

Abstract
Key points
Duchenne muscular dystrophy (DMD) is a fatal muscle wasting disease associated with increased inflammation, oxidative stress and myofibre necrosis.
Cysteine precursor antioxidants such as N‐acetyl cysteine (NAC) and l‐2‐oxothiazolidine‐4‐carboxylate (OTC) reduce dystropathology in the mdx mouse model for DMD, and we propose this is via increased synthesis of the amino acid taurine.
We compared the capacity of OTC and taurine treatment to increase taurine content of mdx muscle, as well as effects on in vivo and ex vivo muscle function, inflammation and oxidative stress.
Both treatments increased taurine in muscles, and improved many aspects of muscle function and reduced inflammation. Taurine treatment also reduced protein thiol oxidation and was overall more effective, as OTC treatment reduced body and muscle weight, suggesting some adverse effects of this drug.
These data suggest that increasing dietary taurine is a better candidate for a therapeutic intervention for DMD.
Abstract
Duchenne muscular dystrophy (DMD) is a fatal muscle wasting disease for which there is no widely available cure. Whilst the mechanism of loss of muscle function in DMD and the mdx mouse model are not fully understood, disruptions in intracellular calcium homeostasis, inflammation and oxidative stress are implicated. We have shown that protein thiol oxidation is increased in mdx muscle, and that the indirect thiol antioxidant l‐2‐oxothiazolidine‐4‐carboxylate (OTC), which increases cysteine availability, decreases pathology and increases in vivo strength. We propose that the protective effects of OTC are a consequence of conversion of cysteine to taurine, which has itself been shown to be beneficial to mdx pathology. This study compares the efficacy of taurine with OTC in decreasing dystropathology in mdx mice by measuring in vivo and ex vivo contractile function and measurements of inflammation and protein thiol oxidation. Increasing the taurine content of mdx muscle improved both in vivo and ex vivo muscle strength and function, potentially via anti‐inflammatory and antioxidant effects of taurine. OTC treatment increased taurine synthesis in the liver and taurine content of mdx muscle, improved muscle function and decreased inflammation. However, OTC was less effective than taurine treatment, with OTC also decreasing body and EDL muscle weights, suggesting that OTC had some detrimental effects. These data support continued research into the use of taurine as a therapeutic intervention for DMD, and suggest that increasing dietary taurine is the better strategy for increasing taurine content and decreasing severity of dystropathology.
Key points
Duchenne muscular dystrophy (DMD) is a fatal muscle wasting disease associated with increased inflammation, oxidative stress and myofibre necrosis.
Cysteine precursor antioxidants such as N‐acetyl cysteine (NAC) and l‐2‐oxothiazolidine‐4‐carboxylate (OTC) reduce dystropathology in the mdx mouse model for DMD, and we propose this is via increased synthesis of the amino acid taurine.
We compared the capacity of OTC and taurine treatment to increase taurine content of mdx muscle, as well as effects on in vivo and ex vivo muscle function, inflammation and oxidative stress.
Both treatments increased taurine in muscles, and improved many aspects of muscle function and reduced inflammation. Taurine treatment also reduced protein thiol oxidation and was overall more effective, as OTC treatment reduced body and muscle weight, suggesting some adverse effects of this drug.
These data suggest that increasing dietary taurine is a better candidate for a therapeutic intervention for DMD.
Abbreviations
- 2ME
2‐mercaptoethanol
- APF
2‐[6‐(4‐aminophenoxy)‐3‐oxo‐3H‐xanthen‐9‐yl]benzoic acid
- CD
cysteine deoxygenase
- CSA
cross‐sectional area
- CSD
cysteine sulfinate decarboxylase
- dF dt–1
maximum rate of force development
- DMD
Duchenne muscular dystrophy
- EDL
extensor digitorum longus
- FLM
BODIPY FL‐N‐(2‐aminoethyl) maleimide
- GAP
glyceraldehyde 3‐phosphate dehydrogenase
- GSH
glutathione
- Lf
optimal fibre length
- Lo
optimal length
- MPO
myeloperoxidase
- NAC
N‐acetylcysteine
- OPA
o‐phthalaldehyde
- OTC
l‐2‐oxothiazolidine‐4‐carboxylate
- Pt
peak isometric twitch force
- ROS
reactive oxygen species
- TA
tibialis anterior
- TauT
taurine transporter protein
- TCA
trichloroacetic acid
- TCEP
tris(2‐carboxyethyl)phosphine
- Texas red
Texas Red C2‐maleimide
- TNF
tumour necrosis factor
- TTP
time to peak twitch force
Introduction
Duchenne muscular dystrophy (DMD) is a lethal, X‐chromosome linked muscle disease affecting about 1 in 3500–6000 boys worldwide (reviewed by Emery, 2002; Bushby et al. 2010). DMD is characterised by severe muscle weakness caused by mutations in the dystrophin gene, which result in the loss of functional dystrophin protein in muscle: this increases susceptibility to sarcolemma damage after muscle contraction, leading to myofibre necrosis, inflammation, regeneration and fibrosis (Petrof et al. 1993; Lapidos et al. 2004; Grounds, 2008; Kharraz et al. 2014). Repeated cycles of widespread myofibre necrosis and progressive failure of regeneration (with replacement of myofibres by fatty and fibrous connective tissue) lead to the loss of muscle mass and function in DMD boys with premature death often due to respiratory or cardiac failure (Reviewed by Biggar, 2006; Bushby et al. 2010).
While the mechanism for loss of muscle function in DMD and the mdx mouse model of the disease is not fully understood, disruptions in intracellular calcium homeostasis caused by sarcolemma instability, inflammation and oxidative stress are implicated (reviewed by Whitehead et al. 2006). Excessive oxidative stress, which occurs in conditions such as chronic inflammation, during strenuous exercise and disease states, has been shown to cause muscle weakness (Smith & Reid, 2006), and is implicated in the pathology of numerous muscular diseases such as muscular dystrophies (Terrill et al. 2013 b). Oxidative stress is caused by increased generation of reactive oxygen species (ROS), or the failure to eliminate ROS by antioxidants (Halliwell & Gutteridge, 2007). While ROS can cause cellular damage by directly and irreversibly damaging macromolecules such as proteins, membrane lipids and DNA, another major cellular consequence of ROS exposure is the oxidation of protein thiol side chains (–SH, in the amino acid cysteine). Protein thiols can undergo numerous reactions, which are dependent on the species and concentration of oxidants they contact (Eaton, 2006). ROS, such as hydrogen peroxide, can cause reversible oxidation (disulphide formation), and the reduction/oxidation (redox) state of thiols is an important regulator of protein function. In skeletal muscle, contractile function, force production and the development of fatigue are directly influenced by the redox state of thiol side chains of contractile proteins (Andrade et al., 1998, 2001; Dalle‐Donne et al. 2003; Tiago et al. 2006; Hertelendi et al. 2008; Prochniewicz et al. 2008; Pinto et al. 2011; Mollica et al. 2012). More reactive oxidants, such as hypochlorous acid, can also cause irreversible oxidation of protein thiols, such as the formation of sulfinic and sulfonic acids (Winterbourn & Hampton, 2008) consequentially leading to damage and dysfunction of proteins (Thomas & Mallis, 2001).
We have established that reversible protein thiol oxidation is increased in muscles of the mdx mouse, and have proposed that protein thiol oxidation contributes to dystropathology (El‐Shafey et al. 2011; Radley‐Crabb et al. 2012; Terrill et al. 2012, 2013 a,b; Iwasaki et al. 2013). Drugs that target protein thiol oxidation such as N‐acetylcysteine (NAC) and l‐2‐oxothiazolidine‐4‐carboxylate (OTC) have been investigated in pre‐clinical studies in the mdx mouse (Whitehead et al. 2008; Terrill et al. 2012, 2013 a; de Senzi Moraes Pinto et al. 2013; Rapucci Moraes et al. 2015). These drugs are derivatives of the amino acid cysteine, and can also increase cysteine content (Sen & Packer, 2000; Zafarullah et al. 2003; Ferreira et al. 2009). Cysteine can itself function directly as a thiol antioxidant, and can also function indirectly to reduce oxidative stress as cysteine is essential for the synthesis of glutathione (GSH), the major intracellular thiol antioxidant (Medved et al. 2004; Dilger & Baker, 2007). Treatment of mdx mice with NAC or OTC decreases muscle pathology, as shown by decreased myofibre necrosis, inflammatory cells and tumour necrosis factor (TNF) levels, and improved grip strength (Whitehead et al. 2008; Terrill et al. 2012, 2013 a; de Senzi Moraes Pinto et al. 2013). Ex vivo exposure of mdx extensor digitorum longus (EDL) muscles to NAC decreased force loss after a series of eccentric (stretched) contractions (Whitehead et al. 2008), suggesting that protein thiol oxidation directly affects contractile function of mdx muscle. While NAC had similar effects to OTC, NAC is readily oxidised and is unstable in drinking water, so OTC appears the better drug for oral treatment of mice.
Interestingly, we unexpectedly showed that the benefits of NAC and OTC administration in mdx mice could not be ascribed to increased content of cysteine nor GSH, as neither treatment was associated with an increase in either cysteine or GSH, in tissues that included muscle, plasma and liver (Terrill et al. 2012, 2013 a). While adequate levels of cysteine are essential for the synthesis of protein and GSH, cysteine homeostasis is tightly regulated in the liver because excess cysteine is toxic in mammals (Stipanuk et al. 2006) and, in the presence of abundant cysteine, the liver readily synthesises the semi‐essential amino acid taurine (2‐aminoethanesulfonic acid) (Stipanuk, 2004). Recently, we showed that OTC treatment of mdx mice leads to a dramatic increase in the taurine content of liver, plasma and muscle (Terrill et al. 2013 a). In a study of systemic taurine homeostasis and metabolism in young mice aged 18 days to 6 weeks (Terrill et al. 2015) we show, in accordance with others, that taurine levels are decreased in young mdx muscles, compared with C57 control mice, before and during the onset of dystropathology (around 3 weeks), implying that taurine deficiency is contributing to disease pathology (McIntosh et al. 1998; Griffin et al. 2001; Terrill et al. 2013 a).
Taurine is found in many tissues and is considered important for the function of skeletal muscle, where it modulates ion channel function, membrane stability and calcium homeostasis (Huxtable, 1992; Bakker & Berg, 2004; Camerino et al. 2004; Warskulat et al. 2004, 2007; Hamilton et al. 2006). Both oral treatment of mice and the ex vivo exposure of mdx EDL muscle to taurine ameliorated the negative mechanical threshold for contraction observed in untreated mdx muscle, indicating a role for taurine in calcium sensitive excitation–contraction coupling in dystrophic muscle (De Luca et al. 2001, 2003; Cozzoli et al. 2011). Additionally, oral treatment increases the forelimb grip strength in mdx mice, indicating improved in vivo strength of taurine‐treated mice (De Luca et al. 2003; Cozzoli et al. 2011).
Both taurine and OTC improve dystrophic muscle function in mdx mice and, because both are approved for use in humans, they are prospective candidates for treatment of DMD. Although we propose that the protective effects of OTC are a consequence of conversion to taurine, it is not certain that taurine is the preferred candidate for further testing, as the synthesis of taurine from OTC may be a superior route for increasing muscle content of taurine in mdx mice. In this study, we comprehensively compare the efficacy of taurine and OTC (in drinking water) to reduce dystrophic pathology using in vivo measures of grip strength as well as ex vivo measurement of contractile function in isolated EDL muscle. Additionally, we examine whether treatment with taurine and OTC affects protein thiol oxidation because of its proposed involvement in dystropathology. As taurine is synthesised endogenously, we examined whether treatment with OTC and taurine could disrupt endogenous synthesis of taurine. Our results show that increasing the taurine content of a diet (via drinking water) is more effective at improving dystrophic muscle function than treatment with OTC.
Methods
All reagents used were obtained from Sigma Aldrich (St Louis, MO, USA) unless otherwise specified.
Ethical approval
All animal experiments were conducted in strict accordance with the guidelines of the National Health and Medical Research Council Code of practice for the care and use of animals for scientific purposes (2004), and the Animal Welfare act of Western Australia (2002), and were approved by the Animal Ethics committee at the University of Western Australia.
Taurine and OTC treatment
All experiments were carried out on dystrophic mdx (C57Bl/10ScSnmdx/mdx) and non‐dystrophic control C57 (C57Bl/10ScSn) mice (the parental strain for mdx). Mice were obtained from the Animal Resource Centre, Murdoch, Western Australia. Mice were maintained at the University of Western Australia on a 12 h light/dark cycle, under standard conditions, with free access to food and drinking water.
Mice were weaned at 18 days, and were given either no treatment, taurine or OTC in the drinking water for 24 days. All mice were sampled at 42 days of age (6 weeks). Both male and female pups were used in the study (with equal numbers of both sex in each group), with no significant sex difference observed for any measurement (data not shown). OTC‐treated mice received a 0.5% (w/v) OTC solution, as previously described (Terrill et al. 2013 a). Taurine‐treated mice received a 2% taurine solution, a dose that was calculated to fall between two previously used doses (De Luca et al. 2003; Cozzoli et al. 2011). Water ingestion and body weights were monitored twice weekly for all mdx treatment groups. No significant differences were observed in the amount of water ingested for any group (data not shown). The approximate consumption of each treatment, based on water consumption, was about 0.8 g kg–1 day–1 for OTC‐treated mice and 4 g kg–1 day–1 for taurine‐treated mice.
Grip strength
The grip strength of all mice was measured using a Chatillon Digital Force Gauge (DFE‐002) and a triangle metal bar, as per the TREAT‐NMD recommended standard protocol ‘Use of grip strength meter to assess limb strength of mdx mice – M.2.2_001’ (http://www.treat‐nmd.eu/research/preclinical/SOPs/). In brief, the mouse was placed on the front of the triangle bar (attached to a force transducer) and pulled gently until released. Each mouse underwent five consecutive grip‐strength trials; the grip strength value for each mouse was recorded as the average of the three trials with the highest force. Average grip strength was presented as total force (g) and also normalised for body weight [force (g)/BW (g)]. Grip strength measurements were performed on three separate occasions during the final week of treatment to accustom mice, but only data from the final testing session were used for analysis.
Tissue collection
After 24 days of treatment the 6‐week‐old mice were anaesthetised via an intraperitoneal (i.p.) injection of sodium pentobarbitone (40 mg kg body weight–1). Anaesthetised mice were placed on a heated plate at 37°C to maintain core body temperature. Following dissection of the EDL muscle, other tissues were removed for further biochemical analysis including the contralateral EDL, tibialis anterior and liver.
Ex vivo muscle function testing was performed as per Ramsey et al. (2010). First, EDL tendons were tied with non‐absorbable black braided surgical silk (Dysilk, Dynek Pty Ltd, Hendon, South Australia) and mounted in an in vitro muscle test system (805A In Vitro Force Transducer System, Aurora Scientific Inc., Aurora, ON, Canada). One EDL tendon was fixed to a stabilised hook and the other attached to a dual force transducer and lever arm. The muscle was then stimulated with an electrical stimulator (701B High Power Bi‐phase Current Stimulator, Aurora Scientific) via parallel electrodes situated on both sides of the suspended muscle.
The organ bath was filled with mammalian Ringer solution: NaCl (121 mm), KCl (5.4 mm), MgSO4.7H2O (1.2 mm), NaHCO3 (25 mm), HEPES (5 mm), glucose (11.5 mm) and CaCl2 (2.5 mm), bubbled with Carbogen and maintained at 25°C. The recording of force and control of the level arm was achieved using Dynamic Muscle Control software and data analysed using Dynamic Control Data Analysis software (Aurora Scientific).
Ex vivo measurements of contractile function
At the start of each experiment the muscle was set to the optimal length (L o) by manually adjusting the lever arm and recording the twitch force response. The length at which the maximum twitch force was recorded was taken as L o. All subsequent measurements of contractile function were performed at L o. The optimal fibre length (L f) in the EDL was calculated from a pre‐defined fibre length to muscle length ratio of 0.44 (Hakim et al. 2013). Evaluation of twitch characteristics included measures of peak isometric twitch force (P t), time to peak twitch force (TTP), half relaxation time (½RT) and the maximum rate of force development (dF dt –1). The force frequency was evaluated by recording the force responses at stimulation frequencies of 5, 10, 20, 30, 50, 80, 100, 120 and 150 Hz. Each stimulus was separated by a 2 min interval to avoid inducing muscle fatigue. The stimulation that produced the greatest force throughout the experiment was recorded as the maximum tetanic force. The susceptibility to stretch‐induced muscle damage was evaluated by measuring the decrease in force production following a series of eccentric contractions of increasing amplitude. The muscle was stimulated to contract isometrically and a stretch was applied after the isometric tension had plateaued (after approximately 300 ms) to induce an eccentric contraction. Successive stretches were applied at amplitudes of 5, 10, 20, 30, 40 and 50% of L f. A final isometric contraction was recorded after the 50% stretch. Each contraction was separated by a 2 min interval and muscle damage was evaluated by the decrease in isometric force after each stretch.
Calculating cross‐sectional area and specific force
Force recordings were normalised relative to muscle cross‐sectional area (CSA). CSA was estimated by dividing the wet muscle mass (g) by L f and the density of mammalian skeletal muscle (1.056 mg mm–3). Specific force (N cm–2) was calculated by dividing the maximum isometric force (N) by the CSA.
HPLC analysis of taurine and cysteine
Taurine and cysteine in liver and tibialis anterior muscle were measured using reversed‐phase HPLC as previously described (Terrill et al. 2013 a). In brief, plasma samples were precipitated by addition of 10 times by weight of 5% trichloroacetic acid (TCA). Frozen tissues were crushed using a mortar and pestle under liquid nitrogen and homogenised in 25 times 5% TCA. After centrifugation, supernatants were removed and stored at −80°C before analysis. Analytes were separated using HPLC with fluorescence detection, with pre‐column derivitisation with o‐phthalaldehyde (OPA) and 2‐mercaptoethanol (2ME). OPA reacts rapidly with amino acids and sulfhydryl groups to yield intensely fluorescent derivatives, and 2ME, a reducing agent, prevents the OPA reagent from oxidising. Supernatants were mixed with iodoacetamide, dissolved in 5% TCA, to a final concentration of 25 mm. An internal standard, o‐phospho‐dl‐serine, dissolved in 5% TCA, was added to a final concentration of 5 mm. Sodium borate was used to adjust the pH to 9. Samples were placed in an autosampler, which was maintained at 4°C. Samples were mixed on a sample loop with a derivitising solution containing 40 mm OPA and 160 mm 2ME in 100 mm sodium borate, pH 12, for 30 s before injection onto the column. Separation was achieved with a C18 column (5 μl, 4.6 × 150 mm, Phenomenex) using a Dionex Ultimate 3000 HPLC system. Mobile phase A consisted of 50 mm potassium phosphate buffer, methanol and tetrahydrofuran (94:3:3). Mobile phase B consisted of 90% methanol, with a gradient increase in B from 0 to 25%. Fluorescence was set at 360 and 455 nm for excitation and emission, respectively. The protein content of liver and muscle samples was quantified by solubilising the pellet in 0.5 m sodium hydroxide, before incubation at 80°C for 15 min. Once fully dissolved, protein concentrations of supernatants were quantified using a Bradford protein assay (Bio‐Rad, Hercules, CA, USA).
Protein extraction and immunoblotting
Frozen livers were crushed using a mortar and pestle under liquid nitrogen and homogenised in 1 ml ice‐cold 1% NP40, 1 mm EDTA in PBS, supplemented with complete EDTA‐free protease inhibitor tablets and PhosSTOP phosphatase inhibitor tablets (Roche Australia, Dee Why, NSW, Australia), and centrifuged at 13,000 g for 10 min. The protein concentration of supernatants was quantified using the Detergent Compatible (DC) protein assay (Bio‐Rad). Samples were resolved on 4–15% SDS‐PAGE TGX gels (Bio‐Rad) and transferred onto nitrocellulose membrane using a Trans Turbo Blot system (Bio‐Rad). Immunoblotting was performed with antibodies to cysteine dioxygenase type 1 (ab53436, Abcam, Cambridge, MA, USA), cysteine sulfinate decarboxylase (ab101847, Abcam) and glyceraldehyde 3‐phosphate dehydrogenase (GAP, 14C10, Cell Signaling, Danvers, MA, USA), all dissolved 1:1000 in 5% BSA. HRP‐conjugated secondary antibodies were from Thermo Fisher Scientific (Waltham, MA, USA). Chemiluminescence signal was captured using the ChemiDoc MP Imaging System (Bio‐Rad). Resultant images were quantified using ImageJ software (Schneider et al. 2012). A common sample was loaded onto each gel to normalise for detection efficiencies across membranes. Glyceraldehyde 3‐phosphate dehydrogenase loading controls were immunoblotted on the same membrane as immunoblotted protein. All representative immunoblots in figures represent samples immunoblotted on the same membrane.
Inflammatory cell presence (MPO)
Myeloperoxidase (MPO) is an enzyme secreted by inflammatory cells such as neutrophils, and MPO activity is a useful biomarker of inflammatory cells in tissues (Winterbourn et al. 2000; Setsukinai et al. 2003). The enzyme MPO catalyses the production of hypochlorous acid from hydrogen peroxide and chloride (Winterbourn & Kettle, 2000) and hypochlorus acid reacts with 2‐[6‐(4‐aminophenoxy)‐3‐oxo‐3H‐xanthen‐9‐yl]benzoic acid (APF) to form the highly fluorescent compound fluorescein, which is measured in this method as per Terrill et al. (2013 a). Briefly, frozen tibialis anterior muscle was ground using a mortar and pestle under liquid nitrogen and homogenised in 0.5% hexadecyltrimethylammonium bromide in PBS. Samples were centrifuged and supernatants diluted in PBS. Human MPO was used as the standard for the assay (Cayman Chemical, Ann Harbor, MI, USA). Aliquots of each experimental sample or MPO standard were pipetted into a 384 well plate, before the addition of APF working solution (20 μm APF and 20 μm hydrogen peroxide in PBS) was added. The plate was incubated at room temperature (protected from light) for 30 min, with the fluorescence being measured every minute using excitation at 485 nm and emission at 515–530 nm. The rate of change of fluorescence for each sample was compared to that of the standards and results were expressed per milligram of protein, quantified using the DC protein assay (Bio‐Rad).
Quantification of protein thiol oxidation
Reduced and oxidised protein thiols were measured in contralateral EDL muscles (the muscle not used for ex vivo muscle function testing) using the two‐tag technique as described previously (Armstrong et al. 2010; El‐Shafey et al. 2011; Radley‐Crabb et al. 2012; Terrill et al. 2012, 2013 a,b). In brief, frozen tissue was crushed under liquid nitrogen, before protein was extracted with 20% TCA/acetone. Protein was solubilised in 0.5% SDS with 0.5 m Tris at pH 7.3 (SDS buffer) and protein thiols were labelled with the fluorescent dye BODIPY FL‐N‐(2‐aminoethyl) maleimide (FLM, Invitrogen, Carlsbad, CA, USA). Following removal of the unbound dye using ethanol, protein was re‐solubilised in SDS buffer, pH 7, and oxidised thiols were reduced with tris(2‐carboxyethyl)phosphine (TCEP) before the subsequent unlabelled reduced thiols were labelled with a second fluorescent dye Texas Red C2‐maleimide (Texas red, Invitrogen). The sample was washed in ethanol and resuspended in SDS buffer. Samples were read using a fluorescence plate reader (Fluostar Optima) with wavelengths set at excitation 485 nm, emission 520 nm for FLM and excitation 595 nm, emission 610 nm for Texas red. A standard curve for each dye was generated using ovalbumin and results were expressed per milligram of protein, quantified using the DC protein assay (Bio‐Rad).
Reduced and oxidised thiols of actin and albumin proteins were quantified using one‐dimensional SDS‐PAGE, as described previously (Terrill et al. 2013 a). Briefly, labelled samples (remaining from the plate assay above) were diluted to equivalent protein concentrations. FLM‐ and Texas red‐labelled ovalbumin standards were combined and both the standards and the samples were diluted by the addition of sample buffer [125 mm Tris, pH 6.8, 4% SDS, 30% (v/v) glycerol, 0.02% bromophenol blue]. Standards and samples were applied to a 12% polyacrylamide gel. Gel electrophoresis was performed using the Bio‐Rad Mini Protean III system. Each fluorescent gel was scanned using the ChemiDoc MP Imaging System (Bio‐Rad) for fluorescence, with wavelengths set at excitation 485 nm, emission 520 nm for FLM and excitation 595 nm, emission 610 nm for Texas red. The bands were quantified by densitometry using ImageJ version 1.41 software using the integrated density function, after first removing the background. To assess the reversible protein thiol oxidation state of specific protein bands, dominant bands were compared against FLM and Texas red using in‐gel standard curves using polynomial regression. The location of actin and albumin on the gels was determined by using Bio‐Rad Precision Plus Protein Kaleidoscope Standards to match previously performed mass spectrometry (MADLI) analysis (Terrill et al. 2013 a).
Statistics
Data were analysed using GraphPad Prism software. One‐way ANOVA tests with post‐hoc (LMS) comparisons were used to identify significant differences between experimental groups. Two‐way repeated measures ANOVAs were used to analyse data for the force‐frequency and stretch‐induced muscle damage protocols. Statistical significance was accepted at P < 0.05. All data are presented as mean ± SEM.
Results
Muscle taurine and cysteine content
To confirm treatment efficacy, taurine (and cysteine) content of tibialis anterior muscles was measured in mice (male and female) aged 6 weeks. At this age there was no significant difference between levels of taurine or cysteine in C57 and mdx muscles (Fig. 1). Taurine content was significantly elevated by about 1.2‐fold in mdx muscle following treatment with both taurine and OTC (Fig. 1 A). Cysteine content was also measured in muscle because OTC is converted to cysteine, but there was no evidence of increased cysteine content with OTC treatment (Fig. 1 B). Taurine treatment also had no effect on cysteine content (Fig. 1 B).
Figure 1. Muscle content of taurine (A) and cysteine (B) in untreated C57, untreated mdx, taurine‐treated mdx and OTC‐treated mdx mice (aged 6 weeks) .
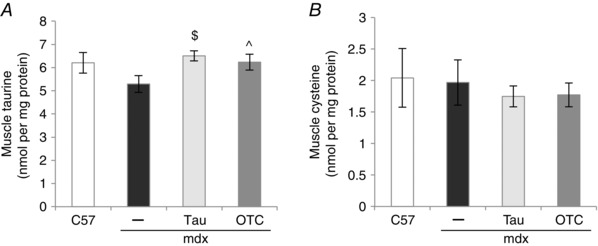
Measurements are for tibialis anterior muscles. Symbols for significant differences (P < 0.05) are: $between untreated mdx and taurine‐treated mdx, ^between untreated mdx and OTC‐treated mdx. Data are presented as mean ± SEM and n = C57 (10), mdx (6), taurine (8) and OTC (8) treated mice.
Liver taurine and cysteine content
Dietary taurine is absorbed and transported to the liver, and the liver is also the primary site of taurine synthesis from cysteine (Stipanuk, 2004; Terrill et al. 2015). We therefore examined the effect of taurine and OTC administration on taurine content of liver. There was no significant difference in liver content of taurine or cysteine between untreated mdx and C57 mice (Fig. 2). However, a striking increase in taurine content of mdx liver, about 4‐fold, was seen after 24 days of taurine administration (Fig. 2 A) and OTC treatment produced about a 2‐fold increase in taurine content of mdx liver (Fig. 2 A). Neither treatment affected the cysteine content of mdx liver (Fig. 2 B).
Figure 2. Liver content of taurine (A) and cysteine (B) in untreated C57, untreated mdx, taurine‐treated mdx and OTC‐treated mdx mice .
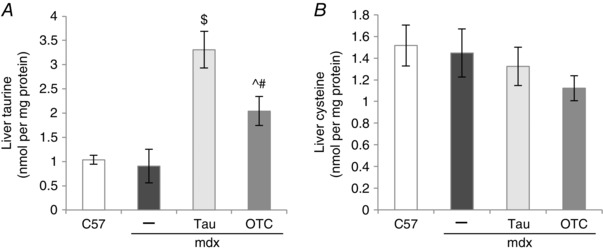
Symbols for significant differences (P < 0.05) are: $between untreated mdx and taurine‐treated mdx, ^between untreated mdx and OTC‐treated mdx, #between OTC‐ and taurine‐treated mdx. Data are presented as mean ± SEM and n = C57 (10), mdx (6), taurine (8) and OTC (8) treated mice.
Liver taurine synthesis
Treatment with taurine and OTC potentially affects endogenous taurine synthesis (Terrill et al. 2015), so the taurine synthetic enzymes cysteine deoxygenase (CD) and cysteine sulfinate decarboxylase (CSD) were quantified using Western blotting (Fig. 3). Liver CD content was similar for C57 and mdx mice (Fig. 3 A). Both taurine and OTC treatment upregulated CD content by over 20‐fold in mdx livers (Fig. 3 A). Liver CSD levels were about 1.35‐fold lower in mdx compared with C57 mice (Fig. 3 B) and taurine and OTC treatments had a striking effect with about a respective 50‐ and 5‐fold decrease in content of CSD in mdx liver (Fig. 3 B). These data indicate that regulation of levels of taurine synthesis enzymes in the mdx liver are sensitive to both taurine and cysteine availability.
Figure 3. Liver content of cysteine deoxygenase (CD, A) and cysteine sulfinate decarboxylase (CSD, B) in untreated C57, untreated mdx, taurine‐treated mdx and OTC‐treated mdx mice .
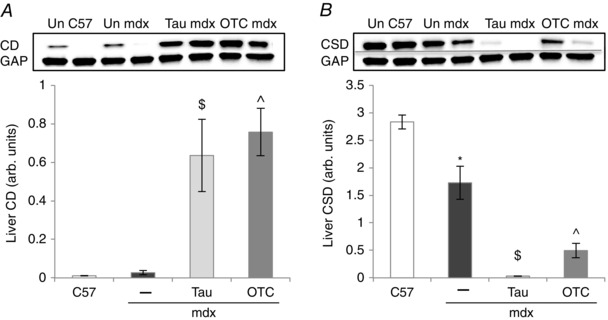
Symbols for significant differences (P < 0.05) are: *between untreated mdx and C57, $between untreated mdx and taurine‐treated mdx, ^between untreated mdx and OTC‐treated mdx. Data are presented as mean ± SEM and n = C57 (10), mdx (6), taurine (8) and OTC (8) treated mice. Representative blots (×2) are shown of CD and CSD. The loading control was glyceraldehyde 3‐phosphate dehydrogenase (GAP).
Phenotypic data
Body, muscle and liver weights were measured to identify potential detrimental effects of treatment with taurine or OTC. While body weights were similar for C57 and mdx mice (Table 1), the OTC‐treated mdx mice were 1.15‐fold lighter than untreated mdx mice. Liver weights were similar across all groups.
Table 1.
Phenotypic data of untreated C57, untreated mdx, taurine‐treated mdx and OTC‐treated mdx, including body and liver weight, and EDL weight, cross sectional area (CSA) and optimal fibre length (Lf)
| Untreated C57 | Untreated mdx | Taurine mdx | OTC mdx | |
|---|---|---|---|---|
| Body weight (g) | 22.22 ± 0.73 | 20.81 ± 0.79 | 21.47 ± 0.74 | 18.31 ± 0.71^# |
| Liver weight (g) | 0.98 ± 0.04 | 1.04 ± 0.08 | 0.98 ± 0.08 | 0.91 ± 0.07 |
| EDL weight (g) | 10.48 ± 0.32 | 10.62 ± 0.97 | 10.44 ± 0.37 | 8.56 ± 0.45^# |
| EDL L f (mm) | 5.60 ± 0.30 | 4.89 ± 0.20* | 5.64 ± 0.17$ | 5.15 ± 0.12 |
| EDL CSA (mm2) | 1.80 ± 0.07 | 2.04 ± 0.13* | 1.75 ± 0.05$ | 1.57 ± 0.07^ |
Symbols for significant differences (P < 0.05) are: *between untreated mdx and C57, $between untreated mdx and taurine‐treated mdx, ^between untreated mdx and OTC‐treated mdx, #between OTC‐ and taurine‐treated mdx. Data are presented as mean ± SEM and n = C57 (10), mdx (6), taurine (8) and OTC (8) treated mice.
The EDL muscle weights were similar for C57 and most mdx mice (Table 1), but OTC treatment caused about a 1.25‐fold decrease in mdx EDL weight. Optimal fibre length (the length at which the maximum twitch force was recorded) was about 1.15‐fold lower in untreated mdx EDL muscle compared with C57 mice (Table 1) and taurine treatment increased optimal fibre length by 1.15‐fold in mdx EDLs to become restored to C57 values. The EDL CSA was about 1.1‐fold higher in mdx muscle compared with C57 muscle (Table 1) and this difference was normalised by taurine treatment. OTC treatment further decreased the CSA of mdx EDL, by 1.25‐fold, to less than normal (C57) values (Table 1).
Together, these data indicate that treatment with OTC and taurine affected mdx phenotype, and that there were differences in the extent of the effects between OTC and taurine.
Grip strength
The grip strength test is a non‐invasive in vivo measure of mouse limb strength. Mdx mice displayed a 1.35‐fold weaker normalised fore limb grip strength compared with C57 mice (Fig. 4 A). This weakness was ameliorated by both taurine and OTC treatment: normalised force was increased 1.35‐ and 1.3‐fold, respectively, in treated mdx mice, to become restored to C57 strength (Fig. 4 A). Because OTC treatment reduced mdx body weight (Table 1), absolute grip strength is also reported (see Discussion for rationale). By this measure, OTC treatment had no effect on total grip strength (Fig 4 B). In contrast, taurine treatment significantly increased, by about 1.5‐fold, total grip strength, although this was still significantly lower than C57 total grip strength.
Figure 4. Forelimb grip strength of untreated C57, untreated mdx, taurine‐treated mdx and OTC‐treated mdx mice (aged 6 weeks) .
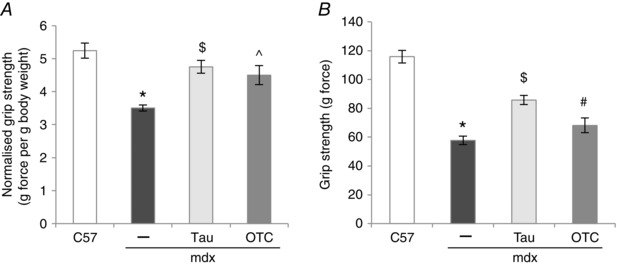
Data are presented as both force normalised to body weight (A) and total force (B). Symbols for significant differences (P < 0.05) are: *between untreated mdx and C57, $between untreated mdx and taurine‐treated mdx, ^between untreated mdx and OTC‐treated mdx, #between OTC‐ and taurine‐treated mdx. Data are presented as mean ± SEM and n = C57 (11), mdx (10), taurine (10) and OTC (11) treated mice.
Ex vivo EDL muscle function
To establish whether taurine and OTC treatment directly affects muscle contractile function, physiological contraction parameters were measured in isolated EDL muscles (Table 2). Specific force (SF) was measured at a low frequency of 20 Hz (SF 20), where mdx EDL muscles produced 1.4‐fold less force than C57 muscle. This effect was ameliorated with taurine treatment, which increased force 1.6‐fold in mdx muscle, to restore force to C57 values. There was no significant effect of OTC treatment on SF of mdx muscle. Twitch characteristics of TTP, half relaxation time (½RT) and the rate of maximal force production (dF dt –1) did not differ significantly between C57 and mdx muscles (Table 2), although peak twitch force (P t) was 1.2‐fold lower in mdx EDL muscles. Treatment with taurine and OTC did not affect the rate of maximal force production nor peak twitch force (Table 2), although taurine increased the time to peak force by 1.2‐fold in mdx muscle to restore TTP to C57 values. Both taurine and OTC treatment increased half relaxation time 1.2‐fold in mdx muscle; half relaxation time of taurine‐treated mdx muscle was also significantly higher than C57 muscle (Table 2).
Table 2.
Contractile parameters of untreated C57, untreated mdx, taurine‐treated mdx and OTC‐treated mdx EDL muscle
| Untreated C57 | Untreated mdx | Taurine mdx | OTC mdx | |
|---|---|---|---|---|
| SF 20 (N cm–2) | 4.18 ± 0.34 | 2.91 ± 0.19* | 4.59 ± 0.52$ | 3.7 ± 0.19 |
| P t (N cm–2) | 2.63 ± 0.09 | 2.18 ± 0.09* | 2.57 ± 0.16 | 2.50 ± 0.15 |
| TTP (ms) | 24.3± 0.7 | 21.7 ± 1.0 | 25.8 ± 1.4$ | 22.1 ± 0.7# |
| ½RT (ms) | 27.3 ± 1.4 | 26.5 ± 1.3 | 31.9 ± 2.0$ | 31.4 ± 1.1^ |
| dF dt –1 (g s–1) | 511± 29 | 514 ± 42 | 490 ± 22 | 456 ± 23 |
Measurements include specific force at 20 Hz (SF 20), peak twitch force (P t), time to peak force (TTP), half relaxation time (½RT) and the rate of maximal force production (dF dt –1). Symbols for significant differences (P < 0.05) are: *between untreated mdx and C57, $between untreated mdx and taurine‐treated mdx, ^between untreated mdx and OTC‐treated mdx, #between OTC‐ and taurine‐treated mdx. Data are presented as mean ± SEM and n = C57 (10), mdx (6), taurine (8) and OTC (8) treated mice.
The isometric force–frequency relationship was recorded for stimulation frequencies of 5–150 Hz (Fig. 5). At frequencies of 50 Hz and higher, mdx EDL muscles produced significantly less (up to 1.4‐fold) specific force than C57 muscle (Fig. 5 A). Taurine significantly increased specific force (up to 1.2‐fold) at frequencies of 30 Hz and higher (Fig. 5 A), and OTC treatment increased (up to 1.15‐fold) specific force at frequencies of 100 Hz and higher in mdx muscle (Fig. 5 A). Taurine‐treated mdx muscles still produced significantly less specific force than C57 muscles at frequencies of 100 Hz, as did OTC‐treated mdx muscles at 50 Hz and higher. When force was not normalised to CSA, mdx EDL muscles still produced significantly less (up to 1.25‐fold) force than C57 muscles (Fig. 5 B). Unlike normalised force, taurine and OTC treatment did not affect total force (Fig. 5 B), but OTC produced less (up to 1.15‐fold) force than taurine‐treated mdx muscle at frequencies of 50 Hz and higher (Fig. 5 B).
Figure 5. Force–frequency curves of untreated C57, untreated mdx, taurine‐treated mdx and OTC‐treated mdx EDL muscle .
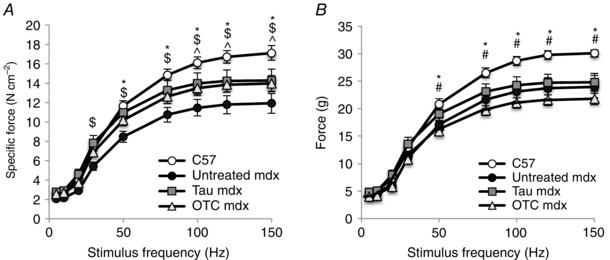
Isometric force produced at frequencies of 5–150 Hz was plotted for both specific force (A) and total force (B). Symbols for significant differences (P < 0.05) are: *between untreated mdx and C57, $between untreated mdx and taurine‐treated mdx, ^between untreated mdx and OTC‐treated mdx. Data are presented as mean ± SEM and n = C57 (10), mdx (6), taurine (8) and OTC (8) treated mice. Data were analysed by two‐way repeated measures ANOVA.
The increased susceptibility of dystrophic muscle to stretch‐induced damage is dependent on the number of repetitions (Sharp et al. 2011) and also the stretch amplitude (Consolino & Brooks, 2004). To examine whether taurine or OTC improved resistance to stretch‐induced damage across a range of stretch amplitudes, EDL muscles were exposed to a series of increasing eccentric contractions. The force deficit after small amplitude stretches was not significantly different between all experimental groups. However, stretches greater than 30% L f caused a larger (up to 2.7‐fold) decrease in force in EDL of mdx mice compared with C57 mice (Fig. 6). OTC treatment had no effect on stretch‐induced damage in mdx muscle. Taurine treatment led to a significantly larger (an additional 1.2‐fold) decrease in force production of mdx muscle at stretches of 40 and 50%, indicating a greater susceptibility to muscle damage from large amplitude stretches after taurine treatment.
Figure 6. Stretch‐induced damage in untreated C57, untreated mdx, taurine‐treated mdx and OTC‐treated mdx EDL muscle .
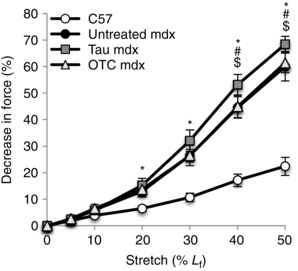
The decrease in maximum force production is plotted following a series of eccentric contractions of increasing amplitude (stretch applied as a % of the optimal fibre length, L f). Symbols for significant differences (P < 0.05) are: *between untreated mdx and C57, $between untreated mdx and taurine‐treated mdx, #between OTC‐ and taurine‐treated mdx. Data are presented as mean ± SEM and n = C57 (10), mdx (6), taurine (8) and OTC (8) treated mice. Data were analysed by two‐way repeated measures ANOVA.
Together these data show that although taurine and OTC improve maximum specific force generation by mdx muscle, neither treatment improved the ability of muscle to resist stretch‐induced damage.
Inflammation and oxidative stress
Both inflammation and oxidative stress have been causally linked to the DMD pathology, so we examined whether taurine and OTC affect these pathogenic pathways. MPO activity and protein thiol oxidation were used as indicators of increased inflammation and oxidative stress, respectively.
MPO activity was about 7‐fold higher in mdx muscle compared with C57 muscle (Fig. 7). Taurine and OTC treatments reduced the MPO activity in mdx muscles by 2.3‐ and 3‐fold, respectively, to be restored to values similar to C57 muscles (Fig. 7).
Figure 7. Myeloperoxidase content of untreated C57, untreated mdx, taurine‐treated mdx and OTC‐treated mdx TA muscle .

Symbols for significant differences (P < 0.05) are: *between untreated mdx and C57, $between untreated mdx and taurine‐treated mdx, ^between untreated mdx and OTC‐treated mdx. Data are presented as mean ± SEM and n = C57 (10), mdx (6), taurine (8) and OTC (8) treated mice.
Protein thiol oxidation was about 1.5‐fold higher in mdx muscle than in C57 muscle (Fig. 8 A). Taurine treatment dramatically decreased protein thiol oxidation by 3‐fold in mdx muscle, and this was also significantly lower than C57 muscle. In contrast, OTC treatment had no effect on thiol oxidation of mdx muscle (Fig. 8 A). Thiol oxidation levels specifically for the proteins actin and albumin were 1.5‐fold higher in mdx compared with C57 muscles (Fig. 8 B and C, respectively). Taurine treatment led to a striking 6‐ and 3‐fold reduction in oxidation of mdx actin and albumin, respectively (Fig. 8 C and D, respectively), which was also significantly lower than C57 muscle, whereas OTC did not affect the thiol oxidation of actin or albumin.
Figure 8. Percentage of protein thiol oxidation (A), thiol oxidation of actin (B) and albumin (C), and total protein thiols (D) in untreated C57, untreated mdx, taurine‐treated mdx and OTC‐treated mdx EDL muscle .
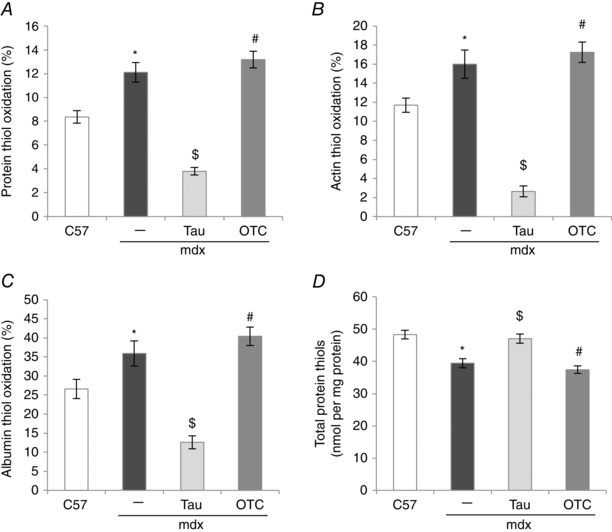
Symbols for significant differences (P < 0.05) are: *between untreated mdx and C57, $between untreated mdx and taurine‐treated mdx, #between OTC‐ and taurine‐treated mdx. Data are presented as mean ± SEM and n = C57 (10), mdx (6), taurine (8) and OTC (8) treated mice.
Total protein thiols were 1.2‐fold lower in mdx muscle compared with C57 muscle and this was ameliorated by taurine treatment (Fig. 8 D). OTC treatment had no effect on total muscle protein thiols.
Discussion
Our major aim was to determine a preferred candidate for a clinical therapeutic intervention for DMD, by increasing taurine content of dystrophic muscles either via direct taurine intake or by increasing taurine synthesis (by elevated cysteine availability) in mdx mice. As has been previously observed, there was no significant difference between taurine content of 6 week mdx and C57 muscle (McIntosh et al. 1998; Griffin et al. 2001; Terrill et al. 2013 a). However our data show that both taurine and OTC treatments led to an increase in taurine content in mdx muscle, and improved skeletal muscle strength and reduced inflammation, but that treatment with taurine was more effective and innocuous compared with OTC. Of particular importance was that taurine (but not OTC) also decreased protein thiol oxidation.
The OTC treatment resulted in reduced body weights and lower muscle weight and smaller CSA of EDL muscles, compared with untreated mdx mice. These effects were not evident for taurine treatment, which suggests that such side effects may be a consequence of OTC metabolism. OTC increases tissue cysteine availability, although excess cysteine is considered toxic, especially in neural tissue, and is implicated in the pathology of numerous conditions, including rheumatoid arthritis, Parkinson's and Alzheimer's [reviewed by Janaky et al. 2000; Stipanuk et al. 2006). Interestingly, we did not detect increased concentrations of cysteine in either liver or muscle. We did, however, observe a dramatic increase in the expression of CD in mdx liver after OTC treatment, an enzyme that catalyses the formation of cysteine sulfinate from cysteine. This suggests that in mdx liver, excess cysteine is being converted to cysteine sulfinate, which is itself toxic (Janaky et al. 2000). The loss of body weight and muscle weight caused by OTC treatment was surprising because OTC is not considered to be toxic or have detrimental side effects, and oral OTC has been used in clinical trials without adverse effects (Leaf & Pace, 1994). There is evidence that route of administration may be a factor. Daily intravenous OTC treatment for 28 days into rats, at double the dose used in our study for mice, did not cause inflammatory liver pathology (Leaf & Pace, 1994), although when the equivalent dose was given as an oral gavage, an inflammatory liver pathology was observed (Leaf & Pace, 1994). This observation indicates that toxicity of OTC could be a consequence of intestinal metabolism. If so, then there is an advantage to treating with taurine because it can be administered orally without causing the side effects evident for oral OTC treatment.
The effect of OTC treatment on dystrophic muscle is complex and appears to be a consequence of independent protective and toxicity effects. The protective effect of OTC is probably a consequence of conversion to taurine, as elevated taurine content was evident in liver and tibialis anterior muscle of OTC‐treated mice. Our previous studies also showed increased taurine (restoration) in mdx quadriceps muscles after OTC treatment of mdx and C57 mice from 6 to 12 weeks of age (Terrill et al. 2013 b). In contrast with taurine treatment, OTC was only partially effective in ameliorating pathogenic processes: while inflammation as measured by MPO was significantly decreased (also shown by Terrill et al. 2013 b), there was no effect on protein thiol oxidation. Previously, we found that OTC treatment (at the same dose used in this study; 0.5%) decreased both MPO and protein thiol oxidation in mdx mice (Terrill et al. 2013 a). The discrepancy in the effects of OTC treatment on protein thiol oxidation may be a consequence of age and stage of pathology, as mice in the present study were treated from 3 to 6 weeks of age during time of peak muscle damage and pathology, whereas in the 2013 study, mice were treated from 6 to 12 weeks of age when the disease is stabilising. The impact of growth is important to consider in the context of manifestation of disease severity across species from mdx mice to DMD boys (Grounds, 2008; Grounds & Shavlakadze, 2011), especially as there are additional metabolic demands in growing dystrophic muscles (Radley‐Crabb et al. 2014). Therefore, the dosing level effective for adult mdx mice may not have been sufficient to counteract the more severe dystropathology evident in younger rapidly growing mice. However, testing higher oral doses of OTC would not appear to be useful given the likelihood of increased toxicity.
Grip strength has been identified as an effective measure for testing the efficacy of therapeutic interventions in mdx mice (Spurney et al. 2009). A particular advantage of grip strength as a preclinical measure is that it is an in vivo strength measure and, as such, can be used as an on‐going assessment for drug efficacy, akin to the non‐invasive 6 min walk test for DMD boys (Spurney et al. 2009; Lynn et al. 2015). Both taurine and OTC treatments were comparably effective in preventing loss of grip strength in mdx mice, when normalised to body weight as has been recommended (Spurney et al. 2009). However, absolute grip strength of OTC‐treated mdx mice was not different from that of untreated mdx mice, and was significantly less for than taurine‐treated mdx mice. This difference in data after normalisation may reflect an obscuring of results due to the decreased body weight of OTC‐treated mice, or it may simply reflect a true increase in force after OTC treatment, despite a decrease in body weight. Therefore, when treatments affect body weight, we suggest reporting absolute grip strengths as we have done, in addition to reporting normalised grip strength.
Consistent with in vivo grip strength measures, treatment with taurine and OTC significantly improved ex vivo measures of contractile function in isolated EDL muscles. Taurine increased specific force production at both maximal and submaximal stimulation frequencies, compared with untreated mdx mice. The increase in submaximal force production may be explained by the small but significant increase in twitch contraction times following taurine treatment, thus allowing greater force summation at the lower stimulation frequencies. Thus, not only does taurine treatment increase maximum force production, it also increases force output at lower, more physiological, levels of muscle activation. Although OTC treatment also enhanced specific force production, this was attributed to a significant reduction in muscle CSA, rather than increased total force production. These findings again emphasise the importance of considering the impact of treatments on both the specific and the total force production.
We show that taurine treatment increased taurine content of muscle (measured for tibialis anterior), resulting in improved grip strength. The molecular mechanism by which taurine protects dystrophic muscle is uncertain as taurine has several actions in tissue, including the control of ion channel function, membrane stability and calcium homeostasis (Huxtable, 1992; Bakker & Berg, 2004; Camerino et al. 2004; Warskulat et al. 2004, 2007; Hamilton et al. 2006; De Luca et al. 2015). Relevant observations are that taurine treatment ameliorated both protein thiol oxidation, a measure of oxidative stress, and MPO, a measure of inflammation, two pathogenic processes that are known to contribute to muscle weakness. Muscles of mdx mice (and DMD boys) are extremely vulnerable to exercise‐induced damage, which is characterised by increased inflammation, oxidative stress and myofibre necrosis (Piers et al. 2011; Radley‐Crabb et al. 2012). Consequently, taurine could be protecting dystrophic muscle by decreasing susceptibility to exercise‐induced damage, with subsequent associated reductions in inflammation and oxidative damage. However, neither taurine nor OTC treatment prevented the increased susceptibility to stretch‐induced damage observed in isolated EDL mdx muscles. Instead, taurine treatment increased susceptibility to immediate stretch‐induced damage ex vivo at stretches of 40% and over. The mechanism and significance of this result is not understood: stretch contractions of over 30% may not be relevant, as these amplitudes do not reflect physiological conditions that occur in vivo. Furthermore, the force deficits observed after large amplitude stretches are likely to reflect the progressive accumulation of damage from previous non‐physiological stretches. Nonetheless, whilst taurine treatment does not prevent the acute loss of contractile function immediately following stretch‐induced damage in mdx muscle (at least ex vivo), it may prevent subsequent pathology by protecting the response to initial injury and thus ameliorating the ‘down‐stream’ inflammatory and oxidative responses, and ultimately lower levels of myofibre necrosis. Additional in vivo studies using physiologically relevant muscle damage protocols (such as treadmill running or repetitive small amplitude stretches) are strongly recommended to evaluate the impact of taurine supplementation on secondary muscle damage and necrosis.
These data indicate that taurine is a potent thiol antioxidant: taurine treatment dramatically reduced reversible thiol oxidation in mdx muscle, as indicated by decreased percentage of thiol oxidation (of whole muscle homogenates, and on specific proteins such as actin and albumin). Interestingly, the percentage of protein thiol oxidation was significantly lower in taurine‐treated mdx muscle than in untreated C57 muscle, indicating that taurine treatment of mdx mice decreases thiol oxidation to below normal physiological levels. The physiological relevance of this as well as the effect of taurine treatment on protein thiol oxidation of healthy muscle remain to be elucidated. Data indicate that taurine may also prevent irreversible thiol oxidation in the presence of ROS, as thiols can be oxidised irreversibly to sulfinic and sulfonic acids (Ghezzi & Bonetto, 2003). The total protein thiols are decreased in mdx muscle, suggesting that protein thiols are being oxidised to other compounds. This decrease in total thiols is ameliorated by taurine treatment, suggesting ROS exposure in mdx muscle is causing both reversible and irreversible protein thiol oxidation, and both are prevented by taurine treatment. The mechanism by which taurine is acting as a thiol antioxidant is unclear, but may be linked to inflammation. In the presence of hydrogen peroxide, MPO oxidises chloride into the highly reactive ROS hypochlorous acid, which reacts with protein thiols. As hypochlorous acid can be scavenged by taurine to form taurine‐chloramine, increased taurine would ameliorate protein thiol oxidation. Taurine‐chloramine itself has been shown to inhibit the pro‐inflammatory mediators TNF and IL‐1β in macrophages and neutrophils (reviewed by Choi et al. 2006). Therefore, the beneficial outcomes of taurine treatment in mdx mice are probably also due to anti‐inflammatory and antioxidant effects.
Taurine is synthesised endogenously, so oral treatment with both taurine and OTC had the potential to affect taurine synthesis. Indeed, both taurine and OTC treatments affected the liver content of CD and CSD, the two enzymes involved in taurine synthesis. CD activity is considered the major controlling factor in the synthesis of taurine from cysteine (Stipanuk, 2004; Terrill et al. 2015). Consistent with this concept, there was a 20‐fold upregulation of CD content in mdx livers and a 2‐fold increase in taurine content following OTC treatment. Interestingly, taurine treatment of mdx mice also increased CD content 20‐fold: this is a novel observation. It is not known whether this large increase in CD with taurine treatment had any toxic effects, such as the increase in cysteine sulfinate, which we propose as a mechanism of toxicity in OTC‐treated mice (described above). While a high protein diet has been shown to affect activity and content of these enzymes, this response is dependent on the sulfur‐containing amino acids cysteine and methionine (Jerkins & Steele, 1991; Bagley & Stipanuk, 1995; Jerkins et al. 1998; Bella et al. 2000). CSD content in liver (in contrast to CD) declined following OTC (5‐fold) and taurine (50‐fold) treatment: the reasons for this are not clear.
A survey of the literature did not provide a ready explanation of how the combined changes in enzyme content might be expected to affect taurine synthesis, and whether the impact of taurine supplementation may vary between mdx and normal animals. An earlier study using rats did not find any changes in activity of either enzyme in the liver following oral taurine supplementation (Loriette et al. 1979). In a study using infant Rhesus monkeys there was no change in activity of either enzyme in liver after taurine supplementation or taurine removal (Sturman et al. 1988). However, a later study in adult rats showed that CSD was subject to end‐product feedback inhibition, as dietary taurine supplementation caused a significant decline in activity, although there was no effect on CD activity (Eppler & Dawson, 2001). Consequently, although our data do show that OTC and taurine treatment are probably affecting taurine synthesis in the liver of mdx mice, this regulation remains to be understood, as does the contribution of de novo taurine synthesis to the increased taurine content in the livers of treated mdx mice. While other studies have administered taurine to mdx mice (De Luca et al. 2003; Cozzoli et al. 2011), the impact on liver metabolism was not assessed. This is a new area that requires further consideration in the context of therapies for growing and adult mdx mice and especially DMD boys.
In conclusion, increasing taurine content of mdx muscle improved both in vivo and ex vivo strength, potentially via anti‐inflammatory and antioxidant effects of taurine, despite an increase in susceptibility to cumulative muscle damage after large amplitude stretches ex vivo. Increasing cysteine availability with OTC treatment increased taurine synthesis in the liver and taurine content of mdx muscles, and reduced inflammation in muscles, with some improvement of in vivo and ex vivo strength. However, the effects of OTC on strength indices were not as convincing as for taurine treatment, as OTC did not reduce protein thiol oxidation, and OTC treatment decreased body and muscle weights, suggesting some toxicity in mdx mice. Our work supports the contention that taurine is a promising candidate for treating DMD (De Luca et al. 2015) and, from a clinical perspective, it is attractive because it is affordable, readily available and appears to have minimal side effects. However, further work is required to validate the level of protection afforded by the drug on parameters such as myofibre necrosis, and to clarify optimal delivery of taurine at different stages of the disease progression. Consideration is required of metabolic and other systemic parameters that can alter the effects (and dosage required) of supplements and drug interventions during the growth of very young and adolescent DMD boys, and these can also be influenced by the amount of surviving muscle tissue during the disease progression. These data support continued research into the use of taurine as a therapeutic intervention for DMD, and suggest that increasing dietary taurine, rather than increasing taurine synthesis, is the better strategy for increasing taurine content of dystrophic muscles to reduce disease severity.
Additional information
Competing interests
All authors have no financial or personal conflict with other people or organisations that could inappropriately influence our work.
Author contributions
J.R.T., G.J.P., M.D.G. and P.G.A. were responsible for study design; J.A.G. performed the grip strength and ex vivo contractile function experiments and analyses; J.R.T. performed all other experiments and analyses. All authors were involved in drafting and critically revising the manuscript. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Acknowledgements
This research was supported by funding from the National Health and Medical Research Council (NHMRC) of Australia and Parent Project Muscular Dystrophy USA.
References
- Andrade FH, Reid MB, Allen DG & Westerblad H (1998). Effect of hydrogen peroxide and dithiothreitol on contractile function of single skeletal muscle fibres from the mouse. J Physiol 509, 565–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade FH, Reid MB & Westerblad H (2001). Contractile response of skeletal muscle to low peroxide concentrations: myofibrillar calcium sensitivity as a likely target for redox‐modulation. FASEB J 15, 309–311. [DOI] [PubMed] [Google Scholar]
- Armstrong AE, Zerbes R, Fournier PA & Arthur PG (2010). A fluorescent dual labeling technique for the quantitative measurement of reduced and oxidized protein thiols in tissue samples. Free Radic Biol Med 50, 510–517. [DOI] [PubMed] [Google Scholar]
- Bagley PJ & Stipanuk MH (1995). Rats fed a low‐protein diet supplemented with sulfur amino‐acids have increased cysteine dioxygenase activity and increased taurine production in hepatocytes. J Nutr 125, 933–940. [DOI] [PubMed] [Google Scholar]
- Bakker AJ & Berg HM (2004). Effect of taurine on sarcoplasmic reticulum function and force in skinned fast‐twitch skeletal muscle fibres of the rat. J Physiol 538, 185–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bella DL, Kwon Y‐H, Hirschberger LL & Stipanuk MH (2000). Post‐transcriptional regulation of cysteine dioxygenase in rat liver. Adv Exp Med Biol 483, 71–85. [DOI] [PubMed] [Google Scholar]
- Biggar W (2006). Duchenne muscular dystrophy. Pediatr Rev 27, 83–88. [DOI] [PubMed] [Google Scholar]
- Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, Kaul A, Kinnett K, McDonald C, Pandya S, Poysky J, Shapiro F, Tomezsko J, Constantin C & Group DMDCCW (2010). Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol 9, 77–93. [DOI] [PubMed] [Google Scholar]
- Camerino DC, Tricarico D, Pierno S, Desaphy JF, Liantonio A, Pusch M, Burdi R, Camerino C, Fraysse B & De Luca A (2004). Taurine and skeletal muscle disorders. Neurochem Res 29, 135–142. [DOI] [PubMed] [Google Scholar]
- Choi HS, Cha Y‐N & Kim C (2006). Taurine chloramine inhibits PMA‐stimulated superoxide production in human neutrophils perhaps by inhibiting phosphorylation and translocation of p47 phox. Int Immunopharmacol 6, 1431–1440. [DOI] [PubMed] [Google Scholar]
- Consolino CM & Brooks SV (2004). Susceptibility to sarcomere injury induced by single stretches of maximally activated muscles of mdx mice. J Appl Physiol 96, 633–638. [DOI] [PubMed] [Google Scholar]
- Cozzoli A, Rolland JF, Capogrosso R, Sblendorio V, Longo V, Simonetti S, Nico B & De Luca A (2011). Evaluation of potential synergistic action of a combined treatment with alpha‐methyl‐prednisolone and taurine on the mdx mouse model of Duchenne muscular dystrophy. Neuropathol Appl Neurobiol 37, 243–256. [DOI] [PubMed] [Google Scholar]
- Dalle‐Donne I, Giustarini D, Rossi R, Colombo R & Milzani A (2003). Reversible S‐glutathionylation of Cys374 regulates actin filament formation by inducing structural changes in the actin molecule. Free Radic Biol Med 34, 23–32. [DOI] [PubMed] [Google Scholar]
- De Luca A, Pierno S & Camerino DC (2015). Taurine: the appeal of a safe amino acid for skeletal muscle disorders. J Transl Med 13, 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Luca A, Pierno S, Liantonio A, Cetrone M, Camerino C, Fraysse B, Mirabella M, Servidei S, Rüegg U & Conte Camerino D (2003). Enhanced dystrophic progression in mdx mice by exercise and beneficial effects of taurine and insulin‐like growth factor‐1. J Pharmacol Exp Ther 304, 453–463. [DOI] [PubMed] [Google Scholar]
- De Luca A, Pierno S, Liantonio A, Cetrone M, Camerino C, Simonetti S, Papadia F & Camerino DC (2001). Alteration of excitation–contraction coupling mechanism in extensor digitorum longus muscle fibres of dystrophic mdx mouse and potential efficacy of taurine. Br J Pharmacol 132, 1047–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Senzi Moraes Pinto R, Ferretti R, Moraes LHR, Neto HS, Marques MJ & Minatel E (2013). N‐Acetylcysteine treatment reduces TNF‐α levels and myonecrosis in diaphragm muscle of mdx mice. Clin Nutr 32, 472–475. [DOI] [PubMed] [Google Scholar]
- Dilger RN & Baker DH (2007). Oral N‐acetyl‐l‐cysteine is a safe and effective precursor of cysteine. J Anim Sci 85, 1712–1718. [DOI] [PubMed] [Google Scholar]
- Eaton P (2006). Protein thiol oxidation in health and disease: techniques for measuring disulfides and related modifications in complex protein mixtures. Free Radic Biol Med 40, 1889–1899. [DOI] [PubMed] [Google Scholar]
- El‐Shafey A, Armstrong A, Terrill J, Grounds M & Arthur P (2011). Screening for increased protein thiol oxidation in oxidatively stress muscle tissue. Free Radic Res 45, 991–999. [DOI] [PubMed] [Google Scholar]
- Emery AE (2002). The muscular dystrophies. Lancet 359, 687–695. [DOI] [PubMed] [Google Scholar]
- Eppler B & Dawson R (2001). Dietary taurine manipulations in aged male Fischer 344 rat tissue: taurine concentration, taurine biosynthesis, and oxidative markers. Biochem Pharmacol 62, 29–39. [DOI] [PubMed] [Google Scholar]
- Ferreira LF, Gilliam LAA & Reid MB (2009). l‐2‐Oxothiazolidine‐4‐carboxylate reverses glutathione oxidation and delays fatigue of skeletal muscle in vitro. J Appl Physiol 107, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghezzi P & Bonetto V (2003). Redox proteomics: identification of oxidatively modified proteins. Proteomics 3, 1145–1153. [DOI] [PubMed] [Google Scholar]
- Griffin J, Williams H, Sang E, Clarke K, Rae C & Nicholson J (2001). Metabolic profiling of genetic disorders: a multitissue 1H nuclear magnetic resonance spectroscopic and pattern recognition study into dystrophic tissue. Anal Biochem 293, 16–21. [DOI] [PubMed] [Google Scholar]
- Grounds M (2008). Two‐tiered hypotheses for Duchenne muscular dystrophy. Cell Mol Life Sci 65, 1621–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grounds M & Shavlakadze T (2011). Impact of growth on properties of sarcolemma of skeletal myofibres: clinical and scientific implications. Bioessays 33, 458–468. [DOI] [PubMed] [Google Scholar]
- Hakim CH, Wasala NB & Duan D (2013). Evaluation of muscle function of the extensor digitorum longus muscle ex vivo and tibialis anterior muscle in situ in mice. J Vis Exp e50183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliwell B & Gutteridge (2007). Free Radicals in Biology and Medicine, vol. 4 Oxford University Press, New York. [Google Scholar]
- Hamilton EJ, Berg HM, Easton CJ & Bakker AJ (2006). The effect of taurine depletion on the contractile properties and fatigue in fast‐twitch skeletal muscle of the mouse. Amino Acids 31, 273–278. [DOI] [PubMed] [Google Scholar]
- Hertelendi Z, Toth A, Borbely A, Galajda Z, van der Velden J, Stienen GJ, Edes I & Papp Z (2008). Oxidation of myofilament protein sulfhydryl groups reduces the contractile force and its Ca2+ sensitivity in human cardiomyocytes. Antioxid Redox Signal 10, 1175–1184. [DOI] [PubMed] [Google Scholar]
- Huxtable R (1992). Physiological actions of taurine. Physiol Rev 72, 101–163. [DOI] [PubMed] [Google Scholar]
- Iwasaki T, Terrill J, Shavlakadze T, Grounds MD & Arthur PG (2013). Visualizing and quantifying oxidized protein thiols in tissue sections: a comparison of dystrophic mdx and normal skeletal mouse muscles. Free Radic Biol Med 65, 1408–1416. [DOI] [PubMed] [Google Scholar]
- Janaky R, Varga V, Hermann A, Saransaari P & Oja S (2000). Mechanisms of l‐cysteine neurotoxicity. Neurochem Res 25, 1397–1405. [DOI] [PubMed] [Google Scholar]
- Jerkins AA, Jones DD & Kohlhepp EA (1998). Cysteine sulfinic acid decarboxylase mRNA abundance decreases in rats fed a high‐protein diet. J Nutr 128, 1890–1895. [DOI] [PubMed] [Google Scholar]
- Jerkins AA & Steele RD (1991). Dietary sulfur amino acid modulation of cysteine sulfinic acid decarboxylase. Am J Physiol 261, E551–555. [DOI] [PubMed] [Google Scholar]
- Kharraz Y, Guerra J, Pessina P, Serrano AL & Munoz‐Canoves P (2014). Understanding the process of fibrosis in Duchenne muscular dystrophy. Biomed Res Int 2014, 965631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapidos KA, Kakkar R & McNally EM (2004). The dystrophin glycoprotein complex – signaling strength and integrity for the sarcolemma. Circ Res 94, 1023–1031. [DOI] [PubMed] [Google Scholar]
- Leaf CD & Pace GW (1994). Development of a novel glutathione repleting agent, l‐2‐oxothiazolidine‐4‐carboxylic acid (Procysteine®). Expert Opin Investig Drugs 3, 1293–1302. [Google Scholar]
- Loriette C, Pasantes‐Morales H, Portemer C & Chatagner F (1979). Dietary casein levels and taurine supplementation. Ann Nutr Metab 23, 467–475. [PubMed] [Google Scholar]
- Lynn S, Aartsma‐Rus A, Bushby K, Furlong P, Goemans N, De Luca A, Mayhew A, McDonald C, Mercuri E & Muntoni F (2015). Measuring clinical effectiveness of medicinal products for the treatment of Duchenne muscular dystrophy. Neuromuscul Disord 25, 96–105. [DOI] [PubMed] [Google Scholar]
- McIntosh L, Granberg KE, Brière KM & Anderson JE (1998). Nuclear magnetic resonance spectroscopy study of muscle growth, mdx dystrophy and glucocorticoid treatments: correlation with repair. NMR Biomed 11, 1–10. [DOI] [PubMed] [Google Scholar]
- Medved I, Brown MJ, Bjorksten AR, Murphy KT, Petersen AC, Sostaric S, Gong X & McKenna MJ (2004). N‐Acetylcysteine enhances muscle cysteine and glutathione availability and attenuates fatigue during prolonged exercise in endurance‐trained individuals. J Appl Physiol 97, 1477–1485. [DOI] [PubMed] [Google Scholar]
- Mollica JP, Dutka TL, Merry TL, Lamboley CR, McConell GK, McKenna MJ, Murphy RM & Lamb GD (2012). S‐Glutathionylation of troponin I (fast) increases contractile apparatus Ca2+ sensitivity in fast‐twitch muscle fibres of rats and humans. J Physiol 590, 1443–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrof BJ, Shrager JB, Stedman HH, Kelly AM & Sweeney HL (1993). Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci USA 90, 3710–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piers A, Lavin T, Radley‐Crabb H, Bakker A, Grounds M & Pinniger G (2011). Blockade of TNF in vivo using cV1q antibody reduces contractile dysfunction of skeletal muscle in response to eccentric exercise in dystrophic mdx and normal mice. Neuromuscul Disord 21, 132–141. [DOI] [PubMed] [Google Scholar]
- Pinto JR, de Sousa VP & Sorenson MM (2011). Redox state of troponin C cysteine in the D/E helix alters the C‐domain affinity for the thin filament of vertebrate striated muscle. Biochim Biophys Acta 1810, 391–397. [DOI] [PubMed] [Google Scholar]
- Prochniewicz E, Spakowicz D & Thomas DD (2008). Changes in actin structural transitions associated with oxidative inhibition of muscle contraction. Biochemistry (Mosc) 47, 11811–11817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radley‐Crabb H, Terrill J, Shavlakadze T, Tonkin J, Arthur P & Grounds MD (2012). A single 30 min treadmill exercise session is suitable for ‘proof‐of concept studies’ in adult mdx mice: A comparison of the early consequences of two different treadmill protocols. Neuromuscul Disord 22, 170–182. [DOI] [PubMed] [Google Scholar]
- Radley‐Crabb HG, Marini JC, Sosa HA, Castillo LI, Grounds MD & Fiorotto ML (2014). Dystropathology increases energy expenditure and protein turnover in the Mdx mouse model of Duchenne muscular dystrophy. PLoS ONE 9, e89277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey KA, Bakker AJ & Pinniger GJ (2010). Fiber‐type dependence of stretch‐induced force enhancement in rat skeletal muscle. Muscle Nerve 42, 769–777. [DOI] [PubMed] [Google Scholar]
- Rapucci Moraes LH, Bollineli RC, Mizobuti DS, dos Reis Silveira L, Marques MJ & Minatel E (2015). Effect of N‐acetylcysteine plus deferoxamine on oxidative stress and inflammation in dystrophic muscle cells. Redox Report 20, 109–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS & Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen CK & Packer L (2000). Thiol homeostasis and supplements in physical exercise. Am J Clin Nutr 72, 653S–669S. [DOI] [PubMed] [Google Scholar]
- Setsukinai K, Urano Y, Kakinuma K, Majima HJ & Nagano T (2003). Development of novel fluorescence probes that can reliably detect reactive oxygen species and distinguish specific species. J Biol Chem 278, 3170–3175. [DOI] [PubMed] [Google Scholar]
- Sharp PS, Bye‐a‐Jee H & Wells DJ (2011). Physiological characterization of muscle strength with variable levels of dystrophin restoration in mdx mice following local antisense therapy. Mol Ther 19, 165–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA & Reid MB (2006). Redox modulation of contractile function in respiratory and limb skeletal muscle. Respir Physiol Neurobiol 151, 229–241. [DOI] [PubMed] [Google Scholar]
- Spurney CF, Gordish‐Dressman H, Guerron AD, Sali A, Pandey GS, Rawat R, Van Der Meulen JH, Cha HJ, Pistilli EE, Partridge TA, Hoffman EP & Nagaraju K (2009). Preclinical drug trials in the mdx mouse: assessment of reliable and sensitive outcome measures. Muscle Nerve 39, 591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stipanuk MH (2004). Role of the liver in regulation of body cysteine and taurine levels: a brief review. Neurochem Res 29, 105–110. [DOI] [PubMed] [Google Scholar]
- Stipanuk MH, Dominy JE, Lee JI & Coloso RM (2006). Mammalian cysteine metabolism: new insights into regulation of cysteine metabolism. J Nutr 136, 1652S–1659S. [DOI] [PubMed] [Google Scholar]
- Sturman J, Messing J, Rossi S, Hofmann A & Neuringer M (1988). Tissue taurine content and conjugated bile acid composition of rhesus monkey infants fed a human infant soy‐protein formula with or without taurine supplementation for 3 months. Neurochem Res 13, 311–316. [DOI] [PubMed] [Google Scholar]
- Terrill JR, Boyatzis A, Grounds MD & Arthur PG (2013. a). Treatment with the cysteine precursor l‐2‐oxothiazolidine‐4‐carboxylate (OTC) implicates taurine deficiency in severity of dystropathology in mdx mice. Int J Biochem Cell Biol 45, 2097–2108. [DOI] [PubMed] [Google Scholar]
- Terrill JR, Grounds MD & Arthur PG (2015). Taurine deficiency, synthesis and transport in the mdx mouse model for Duchenne muscular dystrophy. Int J Biochem Cell Biol 66, 141–148. [DOI] [PubMed] [Google Scholar]
- Terrill JR, Radley‐Crabb HG, Grounds MD & Arthur PG (2012). N‐Acetylcysteine treatment of dystrophic mdx mice results in protein thiol modifications and inhibition of exercise induced myofibre necrosis. Neuromuscul Disord 22, 422–434. [DOI] [PubMed] [Google Scholar]
- Terrill JR, Radley‐Crabb HG, Iwasaki T, Lemckert FA, Arthur PG & Grounds MD (2013. b). Oxidative stress and pathology in muscular dystrophies: focus on protein thiol oxidation and dysferlinopathies. FEBS J 280, 4149–4164. [DOI] [PubMed] [Google Scholar]
- Thomas JA & Mallis RJ (2001). Aging and oxidation of reactive protein sulfhydryls. Exp Gerontol 36, 1519–1526. [DOI] [PubMed] [Google Scholar]
- Tiago T, Simao S, Aureliano M, Martin‐Romero FJ & Gutierrez‐Merino C (2006). Inhibition of skeletal muscle S1‐myosin ATPase by peroxynitrite. Biochemistry (Mosc) 45, 3794–3804. [DOI] [PubMed] [Google Scholar]
- Warskulat U, Flogel U, Jacoby C, Hartwig HG, Thewissen M, Merx MW, Molojavyi A, Heller‐Stilb B, Schrader J & Haussinger D (2004). Taurine transporter knockout depletes muscle taurine levels and results in severe skeletal muscle impairment but leaves cardiac function uncompromised. FASEB J 18, 577–579. [DOI] [PubMed] [Google Scholar]
- Warskulat U, Heller‐Stilb B, Oermann E, Zilles K, Haas H, Lang F & Häussinger D (2007). Phenotype of the taurine transporter knockout mouse. Methods Enzymol 428, 439–458. [DOI] [PubMed] [Google Scholar]
- Whitehead NP, Pham C, Gervasio OL & Allen DG (2008). N‐Acetylcysteine ameliorates skeletal muscle pathophysiology in mdx mice. J Physiol 586, 2003–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead NP, Yeung EW & Allen DG (2006). Muscle damage in mdx (dystrophic) mice: role of calcium and reactive oxygen species. Clin Exp Pharmacol Physiol 33, 657–662. [DOI] [PubMed] [Google Scholar]
- Winterbourn CC & Hampton MB (2008). Thiol chemistry and specificity in redox signaling. Free Radic Biol Med 45, 549–561. [DOI] [PubMed] [Google Scholar]
- Winterbourn CC & Kettle AJ (2000). Biomarkers of myeloperoxidase‐derived hypochlorous acid. Free Radic Biol Med 29, 403–409. [DOI] [PubMed] [Google Scholar]
- Winterbourn CC, Vissers MCM & Kettle AJ (2000). Myeloperoxidase. Curr Opin Hematol 7, 53–58. [DOI] [PubMed] [Google Scholar]
- Zafarullah M, Li WQ, Sylvester J & Ahmad M (2003). Molecular mechanisms of N‐acetylcysteine actions. Cell Mol Life Sci 60, 6–20. [DOI] [PMC free article] [PubMed] [Google Scholar]