

Abstract
Inositol 1,4,5‐trisphosphate receptors (IP3Rs) are a family of ubiquitously expressed intracellular Ca2+ release channels. Regulation of channel activity by Ca2+, nucleotides, phosphorylation, protein binding partners and other cellular factors is thought to play a major role in defining the specific spatiotemporal characteristics of intracellular Ca2+ signals. These properties are, in turn, believed pivotal for the selective and specific physiological activation of Ca2+‐dependent effectors. IP3Rs are also substrates for the intracellular cysteine proteases, calpain and caspase. Cleavage of the IP3R has been proposed to play a role in apoptotic cell death by uncoupling regions important for IP3 binding from the channel domain, leaving an unregulated leaky Ca2+ pore. Contrary to this hypothesis, we demonstrate following proteolysis that N‐ and C‐termini of IP3R1 remain associated, presumably through non‐covalent interactions. Further, we show that complementary fragments of IP3R1 assemble into tetrameric structures and retain their ability to be regulated robustly by IP3. While peptide continuity is clearly not necessary for IP3‐gating of the channel, we propose that cleavage of the IP3R peptide chain may alter other important regulatory events to modulate channel activity. In this scenario, stimulation of the cleaved IP3R may support distinct spatiotemporal Ca2+ signals and activation of specific effectors. Notably, in many adaptive physiological events, the non‐apoptotic activities of caspase and calpain are demonstrated to be important, but the substrates of the proteases are poorly defined. We speculate that proteolytic fragmentation may represent a novel form of IP3R regulation, which plays a role in varied adaptive physiological processes.
Abbreviations
- IP3
inositol 1,4,5‐trisphosphate
- IP3R
inositol 1,4,5‐trisphosphate receptor
- IP3R1
type 1 IP3R
- ER
endoplasmic reticulum
- [Ca2+]i
intracellular calcium concentration
Introduction
Inositol 1,4,5‐trisphosphate receptors (IP3Rs) are a family of ligand‐gated intracellular calcium release channels that are primarily expressed on the endoplasmic reticulum (ER) membrane (Foskett et al. 2007). IP3‐induced Ca2+ release controls a diverse array of cellular processes ranging from cell proliferation, differentiation, fluid secretion and motility to cell fate decisions including apoptosis and autophagy (Berridge, 1993; Joseph & Hajnoczky, 2007; Mikoshiba, 2007; Ivanova et al. 2014). It is thought that the versatility of this signal is largely a result of the spatial and temporal properties imparted by the fine control of IP3‐induced Ca2+ release through regulation of IP3R activity (Fig. 1). IP3R functions as a signal integrator that, depending on the stimuli, encodes the specific properties of the Ca2+ signal necessary to activate distinct downstream effectors with fidelity, specificity and efficiency. IP3R activity is regulated at multiple levels. For example, IP3R can interact with an abundance of intracellular molecules including Ca2+, nucleotides such as ATP and cAMP, and binding proteins, including Bcl‐2, Bok and IRBIT (Bruce et al. 2003; Foskett et al. 2007; Yule et al. 2010; Schulman et al. 2013). Further, IP3R are either homo‐ or heterotetrameric channels assembled from three receptor isoforms with additional splice variants (Nucifora et al. 1996; Joseph et al. 2000; Onoue et al. 2000). The subunit composition of the IP3R can either dominate or contribute to the characteristics of the receptor activity (Alzayady et al. 2013 b; Chandrasekhar et al. 2015). Also, post‐translational modification, such as phosphorylation and redox modification, plays a pivotal role in regulating IP3R activity (Fig. 1) (Betzenhauser & Yule, 2010; Yule et al. 2010; Bansaghi et al. 2014). As a further level of complexity, IP3R type 1 (IP3R1) has also been demonstrated to be a substrate of the cysteine proteases caspase and calpain, two enzymes often associated with cell death (Hirota et al. 1999). Through proteolytic cleavage, IP3R monomers are processed into at least two fragments (Fig. 2); an N‐terminal fragment, which contains the IP3 binding core and majority of the cytoplasmic region (∼210 kDa), and a C‐terminal fragment that comprises the transmembrane helices, the channel pore and the extreme C‐terminus cytoplasmic tail (∼90 kDa). To date, however, the functional consequences of the receptor fragmentation are not established and are the subject of considerable debate.
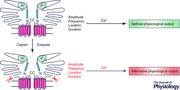
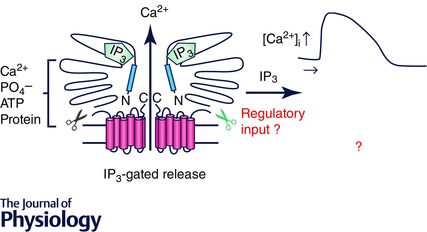
Figure 1. Schematic diagram depicting two monomers of a tetrameric IP3R .
The current model of gating suggests that the signal upon binding of IP3 to the binding core in the N‐terminal is transmitted through an interaction between residues in the suppressor domain (blue region) to the C‐terminal channel domain to result in channel opening and Ca2+ release. The activity of the channel is modulated by numerous cytosolic factors ultimately encoding the particular spatial and temporal characteristics of the intracellular Ca2+ signal. These properties are, in turn, important for the appropriate activation of effectors.
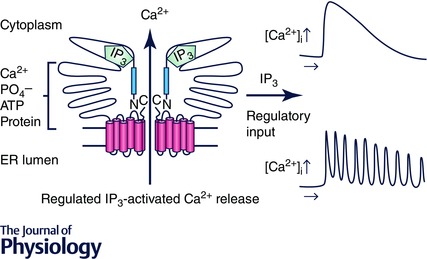
Figure 2. Schematic diagram depicting the hypothetical result of proteolytic cleavage of IP3R1 .
Cleavage by the cysteine proteases, calpain or caspase, results in two major peptide fragments; a large N‐terminal fragment and a C‐terminal remnant containing the channel domain. Loss of peptide continuity and/or diffusion of the cytoplasmic N‐terminal region result in an unregulated ‘leaky channel’.
In this review, we first summarize the major controversies relating to the consequences of receptor cleavage by calpain and caspase. Further, we present and discuss new findings from our lab, which suggest that proteolytic fragmentation of IP3R does not result in disassembly of the protein and does not necessarily disable regulated Ca2+ release, but rather, potentially alters the regulatory properties of the channel. We propose that IP3R fragmentation may represent a novel regulatory mechanism for IP3R1, which may regulate cellular events in addition to cell death.
IP3Rs are proteolytically processed by caspase and calpain
In 1999, Mikoshiba and colleagues first demonstrated that IP3R1 harbours a canonical DEVD substrate motif for caspase cleavage in its primary sequence at amino acids 1888–1891 (Hirota et al. 1999). Consistent with the prediction, IP3R1s were shown to be cleaved into two fragments in Jurkat cells during the process of apoptosis. These data were later confirmed in multiple cell lines including SH‐SY5Y cells, PC‐12 cells, Hela cells, DT‐40 cells and mouse oocytes during ageing, thus establishing that IP3R1s are a bone fide caspase substrate (Haug et al. 2000; Boehning et al. 2003; Assefa et al. 2004; Verbert et al. 2008; Zhang et al. 2011). Similarly, calpain cleaves the IP3R1 in the same region, 26 amino acids downstream of the caspase site, and generates receptor fragments with an almost identical cleavage pattern to that of caspase (Kopil et al. 2011). Importantly, calpain‐mediated IP3R1 fragmentation was shown to occur in rat cerebellum after cardiac arrest providing in vivo evidence of IP3R1 fragmentation in pathological conditions. However, while the idea that IP3R1 is a substrate of both caspase and calpain is widely accepted, the functional consequences of the proteolytic cleavage remain largely unresolved. One prominent idea is the so‐called ‘leaky channel’ theory (Nakayama et al. 2004; Szlufcik et al. 2006). Accordingly, caspase and/or calpain activated during the cell death process cleave the IP3R1 into two prominent fragments. The theory then posits that disruption of receptor continuity following proteolysis abolishes the tight regulation of the channel by IP3 binding in the N‐terminal fragment and this results in essentially an unregulated, constitutively active ‘leaky’ ER channel formed solely from the C‐terminal fragment (Fig. 2). The obvious consequence is that cytosolic calcium is progressively elevated as calcium leaks from the intracellular store. This event could conceivably recruit additional active caspase and calpain, generate more fragmented IP3R1s, and thereby establishe a fatal feed‐forward cycle that can efficiently amplify the death signal. In this scenario the fundamental function of IP3R1 fragmentation is to globally and continuously increase the intracellular calcium concentration ([Ca2+]i) and thereby accelerate the execution of apoptosis.
Evidence supporting the involvement of leaky IP3R channels in apoptosis
Some evidence in support of this idea comes from DT40‐3KO cells, a chicken B‐lymphocyte cell line where all three isoforms of IP3R have been genetically deleted (Sugawara et al. 1997; Assefa et al. 2004; Verbert et al. 2008). This cell line has been widely used in characterizing the activity of IP3Rs because it allows expression of IP3R constructs on an unambiguously null genetic background. Notably, it has been demonstrated that DT40‐3KO is relatively more resistant to programmed cell death than cells expressing IP3R1, indicating a potential role for IP3R1 in mechanisms underlying apoptosis (Jayaraman & Marks, 1997; Assefa et al. 2004; Khan et al. 2007). Interestingly, the sensitivity to apoptosis in DT40‐3KO cells expressing IP3R1 is completely abolished by mutation of the caspase cleavage sequence into a motif rendered non‐cleavable (Assefa et al. 2004). The most direct evidence in support of the general concept comes from measurement of Ca2+ signals following ectopic expression in DT40‐3KO cells of various constructs corresponding to different isolated regions of IP3R1. For example, a construct encoding only the caspase‐cleaved C‐terminal fragment (Δ1–1891), essentially the channel domain ‘stump’, was shown to express appropriately in the ER and enhance Ca2+ leak from intracellular stores. Interestingly, no marked reduction in the ER Ca2+ content was observed in these experiments, presumably as the sarco(endo)plasmic reticulum Ca2+‐ATPase (SERCA) pumping activity largely counteracted the ER leak. The expression of the fragment was also associated with augmentation of both the rate and the extent of apoptosis triggered by staurosporine (Assefa et al. 2004; Verbert et al. 2008). Additional reports generally consistent with the idea of the fragment functioning as a leaky channel include reports that ectopic expression of the channel domain in mouse oocytes decreased the ER Ca2+ content and induced apoptosis (Zhang & Fissore, 2014).
Similarly, a construct encoding the corresponding calpain‐cleaved C‐terminal channel stump (Δ1–1917 IP3R1), when expressed in both the mouse neuroblastoma cell line and primary cortical neurons reduced the ER Ca2+ content and promoted cell death following challenge with detrimental stimuli (Kopil et al. 2011, 2012). While these data provide good evidence that expression of C‐terminal fragments of IP3R can result in Ca2+ leak from the ER, caveats related to the interpretation of these data include that the channel domain was probably overexpressed to various degrees in these studies, and importantly that the channel C‐terminal fragments were expressed in the absence of the N‐terminal cytoplasmic domain of IP3R.
Evidence inconsistent with leaky channels leading to cell death
While the leaky channel theory provides an attractive potential correlation linking IP3R1 fragmentation and unregulated Ca2+ signalling to apoptosis, some observations are not necessarily consistent with this overall scenario. First, clues as to the consequences of fragmentation by intracellular proteases may be offered by earlier data investigating the structure of IP3R by subjecting purified IP3Rs to limited digestion in vitro with the protease trypsin (Joseph et al. 1995; Yoshikawa et al. 1999). A low concentration of trypsin cleaves IP3R1 at four distinct and reproducible sites, resulting in five predictable fragments. These data have been interpreted to indicate that the overall structure of the protein is tightly folded resulting in globular domains with the proteolysis occurring at solvent‐exposed cleavage sites between domains. Four of the resultant fragments are derived from cleavage of the cytosolic portion of IP3R1, while the fifth is the membrane‐associated channel domain, similar to the C‐terminal stump produced by caspase and calpain. Of note, all four tryptic cytosolic fragments were shown to remain associated with the fifth C‐terminal fragment by co‐immunoprecipitation (Joseph et al. 1995; Yoshikawa et al. 1999). Provocatively, IP3R1 in cerebellar microsomes exposed to trypsin or caspase retained significant IP3‐induced calcium release, although it should be noted that a small portion of intact IP3Rs might remain in the preparation (Yoshikawa et al. 1999). These data suggest that while peptide continuity is clearly disrupted following proteolytic processing by trypsin and caspase, IP3R1s, at least initially, retain general domain structure presumably through non‐covalent protein–protein interactions. Perhaps, most importantly, these data also provide early evidence that IP3Rs are not necessarily simply disabled by proteolysis.
Further inconsistences with the idea that Ca2+ leak through cleaved IP3Rs is a causative event in mediating apoptosis have also been reported. Joseph and colleagues reported that expression of a pore mutant IP3R1 in DT40‐3KO cells that cannot conduct Ca2+ is still competent to increase the activation of caspase and augment cell death (Khan et al. 2007). These data suggest that calcium leak per se from the IP3R1s or indeed fragmented IP3R1s is not indispensable for IP3R1‐dependent apoptosis. In addition, IP3R1s may not be the initial caspase target during apoptosis (Elkoreh et al. 2012). The strong apoptotic stimulus staurosporine results in the generation of IP3R1 fragments only after almost complete cleavage of poly ADP‐ribose polymerase, a biomarker of apoptosis, while ‘physiological’ stimuli such as Trail, tumour necrosis factor and UV lead to little IP3R1 fragmentation even though caspase is clearly activated. This finding was confirmed in MCF‐7 cells by Boehning and colleagues (Akimzhanov et al. 2013). Furthermore, in this latter study, an increase of IP3 production was detected in cells treated with staurosporine and scavenging of IP3 production substantially decreased cell death. These data imply that IP3 signalling, but not necessarily IP3R1 fragmentation, plays a major role in apoptosis. Given the earlier data that IP3R fragments remain associated following trypsin‐induced digestion in vitro and possibly retain some function, perhaps the major conceptual difficulty with the leaky channel theory is the assumption that the cytosolic IP3R1 fragments immediately dissociate after caspase or calpain cleavage in vivo and thus exert no further influence over channel function.
Re‐evaluation of the functional consequences of IP3R proteolysis: no evidence for leaky channels
Because of the somewhat contradictory interpretation of data relating to the functional consequences of proteolytic processing of IP3R1s, we have recently revisited these issues in DT40‐3KO expressing IP3R1 (Alzayady et al. 2013 a). First, we investigated a central assumption of the leaky channel hypothesis, namely that the ‘soluble’ N‐terminal fragment of IP3R1 is free to dissociate from the C‐terminal channel domain after proteolytic fragmentation leaving an unregulated channel stump. Our initial experiments demonstrated that, following 12 h treatment with staurosporine, IP3R fragmentation occurs resulting in products consistent with caspase and calpain cleavage profiles (Fig. 3 A). Strikingly, however, both N‐terminal and C‐terminal fragments remained in membrane fractions and not in the cytosolic fraction (Fig. 3 B). Further strong support for the concept that the major fragments remained associated was provided by analysis of fragmented IP3R1s following separation on native, non‐denaturing gels. These experiments revealed that the majority of fragmented IP3R1s, remarkably, migrate at the same molecular weight as the full‐length receptor and thus suggest it largely retains tetrameric architecture after proteolytic fragmentation during apoptosis, at least at this time point (Fig. 3 C). These data also indicate that if the C‐terminal fragment is retained in isolation in the ER membrane, it must occur at a later time point. This would necessitate a multistep process involving more than the initial cleavage event, but also disruption of the interaction between receptor fragments followed by the subsequent removal of cytosolic fragments from the complex.
Figure 3. Exposure of DT40‐3KO cells expressing rat IP3R1 to staurosporine results in the fragmentation of the protein .
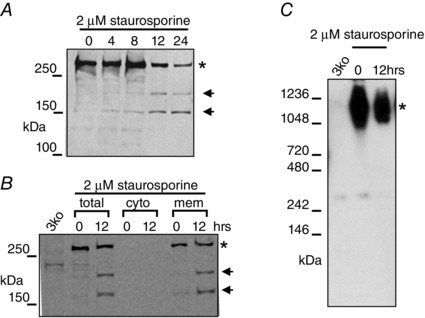
A, Western blot of protein separated by SDS‐PAGE probed with an antibody recognizing the N‐terminus of IP3R1. In B, the N‐terminal fragment remains associated with the membrane fraction. C, Western blot of protein separated under non‐denaturing conditions demonstrates that the majority of the fragmented receptor retains tetrameric structure. Adapted from Alzayady et al. (2013 a) with permission.
To investigate the functional consequences of IP3R1 fragmentation, we constructed dual promoter vectors encoding various combinations of complementary N‐ and C‐terminal fragments of IP3R1 designed based on the in vivo caspase and calpain, and in vitro trypsin, cleavage products and stably expressed the fragments in DT40‐3KO cells (Alzayady et al. 2013 a). The rationale was to mimic the generation of fragmented IP3Rs in a controlled setting with no full‐length endogenous IP3Rs. Intriguingly, both co‐immunoprecipitation and native gel assay show that every pair of complementary IP3R1 fragments expressed in DT40‐3KO cells associates and efficiently assembles into tetrameric IP3R1s (Alzayady et al. 2013 a). Stable expression of either C‐terminal fragments alone or complementary N and C‐terminal fragments did not result in an elevated basal [Ca2+] or decrease in ER store content. Although Ca2+ leak was not measured directly, and thus we cannot formally exclude a minor leak, adequately compensated for by SERCA activity, these data are not consistent with significant increase of basal [Ca2+]i or depletion of ER store mediated by the leak through C‐terminal fragments, at least at the expression level achieved in this study. More importantly, all complemented IP3Rs were functionally regulated by IP3 as stimulation of cells expressing these constructs with Gαq‐coupled protease‐activated receptor 2 (PAR2) agonist resulted in robust Ca2+ release (Fig. 4). These results show unequivocally, that peptide continuity per se is not necessary for IP3‐mediated calcium release and it therefore follows that the IP3R protein is not simply disabled by protease activity. These results are also entirely consistent with conceptually similar experiments performed on ryanodine receptors (RyRs), which share a similar general domain structure to IP3Rs (George et al. 2004). Again, overexpression of the C‐terminal of RyR containing the channel pore resulted in a leaky channel that was unregulated by ligands. However, co‐expression of the N‐terminal portion of RyR reconstituted normal regulated Ca2+ release. Further experiments showed that functional IP3R channels could also be assembled from complementary IP3R2 and IP3R3 fragments and even ‘chimeric’ fragments contributed by different subtypes (Alzayady et al. 2013 a). These data establish a general rule that peptide continuity is not required for gating IP3‐mediated IP3R activation, and in total strongly suggest that contrary to being a leaky intracellular Ca2+ channel, IP3R1s are still at least minimally regulated after receptor fragmentation and are competent to gate Ca2+ release by IP3 (Fig. 5).
Figure 4. Ectopic expression of IP3R1 fragments corresponding to those produced by caspase and calpain proteolysis results in functional Ca2+ release channels .
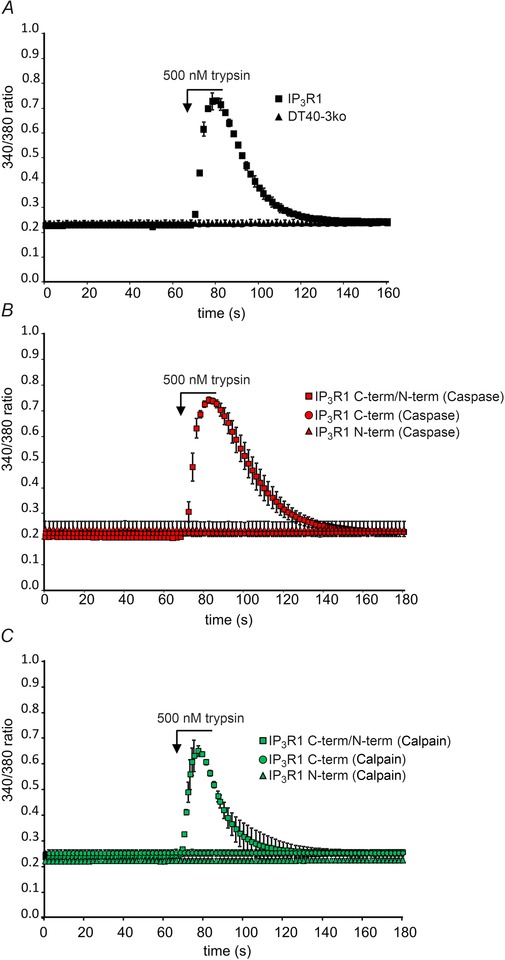
In A, the Gαq‐coupled protease‐activated receptor (PAR) agonist trypsin can stimulate Ca2+ release from DT40‐3KO cells expressing IP3R1, but not the parent DT40‐3KO cells. In B, DT40‐3KO cells were transfected singly, or with a combination of the C‐ terminal or N‐terminal fragments corresponding to those produced by caspase cleavage. PAR agonist stimulation resulted in robust Ca2+ release in cells expressing both fragments. In C, DT40‐3KO cells were transfected singly, or with a combination of the C‐terminal or N‐terminal fragments corresponding to those produced by calpain cleavage. PAR agonist stimulation resulted in robust Ca2+ release in cells expressing both fragments as monitored by an increase in 340/380 fluorescence ratio in cells loaded with Fura‐2. Adapted from Alzayady et al. (2013 a) with permission.
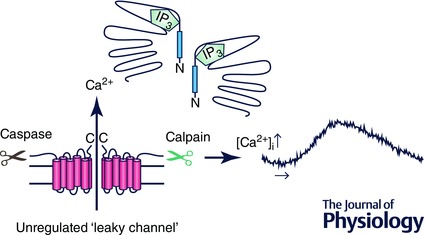
Figure 5. Schematic diagram depicting our working hypothesis based on data in Figs 3 and 4 .
Following proteolytic cleavage of IP3R1, the N and C terminal fragments remain associated through non‐covalent interactions, possibly prominently involving the N‐ and C‐termini. The complemented receptor is not disabled, and can be gated by IP3 following agonist stimulation. We speculate that disruption of peptide continuity may, however, impact the spatio‐temporal characteristics of the Ca2+ signal produced by the cleaved protein.
Proteolytic fragmentation of IP3R: a novel mode of regulation?
Peptide continuity and thus communication of information through the entire length of the IP3R1 peptide chain is clearly not required for IP3 gating of IP3R1. In hindsight, perhaps this is not totally unexpected in light of data that suggest that the conformational change on binding of IP3 occurring in the N‐terminal binding core is communicated to opening of the channel pore in the C‐terminus by a direct physical interaction between the two distant domains (Boehning & Joseph, 2000; Bhanumathy et al. 2012). Nevertheless, although IP3 binding is obviously necessary for Ca2+ release, the overall spatiotemporal characteristics of the Ca2+ signal are dictated by numerous regulatory inputs to the receptor. The structural and molecular basis underlying regulatory input by the majority of other important modulators, such as Ca2+, ATP, phosphorylation and binding partners is much less well defined and may indeed rely on short‐distance allosteric modulation of the channel activity that is dependent on peptide continuity. A particular form of regulation might depend on modulation of IP3 binding or perhaps direct regulation of gating or channel activity – in this scenario the regulatory motif would probably reside in the same fragment as the target of its action. This leads us to speculate that proteolytic fragmentation may constitute a form of IP3R1 regulation in itself. Theoretically, this might occur as IP3R fragmentation results in an alteration (either loss or gain) of a particular set of modulatory inputs regulating channel activity. This change in receptor properties, might in turn be manifested as a distinct Ca2+ release profile, transformed in terms of peak amplitude, duration of signal or frequency of oscillations (Fig. 5). An added level of regulation might occur as the degree of proteolysis is tuned by other cellular input. Of interest, recent evidence suggests that association of Bok, a pro‐apoptotic Bcl‐2 family member interacts with IP3R1 and protects the channel from caspase‐3‐mediated cleavage (Schulman et al. 2013). If only a subset of IP3Rs were subject to proteolytic processing at a particular site, it could be envisioned that any alterations in properties might even occur in a local subcellular region. Given that it is firmly established that particular spatiotemporal Ca2+ release profiles determine the activation of specific downstream pathways, IP3R1 fragmentation may constitute a novel cellular switch resulting ultimately in signalling to different Ca2+‐dependent effectors and entraining distinct cellular outcomes.
Consistent with this idea, cleavage of an increasing number of proteins by caspase and calpain has been shown to alter, rather than abolish, activity. Examples of substrates where activity is altered include the nuclease CAD (Larsen et al. 2010), interleukins (Kuranaga & Miura, 2007) and calcineurin (Mukerjee et al. 2000). Thus, the idea of regulation by proteolysis is an emerging theme (Balcerzak et al. 1995; Sordet et al. 2002; Neumar et al. 2003; Fernando et al. 2005; Connolly et al. 2014; Buffolo et al. 2015). Much work is needed to firmly establish this concept and ongoing work in our laboratory is focusing on determining how IP3R fragmentation alters both the overall spatiotemporal characteristics of the Ca2+ signal and the biophysical properties of the IP3R at the single channel level. We envision that this information may provide clues as to how the distinct downstream effectors are specifically activated by fragmented IP3R1s. Since, IP3R2 and IP3R3 are also subject to proteolytic cleavage, it will also be important to determine if the activity of these subtypes is altered by cleavage (Wojcikiewicz et al. 1999; Diaz & Bourguignon, 2000).
Future perspective: not just cell death. Are fragmented IP3R1s involved in physiological activities of caspase and calpain?
In addition to a potential general role in apoptosis, an exciting possibility is that proteolytic fragmentation of IP3R1s by caspase and calpain may, in fact, be involved in many other physiological events. In recent years, growing evidence suggests that the activity of these two cysteine proteases may also be important for numerous cell events other than cell death (Balcerzak et al. 1995; Sorimachi et al. 1997; Sordet et al. 2002; Neumar et al. 2003; Fernando et al. 2005; Kuranaga & Miura, 2007; Larsen et al. 2010; Zadran et al. 2010; Connolly et al. 2014; Buffolo et al. 2015). Indeed, protease activity has been implicated in adaptive events as diverse as cytoskeletal remodelling (Helfer et al. 2006), synaptic plasticity (Zadran et al. 2010), cell migration (Geisbrecht & Montell, 2004), maturation and differentiation (Sordet et al. 2002; Fernando et al. 2005; Kuranaga & Miura, 2007). In these cases, low level and often local protease activity is necessary, but the substrates of the proteases and therefore the underlying mechanisms are not well defined. Because of the slow turnover of IP3Rs, fragmentation may provide an attractive means of conferring a long‐term modification of the activity of IP3Rs suitable for initiating an adaptive response. Therefore, the possible occurrence and potential functional consequences of fragmented IP3Rs in the aforementioned adaptive events are worthy of further investigation.
Neural development may be an attractive initial model to investigate this general concept. The broad importance of each protein in development is indicated by the knockout of either IP3R1 or caspase in mice, which results in perinatal lethality with severe neurodevelopmental abnormalities (Matsumoto et al. 1996; Matsumoto & Nagata, 1999). Ca2+ release through IP3Rs is reported to be important for neurite sprouting and arborization (Takei et al. 1998; Fiedler & Nathanson, 2011). Although the substrate(s) are not defined, caspase activity is significantly increased during differentiation of neurospheres and pharmacological inhibition alters the expression pattern of neuro‐specific genes resulting in a reduction in neurite extension (Fernando et al. 2005). Moreover, similar results were obtained in a model of PC‐12 cell differentiation, suggesting a general physiological role of caspase in neural differentiation (Vaisid et al. 2005). In summary, there is clear evidence suggesting the activities of caspase and IP3R1 are both essential for neural development. It will be exciting to investigate whether fragmented IP3R1s appear and contribute to these events.
Skeletal muscle myoblast differentiation may be another process where IP3R1 fragmentation may occur and potentially play a role. Elevated caspase and calpain activity have been shown to be important for the transformation of myoblasts to myotubes, highlighting a non‐apoptotic role of the proteases in this process (Balcerzak et al. 1995; Larsen et al. 2010). While the expression and function of IP3Rs in differentiated myotubes are still debated (Moschella et al. 1995; Powell et al. 2001; Tjondrokoesoemo et al. 2013), there is consistent evidence showing that all three isoforms of IP3R are expressed in the myoblast (Arnaudeau et al. 2006; Blaauw et al. 2012; Antigny et al. 2014). Of note, during myoblast differentiation, IP3R1 expression level is increased, and this corresponds with cells exhibiting spontaneous oscillatory calcium signals (Antigny et al. 2014). In addition, evidence from knockdown and overexpression strategies indicates the degree of myoblast differentiation is directly related to the expression level of IP3R1. This strongly suggests that IP3R1s play an essential role in myoblast differentiation. In total, myoblast differentiation provides a physiological platform for a potential cross talk among caspase, calpain and IP3R1. Nevertheless, in common with the case of neural development, establishing a causal link will require demonstrating that IP3Rs are an important substrate of caspase and/or calpain, and its proteolytic fragmentation is necessary for particular step(s) in myoblast differentiation.
Concluding remarks
IP3R1 fragmentation may be a relatively common event, underlying many other physiological events including differentiation of macrophages, osteoblasts and stem cells (Sordet et al. 2002; Mogi & Togari, 2003; Kang et al. 2004; Fujita et al. 2008; Connolly et al. 2014). All these events have common features, including expression of IP3R1 and non‐cell‐death‐related caspase and calpain activities. In summary, accumulating evidence suggests that proteolytic fragmentation by caspase and calpain may be a novel regulatory mechanism for IP3R1. Evidence from the literature warrants further studies to investigate the potential role of IP3R1 fragmentation in both physiological and pathological conditions.
Additional information
Competing interests
None declared.
Funding
This work was supported by NIH (National Institute of Dental and Craniofacial Research) grants DE19245 and DE14756.
Biography
Liwei Wang (left) is a senior graduate student and Kamil Alzayady (right) a Research Assistant Professor working in the Department of Pharmacology and Physiology at The University of Rochester with David Yule (PhD). The lab is interested in how a ubiquitous message, changes in intracellular [Ca2+], activate diverse physiological processes with specificity. A central tenet of our work is that fine tuning of Ca2+ release through inositol 1,4,5‐trisphosphate receptors plays major roles in shaping the spatio‐temporal dynamics of Ca2+ signals which in turn is pivotal for the fidelity of the signal.

This review was presented at the symposium “New Insights into Ca2+ Stores and Intracellular Ca2+ Channels”, which took place at the Gordon Research Conference on Calcium Signalling ‐ Molecular and Cellular Mechanisms in Health and Disease in Maine, USA, 7–12 June, 2015.
References
- Akimzhanov AM, Barral JM & Boehning D (2013). Caspase 3 cleavage of the inositol 1,4,5‐trisphosphate receptor does not contribute to apoptotic calcium release. Cell Calcium 53, 152–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzayady KJ, Chandrasekhar R & Yule DI (2013. a). Fragmented inositol 1,4,5‐trisphosphate receptors retain tetrameric architecture and form functional Ca2+ release channels. J Biol Chem 288, 11122–11134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzayady KJ, Wagner LE 2nd, Chandrasekhar R, Monteagudo A, Godiska R, Tall GG, Joseph SK & Yule DI (2013. b). Functional inositol 1,4,5‐trisphosphate receptors assembled from concatenated homo‐ and heteromeric subunits. J Biol Chem 288, 29772–29784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antigny F, Konig S, Bernheim L & Frieden M (2014). Inositol 1,4,5 trisphosphate receptor 1 is a key player of human myoblast differentiation. Cell Calcium 56, 513–521. [DOI] [PubMed] [Google Scholar]
- Arnaudeau S, Holzer N, Konig S, Bader CR & Bernheim L (2006). Calcium sources used by post‐natal human myoblasts during initial differentiation. J Cell Physiol 208, 435–445. [DOI] [PubMed] [Google Scholar]
- Assefa Z, Bultynck G, Szlufcik K, Nadif Kasri N, Vermassen E, Goris J, Missiaen L, Callewaert G, Parys JB & De Smedt H (2004). Caspase‐3‐induced truncation of type 1 inositol trisphosphate receptor accelerates apoptotic cell death and induces inositol trisphosphate‐independent calcium release during apoptosis. J Biol Chem 279, 43227–43236. [DOI] [PubMed] [Google Scholar]
- Balcerzak D, Poussard S, Brustis JJ, Elamrani N, Soriano M, Cottin P & Ducastaing A (1995). An antisense oligodeoxyribonucleotide to m‐calpain mRNA inhibits myoblast fusion. J Cell Sci 108), 2077–2082. [DOI] [PubMed] [Google Scholar]
- Bansaghi S, Golenar T, Madesh M, Csordas G, RamachandraRao S, Sharma K, Yule DI, Joseph SK & Hajnoczky G (2014). Isoform‐ and species‐specific control of inositol 1,4,5‐trisphosphate (IP3) receptors by reactive oxygen species. J Biol Chem 289, 8170–8181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ (1993). Inositol trisphosphate and calcium signalling. Nature 361, 315–325. [DOI] [PubMed] [Google Scholar]
- Betzenhauser MJ & Yule DI (2010). Regulation of inositol 1,4,5‐trisphosphate receptors by phosphorylation and adenine nucleotides. Curr Top Membr 66, 273–298. [DOI] [PubMed] [Google Scholar]
- Bhanumathy C, da Fonseca PC, Morris EP & Joseph SK (2012). Identification of functionally critical residues in the channel domain of inositol trisphosphate receptors. J Biol Chem 287, 43674–43684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaauw B, Del Piccolo P, Rodriguez L, Hernandez Gonzalez VH, Agatea L, Solagna F, Mammano F, Pozzan T & Schiaffino S (2012). No evidence for inositol 1,4,5‐trisphosphate‐dependent Ca2+ release in isolated fibers of adult mouse skeletal muscle. J Gen Physiol 140, 235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehning D & Joseph SK (2000). Direct association of ligand‐binding and pore domains in homo‐ and heterotetrameric inositol 1,4,5‐trisphosphate receptors. EMBO J 19, 5450–5459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehning D, Patterson RL, Sedaghat L, Glebova NO, Kurosaki T & Snyder SH (2003). Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium‐dependent apoptosis. Nat Cell Biol 5, 1051–1061. [DOI] [PubMed] [Google Scholar]
- Bruce JI, Straub SV & Yule DI (2003). Crosstalk between cAMP and Ca2+ signaling in non‐excitable cells. Cell Calcium 34, 431–444. [DOI] [PubMed] [Google Scholar]
- Buffolo M, Batista Possidonio AC, Mermelstein C & Araujo H (2015). A conserved role for calpains during myoblast fusion. Genesis 53, 417–430. [DOI] [PubMed] [Google Scholar]
- Chandrasekhar R, Alzayady KJ & Yule DI (2015). Using concatenated subunits to investigate the functional consequences of heterotetrameric inositol 1,4,5‐trisphosphate receptors. Biochem Soc Trans 43, 364–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly PF, Jager R & Fearnhead HO (2014). New roles for old enzymes: killer caspases as the engine of cell behavior changes. Front Physiol 5, 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz F & Bourguignon LY (2000). Selective down‐regulation of IP3 receptor subtypes by caspases and calpain during TNFα‐induced apoptosis of human T‐lymphoma cells. Cell Calcium 27, 315–328. [DOI] [PubMed] [Google Scholar]
- Elkoreh G, Blais V, Beliveau E, Guillemette G & Denault JB (2012). Type 1 inositol‐1,4,5‐trisphosphate receptor is a late substrate of caspases during apoptosis. J Cell Biochem 113, 2775–2784. [DOI] [PubMed] [Google Scholar]
- Fernando P, Brunette S & Megeney LA (2005). Neural stem cell differentiation is dependent upon endogenous caspase 3 activity. FASEB J 19, 1671–1673. [DOI] [PubMed] [Google Scholar]
- Fiedler MJ & Nathanson MH (2011). The type I inositol 1,4,5‐trisphosphate receptor interacts with protein 4.1N to mediate neurite formation through intracellular Ca waves. Neurosignals 19, 75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foskett JK, White C, Cheung KH & Mak DO (2007). Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev 87, 593–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita J, Crane AM, Souza MK, Dejosez M, Kyba M, Flavell RA, Thomson JA & Zwaka TP (2008). Caspase activity mediates the differentiation of embryonic stem cells. Cell Stem Cell 2, 595–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisbrecht ER & Montell DJ (2004). A role for Drosophila IAP1‐mediated caspase inhibition in Rac‐dependent cell migration. Cell 118, 111–125. [DOI] [PubMed] [Google Scholar]
- George CH, Jundi H, Thomas NL, Scoote M, Walters N, Williams AJ & Lai FA (2004). Ryanodine receptor regulation by intramolecular interaction between cytoplasmic and transmembrane domains. Mol Biol Cell 15, 2627–2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haug LS, Walaas SI & Ostvold AC (2000). Degradation of the type I inositol 1,4,5‐trisphosphate receptor by caspase‐3 in SH‐SY5Y neuroblastoma cells undergoing apoptosis. J Neurochem 75, 1852–1861. [DOI] [PubMed] [Google Scholar]
- Helfer B, Boswell BC, Finlay D, Cipres A, Vuori K, Bong Kang T, Wallach D, Dorfleutner A, Lahti JM, Flynn DC & Frisch SM (2006). Caspase‐8 promotes cell motility and calpain activity under nonapoptotic conditions. Cancer Res 66, 4273–4278. [DOI] [PubMed] [Google Scholar]
- Hirota J, Furuichi T & Mikoshiba K (1999). Inositol 1,4,5‐trisphosphate receptor type 1 is a substrate for caspase‐3 and is cleaved during apoptosis in a caspase‐3‐dependent manner. J Biol Chem 274, 34433–34437. [DOI] [PubMed] [Google Scholar]
- Ivanova H, Vervliet T, Missiaen L, Parys JB, De Smedt H & Bultynck G (2014). Inositol 1,4,5‐trisphosphate receptor‐isoform diversity in cell death and survival. Biochim Biophys Acta 1843, 2164–2183. [DOI] [PubMed] [Google Scholar]
- Jayaraman T & Marks AR (1997). T cells deficient in inositol 1,4,5‐trisphosphate receptor are resistant to apoptosis. Mol Cell Biol 17, 3005–3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph SK, Bokkala S, Boehning D & Zeigler S (2000). Factors determining the composition of inositol trisphosphate receptor hetero‐oligomers expressed in COS cells. J Biol Chem 275, 16084–16090. [DOI] [PubMed] [Google Scholar]
- Joseph SK & Hajnoczky G (2007). IP3 receptors in cell survival and apoptosis: Ca2+ release and beyond. Apoptosis 12, 951–968. [DOI] [PubMed] [Google Scholar]
- Joseph SK, Pierson S & Samanta S (1995). Trypsin digestion of the inositol trisphosphate receptor: implications for the conformation and domain organization of the protein. Biochem J 307, 859–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang TB, Ben‐Moshe T, Varfolomeev EE, Pewzner‐Jung Y, Yogev N, Jurewicz A, Waisman A, Brenner O, Haffner R, Gustafsson E, Ramakrishnan P, Lapidot T & Wallach D (2004). Caspase‐8 serves both apoptotic and nonapoptotic roles. J Immunol 173, 2976–2984. [DOI] [PubMed] [Google Scholar]
- Khan MT, Bhanumathy CD, Schug ZT & Joseph SK (2007). Role of inositol 1,4,5‐trisphosphate receptors in apoptosis in DT40 lymphocytes. J Biol Chem 282, 32983–32990. [DOI] [PubMed] [Google Scholar]
- Kopil CM, Siebert AP, Foskett JK & Neumar RW (2012). Calpain‐cleaved type 1 inositol 1,4,5‐trisphosphate receptor impairs ER Ca2+ buffering and causes neurodegeneration in primary cortical neurons. J Neurochem 123, 147–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopil CM, Vais H, Cheung KH, Siebert AP, Mak DO, Foskett JK & Neumar RW (2011). Calpain‐cleaved type 1 inositol 1,4,5‐trisphosphate receptor (InsP3R1) has InsP3‐independent gating and disrupts intracellular Ca2+ homeostasis. J Biol Chem 286, 35998–36010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuranaga E & Miura M (2007). Nonapoptotic functions of caspases: caspases as regulatory molecules for immunity and cell‐fate determination. Trends Cell Biol 17, 135–144. [DOI] [PubMed] [Google Scholar]
- Larsen BD, Rampalli S, Burns LE, Brunette S, Dilworth FJ & Megeney LA (2010). Caspase 3/caspase‐activated DNase promote cell differentiation by inducing DNA strand breaks. Proc Natl Acad Sci USA 107, 4230–4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M & Nagata E (1999). Type 1 inositol 1,4,5‐trisphosphate receptor knock‐out mice: their phenotypes and their meaning in neuroscience and clinical practice. J Mol Med (Berl) 77, 406–411. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Nakagawa T, Inoue T, Nagata E, Tanaka K, Takano H, Minowa O, Kuno J, Sakakibara S, Yamada M, Yoneshima H, Miyawaki A, Fukuuchi Y, Furuichi T, Okano H, Mikoshiba K & Noda T (1996). Ataxia and epileptic seizures in mice lacking type 1 inositol 1,4,5‐trisphosphate receptor. Nature 379, 168–171. [DOI] [PubMed] [Google Scholar]
- Mikoshiba K (2007). IP3 receptor/Ca2+ channel: from discovery to new signaling concepts. J Neurochem 102, 1426–1446. [DOI] [PubMed] [Google Scholar]
- Mogi M & Togari A (2003). Activation of caspases is required for osteoblastic differentiation. J Biol Chem 278, 47477–47482. [DOI] [PubMed] [Google Scholar]
- Moschella MC, Watras J, Jayaraman T & Marks AR (1995). Inositol 1,4,5‐trisphosphate receptor in skeletal muscle: differential expression in myofibres. J Muscle Res Cell Motil 16, 390–400. [DOI] [PubMed] [Google Scholar]
- Mukerjee N, McGinnis KM, Park YH, Gnegy ME & Wang KK (2000). Caspase‐mediated proteolytic activation of calcineurin in thapsigargin‐mediated apoptosis in SH‐SY5Y neuroblastoma cells. Arch Biochem Biophys 379, 337–343. [DOI] [PubMed] [Google Scholar]
- Nakayama T, Hattori M, Uchida K, Nakamura T, Tateishi Y, Bannai H, Iwai M, Michikawa T, Inoue T & Mikoshiba K (2004). The regulatory domain of the inositol 1,4,5‐trisphosphate receptor is necessary to keep the channel domain closed: possible physiological significance of specific cleavage by caspase 3. Biochem J 377, 299–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumar RW, Xu YA, Gada H, Guttmann RP & Siman R (2003). Cross‐talk between calpain and caspase proteolytic systems during neuronal apoptosis. J Biol Chem 278, 14162–14167. [DOI] [PubMed] [Google Scholar]
- Nucifora FC Jr, Sharp AH, Milgram SL & Ross CA (1996). Inositol 1,4,5‐trisphosphate receptors in endocrine cells: localization and association in hetero‐ and homotetramers. Mol Biol Cell 7, 949–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onoue H, Tanaka H, Tanaka K, Doira N & Ito Y (2000). Heterooligomer of type 1 and type 2 inositol 1,4,5‐trisphosphate receptor expressed in rat liver membrane fraction exists as tetrameric complex. Biochem Biophys Res Commun 267, 928–933. [DOI] [PubMed] [Google Scholar]
- Powell JA, Carrasco MA, Adams DS, Drouet B, Rios J, Muller M, Estrada M & Jaimovich E (2001). IP3 receptor function and localization in myotubes: an unexplored Ca2+ signaling pathway in skeletal muscle. J Cell Sci 114, 3673–3683. [DOI] [PubMed] [Google Scholar]
- Schulman JJ, Wright FA, Kaufmann T & Wojcikiewicz RJ (2013). The Bcl‐2 protein family member Bok binds to the coupling domain of inositol 1,4,5‐trisphosphate receptors and protects them from proteolytic cleavage. J Biol Chem 288, 25340–25349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sordet O, Rebe C, Plenchette S, Zermati Y, Hermine O, Vainchenker W, Garrido C, Solary E & Dubrez‐Daloz L (2002). Specific involvement of caspases in the differentiation of monocytes into macrophages. Blood 100, 4446–4453. [DOI] [PubMed] [Google Scholar]
- Sorimachi H, Ishiura S & Suzuki K (1997). Structure and physiological function of calpains. Biochem J 328, 721–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara H, Kurosaki M, Takata M & Kurosaki T (1997). Genetic evidence for involvement of type 1, type 2 and type 3 inositol 1,4,5‐trisphosphate receptors in signal transduction through the B‐cell antigen receptor. EMBO J 16, 3078–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szlufcik K, Missiaen L, Parys JB, Callewaert G & De Smedt H (2006). Uncoupled IP3 receptor can function as a Ca2+‐leak channel: cell biological and pathological consequences. Biol Cell 98, 1–14. [DOI] [PubMed] [Google Scholar]
- Takei K, Shin RM, Inoue T, Kato K & Mikoshiba K (1998). Regulation of nerve growth mediated by inositol 1,4,5‐trisphosphate receptors in growth cones. Science 282, 1705–1708. [DOI] [PubMed] [Google Scholar]
- Tjondrokoesoemo A, Li N, Lin PH, Pan Z, Ferrante CJ, Shirokova N, Brotto M, Weisleder N & Ma J (2013). Type 1 inositol (1,4,5)‐trisphosphate receptor activates ryanodine receptor 1 to mediate calcium spark signaling in adult mammalian skeletal muscle. J Biol Chem 288, 2103–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaisid T, Kosower NS & Barnoy S (2005). Caspase‐1 activity is required for neuronal differentiation of PC12 cells: cross‐talk between the caspase and calpain systems. Biochim Biophys Acta 1743, 223–230. [DOI] [PubMed] [Google Scholar]
- Verbert L, Lee B, Kocks SL, Assefa Z, Parys JB, Missiaen L, Callewaert G, Fissore RA, De Smedt H & Bultynck G (2008). Caspase‐3‐truncated type 1 inositol 1,4,5‐trisphosphate receptor enhances intracellular Ca2+ leak and disturbs Ca2+ signalling. Biol Cell 100, 39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcikiewicz RJ, Ernst SA & Yule DI (1999). Secretagogues cause ubiquitination and down‐regulation of inositol 1,4,5‐trisphosphate receptors in rat pancreatic acinar cells. Gastroenterology 116, 1194–1201. [DOI] [PubMed] [Google Scholar]
- Yoshikawa F, Iwasaki H, Michikawa T, Furuichi T & Mikoshiba K (1999). Trypsinized cerebellar inositol 1,4,5‐trisphosphate receptor. Structural and functional coupling of cleaved ligand binding and channel domains. J Biol Chem 274, 316–327. [DOI] [PubMed] [Google Scholar]
- Yule DI, Betzenhauser MJ & Joseph SK (2010). Linking structure to function: Recent lessons from inositol 1,4,5‐trisphosphate receptor mutagenesis. Cell Calcium 47, 469–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zadran S, Bi X & Baudry M (2010). Regulation of calpain‐2 in neurons: implications for synaptic plasticity. Mol Neurobiol 42, 143–150. [DOI] [PubMed] [Google Scholar]
- Zhang N & Fissore RA (2014). Role of caspase‐3 cleaved IP3R1 on Ca2+ homeostasis and developmental competence of mouse oocytes and eggs. J Cell Physiol 229, 1842–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Wakai T & Fissore RA (2011). Caffeine alleviates the deterioration of Ca2+ release mechanisms and fragmentation of in vitro‐aged mouse eggs. Mol Reprod Dev 78, 684–701. [DOI] [PMC free article] [PubMed] [Google Scholar]