal muscle protein content of voltage‐dependent anion channel (VDAC) isoforms 1, 2 and 3 throughout nine sessions of high‐intensity interval exercise .
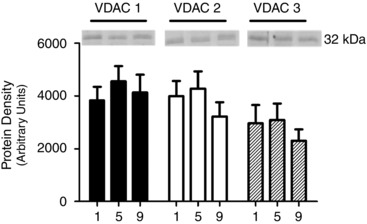
α‐Actinin and β‐tubulin were used as loading controls as described in the Methods section and are represented in Fig. 4. Results are shown as means + SEM; n = 11.
Figure 6. Human skeletal muscle protein content of adenine nucleotide translocase (ANT) isoforms 1 and 2 throughout nine sessions of high‐intensity interval exercise .
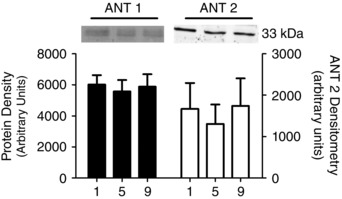
α‐Actinin and β‐tubulin were used as loading controls as described in the Methods section and are represented in Fig. 4. Results are shown as means + SEM; n = 11.
Figure 7. Human skeletal muscle protein content of mitochondrial creatine kinase (mtCK) throughout nine sessions of high‐intensity interval exercise .
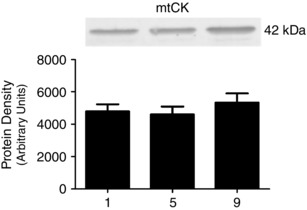
α‐Actinin and β‐tubulin were used as loading controls as described in the Methods section and are represented in Fig. 4. Results are shown as means + SEM; n = 11.
Myosin heavy chain isoforms
Changes in the percentage of type I fibres relative to type II fibres were measured 3 h postexercise at T1, T5 and T9 (Fig. 8). Relative proportions were unchanged over the nine exercise sessions (T1, 41% type I and 59% type II; T5, 34% type I and 66% type II; and T9, 44% type I and 56% type II).
Figure 8. Human skeletal muscle fibre type detected with silver staining measured throughout nine sessions of high‐intensity interval exercise .
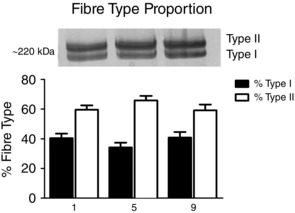
Results are shown as means + SEM; n = 11.
Discussion
This investigation demonstrates the critical importance of modelling in vivo Cr and PCr when determining the effects of exercise on mitochondrial respiratory kinetics in vitro. This is apparent given that the acute effects of exercise were inconsistent across all ±Cr and Cr/PCr conditions, with variable time‐dependent responses observed at specific times throughout training. Specifically, acute exercise had no effect on ADP sensitivity in the presence of ‘exercising’ [Cr:PCr]. Furthermore, and perhaps most strikingly, chronic exercise increased ADP sensitivity when assessed with or without Cr, whereas a decreased sensitivity was seen when in vitro conditions contained ‘exercising’ [Cr:PCr], suggesting that phosphate shuttling may be more sensitive to inhibition by PCr after training. In addition, functional changes were not associated with altered protein contents of known regulators of ADP sensitivity (VDAC, ANT and mtCK), suggesting a complexity in the regulation of energy exchange that has yet to be revealed. These observations, made in human muscle, challenge our understanding of how increased mitochondrial oxidative capacity following chronic exercise is related to metabolic regulation and raises new questions regarding the manner by which respiratory control by ADP is regulated during exercise.
Modelling in vivo [Cr:PCr] in vitro alters the interpretation of how chronic exercise regulates respiratory sensitivity to ADP
The increased sensitivity ±Cr in the present study supports a classic proposal that exercise training improves respiratory sensitivity to ADP in conjunction with increased mitochondrial oxidative capacity (Holloszy, 1967; Dudley et al. 1987). To our knowledge, this is the first study in humans to support this model with direct measurements of ADP‐stimulated respiration in human muscle after endurance exercise training. However, this positive relationship between mitochondrial oxidative capacity and ADP sensitivity is opposite to the decreased sensitivity ±Cr reported previously by one group (Mettauer et al. 2001; Zoll et al. 2002, 2003 a,b). A possible reason for this discrepancy is that ADP sensitivity increases with mitochondrial content early during training (present study) but is somehow decreased with longer‐term training. Nevertheless, our findings ±Cr seemingly support a positive relationship between mitochondrial oxidative capacity and ADP control of respiration (Holloszy, 1967; Dudley et al. 1987).
Although the measurement of respiratory kinetics ±Cr is useful for comparing saturated mtCK‐dependent phosphate shuttling vs. ADP/ATP diffusion kinetics (Ventura‐Clapier et al. 1998; Wallimann et al. 2011), the increases in sensitivity ±Cr are difficult to understand in the context of respiratory control in vivo given that muscle is never devoid of Cr or PCr, as discussed previously (Burelle & Hochachka, 2002). With this in mind, we were surprised to observe reduced ADP sensitivity after training when modelling in vivo exercise [Cr:PCr] in vitro. Given that Cr sensitizes whereas PCr inhibits mtCK‐dependent ADP‐stimulated respiration (Walsh et al. 2001 b), our observations that training increased ADP sensitivity with Cr yet decreased sensitivity with Cr:PCr suggest that the influence of Cr and PCr on mtCK are both enhanced after training, with a net sensitization towards inhibition by PCr. This observation is difficult to explain because there are currently no known mechanisms, to our knowledge, by which exercise may modulate mtCK affinity more to PCr than to Cr. Furthermore, this reduced sensitivity does not support a relationship between increased mitochondrial oxidative capacity and respiratory sensitivity to ADP after training, in contrast to the findings ±Cr.
The reduced sensitivity with Cr:PCr after training is in agreement with reductions seen previously, albeit ±Cr (Mettauer et al. 2001; Zoll et al. 2002, 2003 a,b). These reports of decreased sensitivity ±Cr led to a new proposal that a reduced sensitivity to ADP is beneficial and improves metabolic control by preventing excessive increases in ATP for a given rise in ADPf. Contrary to our hypothesis, the reduced sensitivity with Cr:PCr supports this proposal. However, this observation and proposal are difficult to reconcile with well‐documented improvements in ATP/ADPf after training. Specifically, a reduced sensitivity would seemingly contribute to a greater rise in ADPf and lower ATP/ADPf during exercise. This is clearly not the case, because type I fibres, which are less sensitive to ADP in vitro (Kuznetsov et al. 1996), demonstrate smaller rises in ADPf during contraction ex vivo vs. type II fibres (Dudley et al. 1982, 1987), and endurance training attenuates the rise in ADPf in humans during exercise (Green et al. 1995; Phillips et al. 1996; LeBlanc et al. 2004; Perry et al. 2008). Furthermore, training mitigates the rise in AMP and inosine monophosphate during exercise, which is consistent with smaller rises in ADPf (Baldwin et al. 1999) based on the adenylate kinase equilibrium, which seemingly should not happen with lower ADP sensitivities. Nevertheless, the manner by which decreased ADP sensitivity confers a physiological benefit is unclear. One intriguing possibility is that the decreased sensitivity to ADP post‐training may represent an improved sensitivity to another feedback signal, such as Cr itself, but this possibility would require further investigation.
Using in vivo assessments of PCr resynthesis via 31P magnetic resonance spectroscopy after exercise, it was reported that short‐term training of five sessions improved ADP control of respiratory kinetics in human muscle despite no increase in mitochondrial oxidative capacity (Layec et al. 2013). While these findings suggest that improved control by ADP occurs through a mechanism other than increased mitochondrial content, the increased sensitivity in vivo clearly contrasts the reduced sensitivities we report in vitro with Cr:PCr. Methodological limitations of in vitro measurements should therefore be considered. For example, given that mitochondria are shown to be physically coupled to sites of ATP utilization, including sarcoendoplasmic reticulum, actin–myosin filaments and sarcolemma (Ogata & Yamasaki, 1997), it is plausible that functional coupling involving direct channelling of ADP/ATP or Cr/PCr between mitochondria and ATPases also exists (Seppet et al. 2001). Hence, exogenous addition of ADP during direct measurements of respiration in PmFBs in vitro may not be sufficient to capture altered physical orientations between mitochondria and sites of ATP utilization, as speculated previously (Layec et al. 2013). Any changes in the restriction to ADP diffusion caused by exercise should still cause a similar decrease in ADP sensitivity in vitro in all ±Cr or Cr:PCr conditions. However, this was not the case in the present study. Furthermore, it would be difficult to accept other potential methodological contributions to the decreased sensitivity with Cr:PCr after training given that ±Cr conditions showed the opposite response of increased sensitivity.
The differences in total Cr concentration are a potential limitation of the present study. The additional 2.4 mm PCr in the Cr:PCr conditions (22.4 mm total) vs. +Cr (20 mm) could potentially offer more Cr for rephosphorylation by potential residual cytosolic creatine kinase remaining after permeabilizing and washing the PmFBs. However, the considerable inhibitory effect of this small addition of PCr has been observed previously when total Cr was controlled (Walsh et al. 2001 b), which suggests that the effects seen in the present study are largely attributable to the addition of PCr itself rather than a ∼10% difference in total Cr.
Altered regulation of respiratory sensitivity to ADP following chronic exercise occurs independent of VDAC, ANT or mtCK protein content
Contrary to our hypothesis, the increased ADP sensitivity seen with and without Cr and the decreases observed with Cr:PCr after chronic exercise were not a result of changes in the contents of VDAC, ANT or mtCK. These findings suggest that additional critical regulators of ADP/ATP diffusion or mtCK‐dependent phosphate shuttling have yet to be identified and are not retained during the in vitro assessment of respiratory sensitivity to ADP in PmFBs. For example, VDAC, ANT and mtCK possess sites of phosphorylation, acetylation and redox‐sensitive cysteines (Kaasik et al. 1999; Dolder et al. 2001; Schlattner et al. 2006; Feng et al. 2010; Kerner et al. 2012; Mielke et al. 2014). Mitochondrial creatine kinase becomes more active following an intriguing conformational change from a dimeric to an octameric form, and its association with VDAC and ANT is promoted by calcium (Schlattner & Wallimann, 2000; Schlattner et al. 2001, 2006), which requires tethering to cardiolipin to form proteolipid complexes for phosphate shuttling to occur (Wallimann et al. 2011). Permeability of VDACs can also be modulated by physical interactions with microtubules that are altered by contraction in isolated muscle cells (Guzun et al. 2012). Ultimately, it is our view that determining the mechanism for altered respiratory control by ADP in any condition will be limited until the physiological relevance of post‐translational modifications are characterized further.
Acute exercise does not increase respiratory sensitivity to ADP when modelling in vivo Cr:PCr in vitro
Modelling in vivo exercise [Cr:PCr] suggests that acute exercise does not alter the regulation of energy exchange in human skeletal muscle, in contrast to previous reports (Tonkonogi et al. 1998, 1999; Perry et al. 2012). A key novelty of the present study is the use of [Cr:PCr] found in humans during similar exercise (Perry et al. 2008). Previous reports of increased sensitivity were in the presence of Cr, suggesting that mtCK is acutely regulated by exercise (Tonkonogi et al. 1998, 1999; Perry et al. 2012). Furthermore, the unaltered ADP sensitivity with 20 mm Cr and 2.4 mm PCr in the present study suggests that any potential increases in Cr‐dependent mtCK activity are masked by PCr. Another possibility for these mixed results of acute responses to exercise may be the intensity of exercise itself. For example, decreases in sensitivity were observed following 2 h of exercise at a moderate intensity (60% ) using ‘resting’ muscle [Cr:PCr] (Perry et al. 2012), whereas increased Cr‐dependent sensitivity was seen in the present study and previous work at higher intensities (75–130% ) of various durations (up to 71 min; Tonkonogi et al. 1998, 1999). Collectively, these studies might suggest that intense challenges to energy homeostasis (>75% ) increase ADP sensitivity seen ±Cr, but the reason why this is not detectable with Cr:PCr is unclear. Ultimately, the ‘in vivo’ Cr:PCr conditions do not support the possibility that exercise acutely regulates ADP sensitivity in the untrained state (T1). However, the variable responses at T5 and T9 suggest a complexity of integrating regulatory signals that is not fully understood but appears to occur dynamically as muscle adapts to progressive training.
The present study also used blebbistatin, a myosin II ATPase inhibitor, to prevent ADP‐induced rigor and potential in vitro effects of contraction sensitizing mitochondria to ADP (Perry et al. 2011, 2013). While previous work has shown that the contractile state of PmFBs may change the interpretation of how exercise affects ADP sensitivity (Perry et al. 2012), this may not be the case in the present study given that sensitivity increased in one condition (−Cr), with no change in other conditions (+Cr and Cr:PCr). Furthermore, we repeated all respiratory assessments in the presence and absence of blebbistatin on three participants and found similar trends of decreased K mapp with training in −Cr and acute decreases in K mapp in −Cr (Table 1). Collectively, we believe that the divergent effects of exercise are not influenced by the use of blebbistatin.
Perspectives and conclusion
Mitochondrial respiratory control by ADP is likely to incorporate multiple regulatory inputs that remain to be modelled fully, as discussed above. As such, modelling K mapp to ADP using in vitro respiratory assessments may not reflect control of respiration in vivo. Indeed, in vivo assessments of respiration require a higher‐order model (Jeneson et al. 2009), and such approaches have shown a greater ‘co‐operativity’ (higher Hill coefficient) after training (Layec et al. 2013). The increased K mapp in the presence of Cr:PCr and decreased K mapp +Cr or −Cr in the present study suggest that a more complex regulation of respiration exists in the trained state. While in vitro modelling of the primary regulators of this complex system requires further understanding of key control parameters, the divergent responses in K mapp might suggest that more signals are integrated into the control of respiration during exercise once muscle has acclimatized to training. For example, potential respiratory control by calcium (Korzeniewski, 2015), adenylate kinase (Gueguen et al. 2005) and inorganic phosphate (the remaining balance of the mtCK reaction) could be modelled. Likewise, consideration should be given to the effects of training on intracellular oxygen tension given the influence that [O2] has on the regulation of mitochondrial respiration even at submaximal intensity (Haseler et al. 1998), which is not reflected when using supraphysiological [O2] in vitro. Improved blood flow after training must also be taken into account (Phillips et al. 1996), particularly considering that mitochondrial respiration in trained muscle appears more sensitive to O2 availability (Haseler et al. 1999). Overall, such examples might suggest that training does not simply entail a greater mitochondrial sensitivity for ADP, but perhaps a more complex and integrated regulation of the mitochondrial respiration via multiple signals.
An intriguing possibility is that mitochondrial content per se is not a critical regulator with respect to probabilities of interacting with a rise in cytosolic ADPf (Holloszy, 1967; Holloszy & Coyle, 1984; Dudley et al. 1987; Hood, 2001). In other words, it may be that a greater mitochondrial content will not increase ADP sensitivity across the entire cell simply because of physical constraints in aligning mitochondria with specific locales of ATP utilization. This seems possible when considering the extremely close physical coupling of myofilaments and sarcolemma with mitochondria (Ogata & Yamasaki, 1997) and given the apparent lack of substantial cytosolic compartment seen in electron microscopic images of skeletal muscle owing to the abundance of contractile filaments. Rather, the greater mitochondrial content after training may instead occur in proportion to increased sites of energy utilization with little consequence for the overall sensitivity to a rise in ADPf elsewhere in the cell. While this or other additional regulatory factors discussed above may influence respiratory control by ADP, the apparent inverse relationship between mitochondrial content and in vitro respiratory sensitivity post‐training remains mysterious.
In conclusion, training‐induced increases in mitochondrial respiratory sensitivity to ADP in vitro detected with and without Cr are not evident with [Cr:PCr] conditions known to occur in vivo during high‐intensity exercise. This suggests that PCr suppression of phosphate shuttling may be enhanced after endurance training, but reconciling this possibility with known improvements in energy homeostasis remains unclear. Acute high‐intensity exercise increased ADP sensitivity without Cr, but this was not observed with Cr:PCr, contrasting previous findings seen with and without Cr and at different intensities and durations. Collectively, these findings do not align with classic proposals that a greater mitochondrial oxidative capacity improves respiratory control by ADP and highlight a considerable gap in understanding post‐translational regulation of respiratory control by ADP that must now be explored intensively. The divergent results also underscore how controlling critical in vitro experimental parameters influencing respiratory control by ADP must be carefully considered when investigating mitochondrial responses to exercise in humans.
Additional information
Competing interests
None declared.
Author contributions
C.G.R.P., M.Y., M.C.H. and J.N. contributed to the rationale and study design. C.G.R.P., M.Y., M.C.H. and R.L. conducted clinical trials and experiments and analysed all data. All authors contributed to the interpretation of the data and manuscript preparation. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
Funding was provided to C.G.R.P. by the National Science and Engineering Research Council (#436138‐2013), with infrastructure supported by Canada Foundation for Innovation and the Ontario Research Fund. M.Y. and J.N. were funded by CIF (Sweden Council of International Fellowship and Karolinska Institutet Research Fund). M.C.H. was supported by a Canadian Institutes of Health Research CGS‐M scholarship.
Acknowledgements
We thank the study participants for their considerable efforts and dedication. We also thank Dr Angelo Belcastro, York University for access to his human clinical laboratory, Dr Giuseppe DeVito, University College Dublin for expert advice in microbiopsy procedures and Drs Brennan Smith and James Lally for their preliminary review of this manuscript. All experiments were performed at York University, Toronto, ON, Canada.
M.Y. and M.C.H. made equal contributions to this investigation.
References
- Aliev M, Guzun R, Karu‐Varikmaa M, Kaambre T, Wallimann T & Saks V (2011). Molecular system bioenergics of the heart: experimental studies of metabolic compartmentation and energy fluxes versus computer modeling. Int J Mol Sci 12, 9296–9331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson EJ, Yamazaki H & Neufer PD (2007). Induction of endogenous uncoupling protein 3 suppresses mitochondrial oxidant emission during fatty acid‐supported respiration. J Biol Chem 282, 31257–31266. [DOI] [PubMed] [Google Scholar]
- Anmann T, Guzun R, Beraud N, Pelloux S, Kuznetsov AV, Kogerman L, Kaambre T, Sikk P, Paju K, Peet N, Seppet E, Ojeda C, Tourneur Y & Saks V (2006). Different kinetics of the regulation of respiration in permeabilized cardiomyocytes and in HL‐1 cardiac cells: importance of cell structure/organization for respiration regulation. Biochim Biophys Acta 1757, 1597–1606. [DOI] [PubMed] [Google Scholar]
- Baldwin J, Snow RJ, Carey MF & Febbraio MA (1999). Muscle IMP accumulation during fatiguing submaximal exercise in endurance trained and untrained men. Am J Physiol Regul Integr Comp Physiol 277, R295–R300. [DOI] [PubMed] [Google Scholar]
- Burelle Y & Hochachka PW (2002). Endurance training induces muscle‐specific changes in mitochondrial function in skinned muscle fibers. J Appl Physiol 92, 2429–2438. [DOI] [PubMed] [Google Scholar]
- Caron M‐A, Charette SJ, Maltais F & Debigaré R (2011). Variability of protein level and phosphorylation status caused by biopsy protocol design in human skeletal muscle analyses. BMC Res Notes 4, 488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolder M, Wendt S & Wallimann T (2001). Mitochondrial creatine kinase in contact sites: interaction with porin and adenine nucleotide translocase, role in permeability transition and sensitivity to oxidative damage. Biol Signals Recept 10, 93–111. [DOI] [PubMed] [Google Scholar]
- Dudley GA, Abraham WM & Terjung RL (1982). Influence of exercise intensity and duration on biochemical adaptations in skeletal muscle. J Appl Physiol 53, 844–850. [DOI] [PubMed] [Google Scholar]
- Dudley GA, Tullson PC & Terjung RL (1987). Influence of mitochondrial content on the sensitivity of respiratory control. J Biol Chem 262, 9109–9114. [PubMed] [Google Scholar]
- Feng J, Lucchinetti E, Enkavi G, Wang Y, Gehrig P, Roschitzki B, Schaub MC, Tajkhorshid E, Zaugg K & Zaugg M (2010). Tyrosine phosphorylation by Src within the cavity of the adenine nucleotide translocase 1 regulates ADP/ATP exchange in mitochondria. Am J Physiol Cell Physiol 298, C740–C748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollnick PD & King DW (1969). Effect of exercise and training on mitochondria of rat skeletal muscle. Am J Physiol 216, 1502–1509. [DOI] [PubMed] [Google Scholar]
- Green HJ, Jones S, Ball‐Burnett M, Farrance B & Ranney D (1995). Adaptations in muscle metabolism to prolonged voluntary exercise and training. J Appl Physiol 78, 138–145. [DOI] [PubMed] [Google Scholar]
- Gueguen N, Lefaucheur L, Ecolan P, Fillaut M & Herpin P (2005). Ca2+‐activated myosin‐ATPases, creatine and adenylate kinases regulate mitochondrial function according to myofibre type in rabbit. J Physiol 564, 723–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzun R, Gonzalez‐Granillo M, Karu‐Varikmaa M, Grichine A, Usson Y, Kaambre T, Guerrero‐Roesch K, Kuznetsov A, Schlattner U & Saks V (2012). Regulation of respiration in muscle cells in vivo by VDAC through interaction with the cytoskeleton and MtCK within Mitochondrial Interactosome. Biochim Biophys Acta 1818, 1545–1554. [DOI] [PubMed] [Google Scholar]
- Haseler LJ, Hogan MC & Richardson RS (1999). Skeletal muscle phosphocreatine recovery in exercise‐trained humans is dependent on O2 availability. J Appl Physiol 86, 2013–2018. [DOI] [PubMed] [Google Scholar]
- Haseler LJ, Richardson RS, Videen JS & Hogan MC (1998). Phosphocreatine hydrolysis during submaximal exercise: the effect of J Appl Physiol 85, 1457–1463. [DOI] [PubMed] [Google Scholar]
- Henriksson J (1977). Training induced adaptation of skeletal muscle and metabolism during submaximal exercise. J Physiol 270, 661–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloszy JO (1967). Biochemical adaptations in muscle: effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J Biol Chem 242, 2278–2282. [PubMed] [Google Scholar]
- Holloszy JO & Coyle EF (1984). Adaptations of skeletal muscle to endurance exercise and their metabolic consequences. J Appl Physiol 56, 831–838. [DOI] [PubMed] [Google Scholar]
- Hood DA (2001). Invited review: contractile activity‐induced mitochondrial biogenesis in skeletal muscle. J Appl Physiol 90, 1137–1157. [DOI] [PubMed] [Google Scholar]
- Hughes MC, Ramos SV, Turnbull PC, Nejatbakhsh A, Baechler BL, Tahmasebi H, Laham R, Gurd BJ, Quadrilatero J, Kane DA & Perry CGR (2015). Mitochondrial bioenergetics and fibre type assessments in microbiopsy vs Bergstrom percutaneous sampling of human skeletal muscle. Front Physiol 6, 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley BF, Nemeth PM, Martin WH, Hagberg JM, Dalsky GP & Holloszy JO (1986). Muscle triglyceride utilization during exercise: effect of training. J Appl Physiol 60, 562–567. [DOI] [PubMed] [Google Scholar]
- Jeneson JAL, Schmitz JPJ, van den Broek NMA, van Riel NAW, Hilbers PAJ, Nicolay K & Prompers JJ (2009). Magnitude and control of mitochondrial sensitivity to ADP. Am J Physiol Endocrinol Metab 297, E774–E784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaasik A, Minajeva A, De Sousa E, Ventura‐Clapier R & Veksler V (1999). Nitric oxide inhibits cardiac energy production via inhibition of mitochondrial creatine kinase. FEBS Lett 444, 75–77. [DOI] [PubMed] [Google Scholar]
- Kane DA, Lin C‐T, Anderson EJ, Kwak H‐B, Cox JH, Brophy PM, Hickner RC, Neufer PD & Cortright RN (2011). Progesterone increases skeletal muscle mitochondrial H2O2 emission in nonmenopausal women. Am J Physiol Endocrinol Metab 300, E528–E535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay L, Nicolay K, Wieringa B, Saks V & Wallimann T (2000). Direct evidence for the control of mitochondrial respiration by mitochondrial creatine kinase in oxidative muscle cells in situ . J Biol Chem 275, 6937–6944. [DOI] [PubMed] [Google Scholar]
- Kerner J, Lee K, Tandler B & Hoppel CL (2012). VDAC proteomics: post‐translation modifications. Biochim Biophys Acta 1818, 1520–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiens B, Essen‐Gustavsson B, Christensen NJ & Saltin B (1993). Skeletal muscle substrate utilization during submaximal exercise in man: effect of endurance training. J Physiol 469, 459–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn TA & Myburgh KH (2006). Electrophoretic separation of human skeletal muscle myosin heavy chain isoforms: the importance of reducing agents. J Physiol Sci 56, 355–360. [DOI] [PubMed] [Google Scholar]
- Korzeniewski B (2015). ‘Idealized’ state 4 and state 3 in mitochondria vs. rest and work in skeletal muscle. PLoS ONE 10, e0117145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsov AV, Tiivel T, Sikk P, Kaambre T, Kay L, Daneshrad Z, Rossi A, Kadaja L, Peet N, Seppet E & Saks VA (1996). Striking differences between the kinetics of regulation of respiration by ADP in slow‐twitch and fast‐twitch muscles in vivo . Eur J Biochem 241, 909–915. [DOI] [PubMed] [Google Scholar]
- Kuznetsov AV, Veksler V, Gellerich FN, Saks V, Margreiter R & Kunz WS (2008). Analysis of mitochondrial function in situ in permeabilized muscle fibers, tissues and cells. Nat Protocols 3, 965–976. [DOI] [PubMed] [Google Scholar]
- Layec G, Haseler LJ, Hoff J, Hart CR, Liu X, Le Fur Y, Jeong EK & Richardson RS (2013). Short‐term training alters the control of mitochondrial respiration rate before maximal oxidative ATP synthesis. Acta Physiol (Oxf) 208, 376–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBlanc PJ, Howarth KR, Gibala MJ & Heigenhauser GJF (2004). Effects of 7 wk of endurance training on human skeletal muscle metabolism during submaximal exercise. J Appl Physiol 97, 2148–2153. [DOI] [PubMed] [Google Scholar]
- Mettauer B, Zoll J, Sanchez H, Lampert E, Ribera F, Veksler V, Bigard X, Mateo P, Epailly E, Lonsdorfer J & Ventura‐Clapier R (2001). Oxidative capacity of skeletal muscle in heart failure patients versus sedentary or active control subjects. J Am Coll Cardiol 38, 947–954. [DOI] [PubMed] [Google Scholar]
- Mielke C, Lefort N, McLean CG, Cordova JM, Langlais PR, Bordner AJ, Te JA, Ozkan SB, Willis WT & Mandarino LJ (2014). Adenine nucleotide translocase is acetylated in vivo in human muscle: modeling predicts a decreased ADP affinity and altered control of oxidative phosphorylation. Biochemistry 53, 3817–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill HM, Maarbjerg SJ, Crane JD, Jeppesen J, Jørgensen SB, Schertzer JD, Shyroka O, Kiens B, van Denderen BJ, Tarnopolsky MA, Kemp BE, Richter EA & Steinberg GR (2011). AMP‐activated protein kinase (AMPK) β1β2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc Natl Acad Sci USA 108, 16092–16097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata T & Yamasaki Y (1997). Ultra‐high‐resolution scanning electron microscopy of mitochondria and sarcoplasmic reticulum arrangement in human red, white, and intermediate muscle fibers. Anat Rec 248, 214–223. [DOI] [PubMed] [Google Scholar]
- Perry CGR, Heigenhauser GJF, Bonen A & Spriet LL (2008). High‐intensity aerobic interval training increases fat and carbohydrate metabolic capacities in human skeletal muscle. Appl Physiol Nutr Metab 33, 1112–1123. [DOI] [PubMed] [Google Scholar]
- Perry CGR, Kane DA, Herbst EAF, Mukai K, Lark DS, Wright DC, Heigenhauser GJF, Neufer PD, Spriet LL & Holloway GP (2012). Mitochondrial creatine kinase activity and phosphate shuttling are acutely regulated by exercise in human skeletal muscle. J Physiol 590, 5475–5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry CGR, Kane DA, Lanza IR & Neufer PD (2013). Methods for assessing mitochondrial function in diabetes. Diabetes 62, 1041–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry CGR, Kane DA, Lin C‐T, Kozy R, Cathey BL, Lark DS, Kane CL, Brophy PM, Gavin TP, Anderson EJ & Neufer PD (2011). Inhibiting myosin‐ATPase reveals dynamic range of mitochondrial respiratory control in skeletal muscle. Biochem J 437, 215–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips SM, Green HJ, Tarnopolsky MA, Heigenhauser GJ & Grant SM (1996). Progressive effect of endurance training on metabolic adaptations in working skeletal muscle. Am J Physiol Endocrinol Metab 270, E265–E272. [DOI] [PubMed] [Google Scholar]
- Saks VA, Khuchua ZA, Vasilyeva EV, Belikova OY & Kuznetsov AV (1994). Metabolic compartmentation and substrate channelling in muscle cells. Mol Cell Biochem 133–134, 155–192. [DOI] [PubMed] [Google Scholar]
- Saks VA, Kuznetsov AV, Khuchua ZA, Vasilyeva EV, Belikova JO, Kesvatera T & Tiivel T (1995). Control of cellular respiration in vivo by mitochondrial outer membrane and by creatine kinase. A new speculative hypothesis: possible involvement of mitochondrial‐cytoskeleton interactions. J Mol Cell Cardiol 27, 625–645. [DOI] [PubMed] [Google Scholar]
- Schlattner U, Dolder M, Wallimann T & Tokarska‐Schlattner M (2001). Mitochondrial creatine kinase and mitochondrial outer membrane porin show a direct interaction that is modulated by calcium. J Biol Chem 276, 48027–48030. [DOI] [PubMed] [Google Scholar]
- Schlattner U, Mockli N, Speer O, Werner S & Wallimann T (2002). Creatine kinase and creatine transporter in normal, wounded, and diseased skin. J Invest Dermatol 118, 416–423. [DOI] [PubMed] [Google Scholar]
- Schlattner U, Tokarska‐Schlattner M & Wallimann T (2006). Mitochondrial creatine kinase in human health and disease. Biochim Biophys Acta 1762, 164–180. [DOI] [PubMed] [Google Scholar]
- Schlattner U & Wallimann T (2000). Octamers of mitochondrial creatine kinase isoenzymes differ in stability and membrane binding. J Biol Chem 275, 17314–17320. [DOI] [PubMed] [Google Scholar]
- Seppet EK, Kaambre T, Sikk P, Tiivel T, Vija H, Tonkonogi M, Sahlin K, Kay L, Appaix F, Braun U, Eimre M & Saks VA (2001). Functional complexes of mitochondria with Ca,MgATPases of myofibrils and sarcoplasmic reticulum in muscle cells. Biochim Biophys Acta 1504, 379–395. [DOI] [PubMed] [Google Scholar]
- Tonkonogi M, Harris B & Sahlin K (1998). Mitochondrial oxidative function in human saponin‐skinned muscle fibres: effects of prolonged exercise. J Physiol 510, 279–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonkonogi M, Walsh B, Tiivel T, Saks V & Sahlin K (1999). Mitochondrial function in human skeletal muscle is not impaired by high intensity exercise. Pflugers Arch 437, 562–568. [DOI] [PubMed] [Google Scholar]
- Veksler VI, Kuznetsov AV, Sharov VG, Kapelko VI & Saks VA (1987). Mitochondrial respiratory parameters in cardiac tissue: a novel method of assessment by using saponin‐skinned fibers. Biochim Biophys Acta 892, 191–196. [DOI] [PubMed] [Google Scholar]
- Ventura‐Clapier R, Kuznetsov A, Veksler V, Boehm E & Anflous K (1998). Functional coupling of creatine kinases in muscles: species and tissue specificity. Mol Cell Biochem 184, 231–247. [PubMed] [Google Scholar]
- Wallimann T, Tokarska‐Schlattner M & Schlattner U (2011). The creatine kinase system and pleiotropic effects of creatine. Amino Acids 40, 1271–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh B, Tonkonogi M & Sahlin K (2001. a). Effect of endurance training on oxidative and antioxidative function in human permeabilized muscle fibres. Pflügers Archiv 442, 420–425. [DOI] [PubMed] [Google Scholar]
- Walsh B, Tonkonogi M, Söderlund K, Hultman E, Saks V & Sahlin K (2001. b). The role of phosphorylcreatine and creatine in the regulation of mitochondrial respiration in human skeletal muscle. J Physiol 537, 971–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss M, Smeitink J, Wevers RA & Wallimann T (1992). Mitochondrial creatine kinase: a key enzyme of aerobic energy metabolism. Biochim Biophys Acta 1102, 119–166. [DOI] [PubMed] [Google Scholar]
- Zoll J, Koulmann N, Bahi L, Ventura‐Clapier R & Bigard A‐X (2003. a). Quantitative and qualitative adaptation of skeletal muscle mitochondria to increased physical activity. J Cell Physiol 194, 186–193. [DOI] [PubMed] [Google Scholar]
- Zoll J, N'Guessan B, Ribera F, Lampert E, Fortin D, Veksler V, Bigard X, Geny B, Lonsdorfer J, Ventura‐Clapier R & Mettauer B (2003. b). Preserved response of mitochondrial function to short‐term endurance training in skeletal muscle of heart transplant recipients. J Am Coll Cardiol 42, 126–132. [DOI] [PubMed] [Google Scholar]
- Zoll J, Sanchez H, N'Guessan B, Ribera F, Lampert E, Bigard X, Serrurier B, Fortin D, Geny B, Veksler V, Ventura‐Clapier R & Mettauer B (2002). Physical activity changes the regulation of mitochondrial respiration in human skeletal muscle. J Physiol 543, 191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]