

Abstract
Key points
Neonatal sustained hypoxia exposure modifies brainstem microglia and serotonin expression.
The altered brainstem neurochemistry is associated with impaired ventilatory responses to acute hypoxia and mortality.
The deleterious effects of sustained hypoxia exposure can be prevented by an inhibitor of activated microglia.
These observations demonstrate a potential cause of the brainstem serotonin abnormalities thought to be involved in sudden infant death syndrome.
Abstract
We showed previously that the end of the second postnatal week (days P11–15) represents a period of development during which the respiratory neural control system exhibits a heightened vulnerability to sustained hypoxia (SH, 11% O2, 5 days) exposure. In the current study, we investigated whether the vulnerability to SH during the same developmental time period is associated with changes in brainstem serotonin (5‐HT) expression and whether it can be prevented by the microglia inhibitor minocycline. Using whole‐body plethysmography, SH attenuated the acute (5 min) hypoxic ventilatory response (HVR) and caused a high incidence of mortality compared to normoxia rats. SH also increased microglia cell numbers and decreased 5‐HT immunoreactivity in the nucleus of the solitary tract (nTS) and dorsal motor nucleus of the vagus (DMNV). The attenuated HVR, mortality, and changes in nTS and DMNV immunoreactivity was prevented by minocycline (25 mg kg−1/2 days during SH). These data demonstrate that the 5‐HT abnormalities in distinct respiratory neural control regions can be initiated by prolonged hypoxia exposure and may be modulated by microglia activity. These observations share several commonalities with the risk factors thought to underlie the aetiology of sudden infant death syndrome, including: (1) a vulnerable neonate; (2) a critical period of development; (3) evidence of hypoxia; (4) brainstem gliosis (particularly the nTS and DMNV); and (5) 5‐HT abnormalities.
Key points
Neonatal sustained hypoxia exposure modifies brainstem microglia and serotonin expression.
The altered brainstem neurochemistry is associated with impaired ventilatory responses to acute hypoxia and mortality.
The deleterious effects of sustained hypoxia exposure can be prevented by an inhibitor of activated microglia.
These observations demonstrate a potential cause of the brainstem serotonin abnormalities thought to be involved in sudden infant death syndrome.
Abbreviations
- 5‐HT
serotonin
- CB
carotid body
- CN
cuneate nucleus
- DMNV
dorsal motor nucleus of the vagus
- HCVR
hypercapnic ventilatory response
- HVR
hypoxic ventilatory response
- LPS
lipopolysaccharide
- nAmb
nucleus ambiguus
- NK‐1R
neurokinin‐1 receptor
- nTS
nucleus of the solitary tract
- preBötC
pre‐Botzinger complex
- ROb
raphe obscurus
- SH
sustained hypoxia
- SIDS
sudden infant death syndrome
- XII
hypoglossal motor nucleus
Introduction
Postnatal maturation of brainstem respiratory neural control regions involves changes in the expression of several neurochemical factors, particularly during the second postnatal week of life in the rat (approximately postnatal day (P) 12 of age) (Liu & Wong‐Riley, 2005; Wong‐Riley & Liu, 2005; Liu & Wong‐Riley, 2010 a, 2010 c). The changes that occur during this critical period of development, although apparently necessary for adequate maturation of respiratory neural control, also coincide with a heightened vulnerability to environmental challenges. Specifically, we recently showed that sustained hypoxia exposure (SH, 11% O2, 5 days) encompassing the critical period (between P11 and 15) attenuated the acute hypoxic (HVR) and hypercapnic (HCVR) ventilatory response, and caused an unusually high degree of mortality that occurred several days (at ∼P18) after SH exposure ended (Mayer et al. 2014). The cause for the deleterious effects of SH on respiratory control is unknown, so in the current study we investigated whether it involves a disturbance in brainstem neurochemistry.
There are considerable changes in constitutive expression of various neurotransmitters that define critical stages of postnatal brainstem development (Liu & Wong‐Riley, 2005). Serotonin (5‐HT), GABA and glutamate expression, for example, change abruptly in the rhythmogenic network (pre‐Bötzinger complex, preBötC) and the nucleus of the solitary tract (nTS) (Liu & Wong‐Riley, 2002, 2010 b,c) where carotid body chemoafferents terminate (Finley & Katz, 1992). The reasons for the unique changes in brainstem neurochemistry that occur during the critical stage of development are largely unknown, although they appear necessary for ‘normal’ maturation of the respiratory neural control system. The changes in brainstem 5‐HT expression are of particular interest because of 5‐HT's well‐known modulatory effects on HVR (Richter et al. 1999) and HCVR (Li et al. 2006; Penatti et al. 2006; Hodges & Richerson, 2010 a,b). Genetic disruption of brainstem 5‐HT expression increased the incidence of apnoea and resulted in death several days after birth (Hodges et al. 2009). Furthermore, 5‐HT abnormalities in brainstem cardio‐respiratory control regions, including the dorsal motor nucleus of the vagus (DMNV) and the nTS, have been proposed to underlie the respiratory disturbances and mortality associated with sudden infant death syndrome (SIDS) (Bejjani et al. 2013; Machaalani & Waters, 2014). Collectively, these data indicate that a disturbance in brainstem (e.g. 5‐HT) neurochemistry during critical periods of development could underlie the heightened vulnerability of the neonate to hypoxia exposure (Mayer et al. 2014). Brain tyrosine hydroxylase levels were reduced at 2–3 weeks of age in rats born at altitude (Joseph et al. 2000), indicating at least the dopaminergic system is uniquely vulnerable to prolonged hypoxia exposure during development.
The respiratory control system is also vulnerable to pro‐inflammatory challenges during critical stages of development. Rats treated with an intraperitoneal injection of lipopolysaccharide (LPS) at P10 exhibited an attenuated HVR, which was associated with a high incidence of mortality, neither of which was observed in younger (P5) or older (P20) rats treated with the same dose of LPS (Rourke et al. 2014). Of interest was the similarity in the timing of the heightened vulnerability between LPS (P10) and SH (P11) exposure and the consistent effects on the acute HVR and lethality (Mayer et al. 2014). One explanation for the heightened vulnerability during the critical period is a microglial‐mediated disturbance in brainstem neurochemistry, particularly in key respiratory neural control regions. Brain 5‐HT levels were reduced in a neonatal rat model of hypoxia/ischaemia injury, but these were prevented by treatment with the inhibitor of activated microglia, minocycline, and the anti‐inflammatory agent, ibuprofen, demonstrating CNS neurochemistry can be regulated by microglia activity and pro‐inflammatory responses (Wixey et al. 2011 a,b, 2012). Therefore, we investigated whether the attenuated HVR and lethality of SH exposure during the critical period of development are associated with decreased 5‐HT expression in key brainstem respiratory control regions, and whether they can be prevented by the inhibitor of activated microglia, minocycline.
Methods
Ethical approval
Experiments were performed on neonatal male Lewis rats (Charles River, Wilmington, MA, USA; colony PO6) maintained under standard housing conditions on a 12:12 h light–dark cycle. Pregnant dams were provided with standard bedding, nesting material and food and water ad libitum. All procedures were carried out in accordance with the National Institutes of Health (NIH) guidelines for care and use of laboratory animals and were approved by the Animal Care and Use Committee at Case Western Reserve University. The experiments reported in the present study comply with the policies and regulations of The Journal of Physiology (Drummond, 2009).
SH exposure and minocycline treatment
Time‐pregnant rats were housed in the animal facility of the institution, and allowed to give birth naturally. When the pups reached P11, the litters were randomly assigned to groups that received room air or SH (11% O2) for 5 consecutive days (24 h per day) beginning at 09.00 h. Within each litter, rat pups were also randomly assigned to be treated with (n = 15) or without (n = 13) minocycline. Age‐matched rats that received saline (n = 12) or minocycline (n = 8), but were not exposed to SH, served as normoxia controls. SH was achieved by placing the mother and pups inside a plexiglass chamber connected to adjustable rotameters for mixing air and nitrogen. Airflow through the chamber was maintained at ∼4 l min−1 and O2 levels were monitored (TED 60T, Teledyne Analytical Instruments, City of Industry, CA, USA) and adjusted if necessary to maintain a constant appropriate level of SH. Rats received a 25 mg kg−1 subcutaneous injection of minocycline or saline of equivalent volume immediately before (at P11), once during (P13) and again at the end (P15) of SH exposure (a total of three injections throughout the SH exposure period). This dose of minocycline was chosen based on the literature (Wixey et al. 2011 b). For the injections given during the exposure, chambers were briefly opened, and rats were removed, weighed and injected with the appropriate dose of minocycline (or saline). At the end of the day 5 of exposure, the rats were removed from the chamber and allowed to recover in room air overnight and then assessed for acute HVR and HCVR using whole‐body plethysmography. The HVR and HCVR of SH‐treated rats were compared to age‐matched normoxic raised rats that also received saline or minocycline. After plethysmography measurements were made, a subset of rats were killed with an overdose injection (intraperitoneal) of urethane (3 g kg−1), perfused with paraformaldehyde and brainstems removed for later immunohistochemical analysis of microglia (Iba1) and serotonin expression. Additional rats were used to assess whether minocycline also prevented mortality after SH exposure; these rats did not undergo measurements of plethysmography and were simply used to assess mortality. In these experiments, rats were allowed to recover in room air until weaning (∼P28). In the instances in which the animals died, death was sudden, unexplained even after a veterinary consult and occurred without any apparent signs of distress or discomfort approximately 3 days after SH exposure. Specifically, the rats exposed to SH typically died during the night of day 17, and thus for ethical reasons determination of mortality was kept to a minimum number of animals and because of a lack of an explanation for the death.
Whole‐body plethysmography
The day after the rats were removed from SH, individual pups were separated from the litter and placed inside a custom‐made Perspex plethysmograph chamber as described previously (Mayer et al. 2013, 2014). Temperature inside the chamber was maintained (∼28°C) at all ages by adjusting a water bath (Isotemp 3013S, Fisher Scientific, PA, USA) that circulated water to a heat pad positioned underneath the plethysmograph. Airflow through the chamber was held constant (450 ml min−1) using a mass flow controller (0–2 l min–1; Aalborg, NY, USA). Prior to each experiment, the chamber was assessed for adequate seal by observing stability of the square pressure change following injection of a calibration volume (50 μl) using a glass micro‐syringe (Hamilton, Harvard Apparatus, Holliston, MA, USA). The same injection volume was used later for calibration of tidal volume changes associated with breathing. Rectal temperature was monitored continuously throughout the experiment with a fine temperature thermocouple (Physitemp, Clifton, NJ, USA), which was held securely in place with tape adhered to the base of the tail. Measurement of minute ventilation () was made during baseline, hypoxia and hypercapnia. Rats were allowed ∼25 min to acclimatise to the plethysmograph before receiving 10% O2 (5 min), followed by hypercapnia (5% CO2, 5 min). Ventilation was measured when the plethysmograph was sealed during the last 30 s of each exposure. Chambers were sealed by turning stopcocks upstream and downstream of the plethysmograph. The corresponding pressure signal associated with breathing during the time the chamber was sealed was calibrated to calculate tidal volume as previously described (Drorbaugh & Fenn, 1955; Mayer et al. 2013). The coefficient of variation (CV) was calculated from the baseline period breathing room air immediately prior to receiving acute hypoxia.
Immunohistochemistry
Rats were killed via a urethane overdose and perfused transcardially with 0.9% saline in 0.1 m phosphate buffer, pH 7.4, followed by ice‐cold 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4. The brains were quickly removed, post‐fixed in the same fixative for 2 h at 4°C, and cryoprotected in 30% sucrose until they lost buoyancy (∼48 h). The brainstems were then embedded in OCT compound (Sakura Finetek, Alphen aan Den Rijn, Netherlands), frozen and stored at −80°C until sectioning. Sections were cut using a Leica CM1850 cryostat. Serial coronal brainstem sections were then cut at 20 μm and mounted on gelatin‐coated slides and stored at −20°C until immunohistochemical staining was performed. Brainstem sections were collected from the caudal region of the area postrema and extended through to the merging of the central canal with the 4th ventricle. Brainstem regions of interest included the nTS, DMNV and the raphe obscurus (ROb) because of their suspected role in SIDS (Kinney, 2009; Kinney et al. 2009; Kinney & Thach, 2009; Bejjani et al. 2013). Analysis was also extended to include a diversity of other brain regions with various functions to ascertain whether there are selective effects of SH on neurochemical expression that occur within distinct regions. These brain regions include the hypoglossal motor nucleus (XII, a respiratory motor region), the cuneate nucleus (CN, a non‐respiratory region that receives inputs from the digits of the front paws), the nucleus ambiguus (nAmb, a multifunctional region that includes parasympathetic neurons for the heart) and the rhythmogenic network, the preBötC.
For staining, the slides were first thawed at room temperature for 45 min, at which time a hydrophobic barrier (pap pen) was applied to the outer surface of the slide. Slides were then rehydrated in PBS (pH 7.4) for three washes lasting 5 min each. Antigen retrieval was then performed using 80°C citrate buffer (10 mm citric acid, 0.05% Tween 20, pH 6.0). Slides were then permeabilised with three washes in PBS containing 0.01% Triton X‐100 (PBST), incubated for 1 h in blocking buffer (PBS containing 5% BSA, 5% normal donkey serum and 0.01% Triton X‐100), and then incubated overnight at room temperature in either 1:5000 rabbit anti‐Iba1 (Wako, Tokyo, Japan) antibody or a 1:500 rabbit anti 5‐HT (Chemicon, Temecula, CA, USA) diluted in blocking buffer for microglia or 5‐HT, respectively. A subset of slides with either 5‐HT or Iba‐1 antibodies were co‐incubated with a guinea pig anti‐neurokinin‐1 receptor (NK‐1R; 1:500, EMD Millipore, Billerica, MA, USA) antibody to aid in the localisation of the preBötC and nAmb (Stornetta et al. 2003). After rinsing in PBST, slides were incubated for 2 h with secondary donkey anti‐rabbit Alexa fluor 488 (1:500) diluted in blocking buffer. In NK‐R1 co‐labelling experiments, a donkey anti‐guinea pig Alexa fluor 633 (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA) was used in addition to the Alexa fluor 488 secondary antibody. After rinsing in PBS, slides were cover‐slipped with Prolong Gold anti‐fading medium (Life Technologies, Carlsbad, CA, USA). Tissue processing and staining with each antibody for all animals and sections from each treatment group were performed on the same day.
Tissue sections were photographed for quantification using a Leica TCS SP2 at 20× magnification at 512 × 512 resolution. However, 40× images at 1024 × 1024 resolution were also taken to provide representative images within specific brain regions for illustrative purposes. An argon laser beam allowed imaging of 488 nm emission. Gain and offset of the photomultiplier tube were adjusted to allow only a few saturated pixels, as per the manufacturer's recommendations. Images were captured as single optical slices (xy) located at the approximate z‐axis middle of a tissue section. Digital images for analysis between treatment groups for a given brain region were performed on the same day on compressed z‐stacks of the images. Brain regions of interest were located according to the atlas of Paxinos & Watson (1998), and included the caudal nTS, adjacent DMNV, XII motor nucleus, the preBötC, ROb, and CN. Semi‐quantification of Iba1 and 5‐HT expression was performed via fluorescence densitometry including microglia cell counts within each brain region for all four treatment groups using three sections per animal.
5‐HT and Iba‐1 quantification
Densitometry was performed on 20× images by photographing (Retiga EXI camera) an area within each brain region at the same light intensity and the same exposure time with QImaging software. Images were loaded into ImageJ and the background was corrected to the same level for all images. A circle (100 pixels in diameter for nTS, XII and CN, and 50 pixels in diameter for DMNV, preBötC and adjacent nAmb) was then placed in the pertinent brainstem region and the average pixel intensity within the circle was measured and defines the area of interest (AOI). Measurements were collected for three anatomically equivalent sections in each animal and then averaged to yield n = 1 per animal. The investigator was then unblinded and the results compared between the four treatment groups.
Iba1‐positive cells were automatically quantified by a blinded experimenter using the open source NIH software Image J and its ‘analyse particles’ function to define a threshold and particle size/circularity for each section that enforced recognition of microglia located completely within the section and focal plane. The same particle size and threshold was used to analyse every section across all animals and treatment groups. A similar approach was used to quantify the number of 5‐HT‐positive neurons in the ROb. The ROb was identified based on the presence of large 5‐HT neuron cell bodies and according to the Paxinos & Watson (1998) atlas. 5‐HT neuron counts are expressed as the number of neurons per brainstem section. The expression of 5‐HT within the ROb was also quantified within an outline of the region also in accordance with the atlas; the same outline was used as a template for the AOI in all sections across all treatment groups. The investigator was then unblinded and the results were averaged from the three cryo‐sections to yield n = 1 per animal and expressed as mean ± SD. 5‐HT and Iba1 analysis within preBötC and nAmb were performed by identifying the clustering of NK‐1R‐immunoreactive neurons in the rostral ventral lateral medulla, which is anatomically indicative of the preBötC.
Statistical analysis
Statistical comparisons were made between treatment groups using two‐way ANOVA and a Student–Newman–Keuls post hoc analysis. Differences were considered significant at P < 0.05. All values are expressed as mean ± 1 SD.
Results
Baseline rates of ventilation, mortality and breathing variability
Body weights and values for baseline breathing parameters are provided in Table 1. SH significantly reduced body weight (BW) in both saline (22.6 ± 4.9 g; P < 0.001) and minocycline (24.1 ± 5.0 g; P = 0.003) treated rats compared to normoxia (35.2 ± 3.1) controls. Interestingly, compared to saline treatment, minocycline also tended to decrease BW in normoxia (30.8 ± 1.6 g; P = 0.04) but not SH rats. Resting body temperature was the same between all treatment groups, although baseline minute ventilation () and tidal volume (V T) were significantly higher for all SH rats compared to normoxia controls. Breathing frequency (f B) was similar between groups (Table 1).
Table 1.
Body weight (BW), body temperature (T b) and baseline ventilatory variables in P16 rats after 5 days of SH exposure (P11–15); mortality was determined from the number of rats that had survived through to weaning (P28)
| Variable | |||||||
|---|---|---|---|---|---|---|---|
| Treatment | BW (g) | T b (°C) | V E (ml g−1 min−1) | V T (ml g−1) | f B (breaths min–1) | Mortality (%) | |
| Normoxia | Saline (n = 12) | 35.2 ± 3.1 | 36.9 ± 0.4 | 1.06 ± 0.23 | 0.0077 ± 0.0013 | 137.8 ± 12.9 | 0 |
| Minocycline (n = 8) | 30.8 ± 1.6* | 36.9 ± 0.3 | 1.04 ± 0.16 | 0.0078 ± 0.0009 | 132.6 ± 12.7 | 0 | |
| SH | Saline (n = 12) | 22.6 ± 4.9* | 36.5 ± 0.9 | 1.51 ± 0.46* | 0.0113 ± 0.0019* | 132.8 ± 27.8 | 50 |
| Minocycline (n = 15) | 24.1 ± 5.0* | 37.1 ± 0.4 | 1.47 ± 0.39* | 0.0109 ± 0.0025* | 137.4 ± 24.6 | 12 | |
Values are mean ± 1SD.
*Significant difference from normoxia saline‐treated rats (P < 0.05).
There was a considerable degree of mortality in SH‐treated rats (Table 1), which typically occurred at ∼P18, ∼3 days after being removed from SH (at the end of P15) and 2 days after measurements of plethysmography (P16). Only 1 of the 8 rats treated with minocycline expired following SH, reducing the incidence of mortality from 50% to ∼12%.
To determine if SH affected variability of breathing, the coefficient of variation (CV) of f B was determined during baseline (Fig. 1 A). SH increased the CV of f B (0.27 ± 0.18; P = 0.04) compared with normoxia saline (0.14 ± 0.12) rats. The CV was no longer significantly elevated in SH rats treated with minocycline (0.16 ± 0.13). The CV in normoxia rats treated with minocycline (0.12 ± 0.05) was not different from saline rats. Representative breathing traces are also provided (Fig. 1 B–D).
Figure 1. Variability of breathing frequency and respiratory pattern following SH and minocycline treatment .
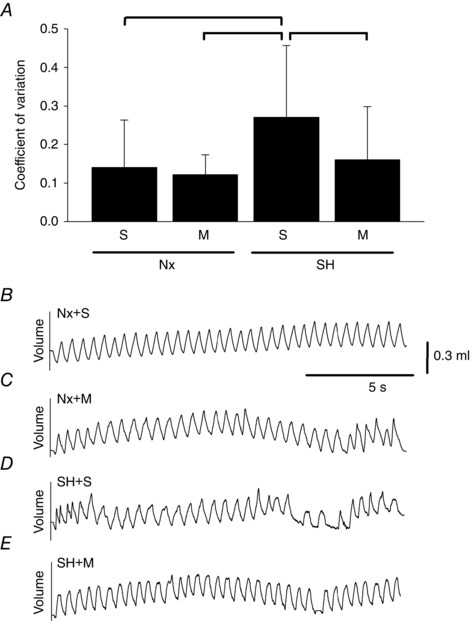
Values for the coefficient of variation (A) and representative respiratory traces (B–E) of P16 rats breathing room air after being raised in normoxia (Nx) or exposed to SH between P11 and P15, and treated with minocycline (M) or saline (S; control). Values in A are means ± SD; P < 0.05.
Hypoxic and hypercapnic ventilatory responses
SH exposure significantly attenuated the acute HVR (expressed as a delta (Δ) baseline; 0.12 ± 0.45 ml g−1 min−1; P = 0.001) compared to normoxia controls (0.63 ± 0.31 ml g−1 min−1; Fig. 2 A). The attenuated HVR of SH rats was caused by a reduced V T (−0.0008 ± 0.0017 ml g−1; P = 0.001; Fig. 2 B) and a trend (P = 0.07) toward a reduction in f B (21.5 ± 28.1 breaths min−1) although it was not significantly different from normoxia control (37.1 ± 20.4 breaths min−1) rats (Fig. 2 C). The HVR of SH rats treated with minocycline (0.39 ± 0.31 ml g−1 min−1) was not significantly different from normoxia, suggesting minocycline prevented the attenuated SH‐induced HVR. In the SH rats, minocycline prevented the trend for decreased f B (53.8 ± 22.3 breaths min−1) caused by SH, whereas V T remained reduced (−0.0011 ± 0.0022 ml g−1) compared to normoxia rats. In fact, f B following minocycline in SH rats was higher than in the saline group (P < 0.001). Minocycline did not affect breathing compared to saline treatment in room air rats. The breathing pattern and the magnitude of HCVR was similar between normoxia and SH rats (Fig. 3). Compared to saline‐treated rats, minocycline also did not affect HCVR in either normoxia or SH rats.
Figure 2. Hypoxic ventilatory responses following SH and minocycline treatment .
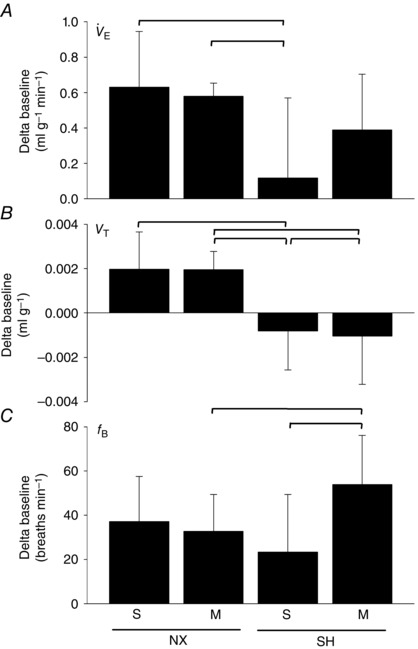
Ventilation (A; ), tidal volume (B; V T) and breathing frequency (C; f B) of P16 rats during acute hypoxia (10% O2, 5 min) after being raised in normoxia (NX) or exposed to SH between P11 and P15, and treated with minocycline (M) or saline (S; control). Note the attenuated HVR (A) following SH compared to control rats, which was prevented by minocycline treatment. Values are means ± SD and expressed as a delta baseline; P < 0.05.
Figure 3. Hypercapnic ventilatory responses following SH and minocycline treatment .
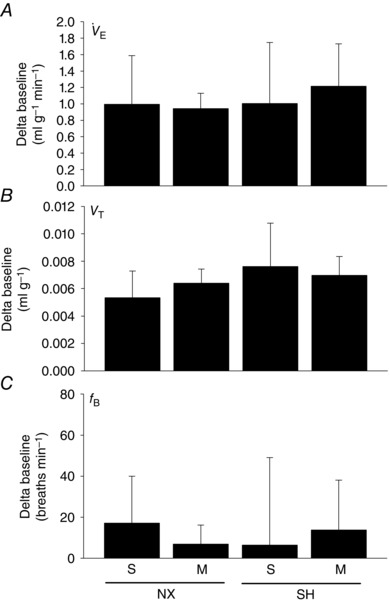
Ventilation (A; ), tidal volume (B; V T) and breathing frequency (C; f B) of P16 rats during acute hypercapnia (5% CO2, 5 min) after being raised in normoxia (NX) or exposed to SH between P11 and P15, and treated with minocycline (M) or saline (S; control). Values are means ± SD and expressed as a delta baseline; P < 0.05.
Brainstem microglia and 5‐HT immunoreactivity
nTS and DMNV
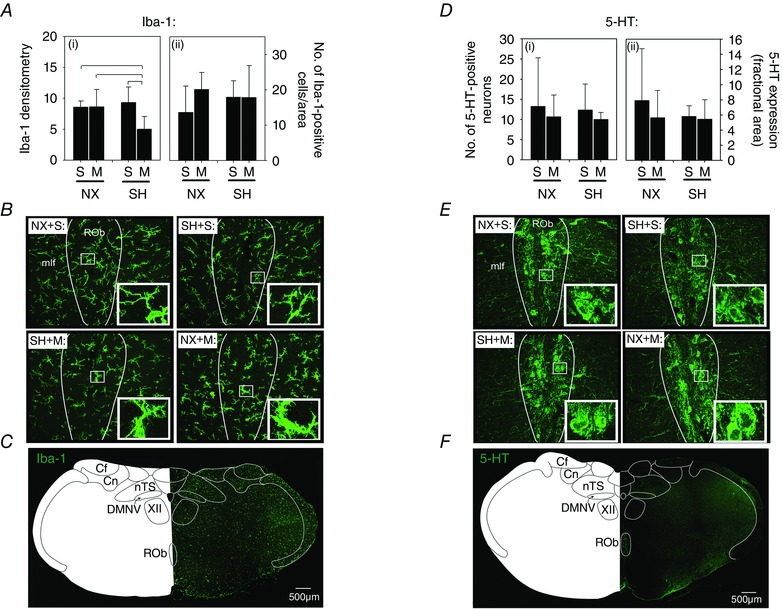
SH increased Iba‐1 densitometry compared to normoxia control rats in both the nTS and the DMNV (Fig. 4). Furthermore, SH also increased the number of Iba‐1‐positive (i.e. microglia) cells in the nTS (89.9 ± 8.8 cells per AOI; P < 0.001) and the DMNV (48.1 ± 10.6 cells per AOI; P < 0.001) compared to normoxia rats (nTS: 61.0 ± 17.7 cells per AOI, DMNV: 25.5 ± 9.1 cells per AOI; Fig. 4 A and B). The increased Iba‐1 expression and the number of microglia following SH in both brainstem regions were prevented by minocycline treatment (nTS: 72.8 ± 9.6 cells per AOI, DMNV: 34.8 ± 8.8 cells per AOI). However, in SH rats, there was only a moderate reduction (P = 0.06) in Iba‐1 densitometry compared to saline rats (Fig. 4 A). Minocycline did not affect Iba‐1 densitometry in normoxia control rats in either the nTS or the DMNV, with the exception that it increased the number of Iba‐1‐positive cells in the nTS (Fig. 4 A; P = 0.02).
Figure 4. Microglia (Iba‐1) expression in the nTS and DMNV following SH and minocycline treatment .
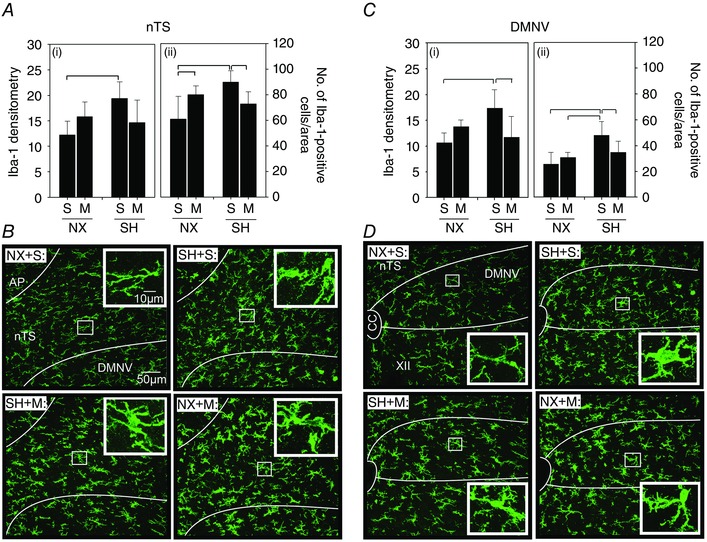
Changes in expression of the microglia marker Iba‐1 in the (A) nTS and (C) DMNV of P16 rats after being raised in normoxia (NX) or exposed to SH between P11 and P15, and treated with minocycline (M) or saline (S; control). Representative images of the (B) nTS and (D) DMNV of rats from each treatment group are also provided. Iba‐1 expression was assessed via fluorescence densitometry (i) and by the number of Iba‐1‐positive cells per AOI (ii). Note the increased Iba‐1 expression in the nTS and the DMNV following SH compared to control rats, which was prevented by minocycline treatment. Inset: higher magnification of microglia within each representative treatment group for nTS and DMNV. A low‐magnification image to illustrate orientation of the nTS and DMNV is provided in Fig. 6 C. Values are means ± SD; P < 0.05.
SH decreased 5‐HT immunoreactivity in the nTS (P = 0.01) and DMNV (P = 0.003) compared to normoxia control rats (Fig. 5), which inversely correlated with the increased microglia expression in the same brain regions (see Fig. 4). Minocycline prevented the decreased 5‐HT expression in SH‐treated rats in both the nTS and the DMNV (Fig. 5 A and C). In fact, 5‐HT immunoreactivity in SH rats treated with minocycline was significantly higher than for corresponding saline‐injected rats in both brainstem regions (nTS, P = 0.02; DMNV, P = 0.007). Minocycline did not affect 5‐HT expression in normoxia control rats in either the nTS or the DMNV (Fig. 5 A and C).
Figure 5. Serotonergic expression in the nTS and DMNV following SH and minocycline treatment .
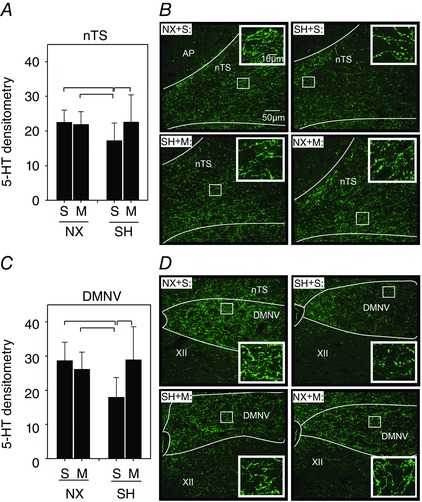
Changes in densitometric expression of serotonin (5‐HT) in the (A) nTS and (C) DMNV of P16 rats after being raised in normoxia (NX) or exposed to SH between P11 and P15, and treated with minocycline (M) or saline (S; control). Representative images (B) of the nTS and (D) DMNV of rats from each treatment group are also provided. Note the decreased 5‐HT expression in the nTS and the DMNV following SH compared to control rats, which were prevented by minocycline treatment. Inset: higher magnification of 5‐HT within each representative treatment group for nTS and DMNV. A low‐magnification image to illustrate the anatomical orientation of the nTS and DMNV is provided in Fig. 6 F. Values are means ± SD and expressed as arbitrary units of mean fluorescence intensity within the AOI; P < 0.05.
Raphe obscurus
SH exposure did not affect Iba‐1 densitometry or the number of microglia in the ROb compared to normoxia control rats (Fig. 6 A). However, within the SH‐treated rats, minocycline reduced Iba‐1 densitometry compared to saline‐treated rats (P = 0.004; Fig. 6 A). There was no difference in the number of 5‐HT neurons between treatment groups (Fig. 6 D). Notably, there was considerable variability in the normoxia control group in which 2 of the 7 rats exhibited in excess of 30 cells per area, which was more than 5‐fold greater than the median (6.3 cells per AOI) for this group. These two rats reduced the statistical power to detect significant differences between groups because when a re‐analysis was performed in which we intentionally excluded these two rats, the increased number of 5‐HT neurons in the SH group was now statistically higher than control (data not shown). Minocycline did no appear to affect the number of 5‐HT neurons in either normoxia or SH‐treated rats (Fig. 6 D). The expression of 5‐HT within the ROb was also not different between treatment groups (Fig. 6 D).
Figure 6. Microglia (Iba‐1) and serotonergic expression in the ROb following SH and minocycline treatment .
Changes in expression of (A) Iba‐1 and (D) 5‐HT in the RoB of P16 rats after being raised in normoxia (NX) or exposed to SH between P11 and P15, and treated with minocycline (M) or saline (S; control). Representative images of (B) (Iba‐1) and (E) 5‐HT for each treatment group are also provided. Iba‐1 expression was assessed via mean fluorescence intensity (i) and by the number of Iba‐1‐positive cells per AOI (ii). Note that SH did not affect Iba‐1 expression compared to control rats with the exception that there was less expression of Iba‐1 following minocycline in SH‐treated rats. There was also no difference in the number of 5‐HT‐positive neurons or amount of 5‐HT in the ROb following SH. Inset: higher magnification of Iba‐1 (B) and 5‐HT (E) within each representative treatment group. Anatomical orientation of the brainstem regions is also provided for both Iba‐1 (C) and 5‐HT (F). Values are means ± SD and expressed as arbitrary units of mean fluorescence intensity (Iba‐1), cells per AOI (Iba‐1 and 5‐HT neuron counts) or pixels per AOI (5‐HT); P < 0.05.
PreBötC and nAmb
Double labelling of Iba‐1 and 5‐HT with NK‐1R was performed to identify the preBötC and nAmb. There was no effect of SH on Iba‐1 densitometry or the number of microglia in either the preBötC (Fig. 7 A) or the nAmb (Fig. 7 B) compared to normoxia control rats. However, in rats exposed to SH, there was a slightly greater number of Iba‐1‐positive cells in the preBötC following minocycline treatment (30.8 ± 6.7 cells per AOI) compared to saline rats (21.3 ± 7.9 cells per AOI, P = 0.04; Fig. 7 A). There was also a slightly higher Iba‐1 densitometry in the preBötC in normoxia rats treated with minocycline compared to saline (P = 0.04; Fig. 7 A). Neither minocycline or SH exposure affected Iba‐1 immunoreactivity in the nAmb (Fig. 7 B). Similarly, there was no effect of SH or minocycline on 5‐HT immunoreactivity in the preBötC or the nAmb (Fig. 8).
Figure 7. Microglia (Iba‐1) expression in the nAmb and preBotC following SH and minocycline treatment .
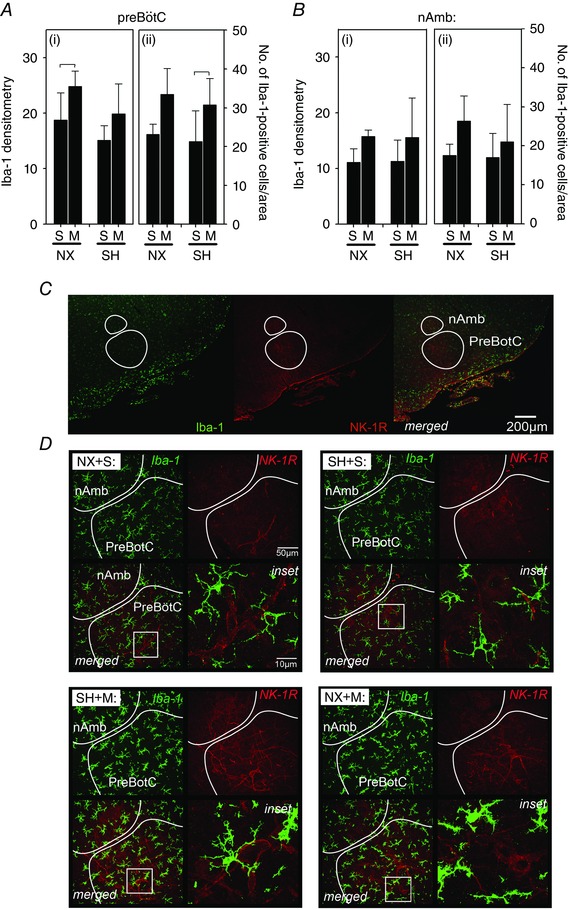
Changes in expression of Iba‐1 in the (A) preBötC and (B) nAmb of P16 rats after being raised in normoxia (NX) or exposed to SH between P11 and P15, and treated with minocycline (M) or saline (S; control). D, representative images of both brain regions from rats in each treatment group. Note that double‐labelling of neurons with an antibody specific for the neurokinin‐1 receptor (NK‐R1) was used to anatomically identify the preBötC and nAmb (C). Iba‐1 is expressed in arbitrary units as mean fluorescence intensity (i) and by the number of Iba‐1‐positive cells per AOI (ii). SH did not affect Iba‐1 expression in the preBötC or the nAmb; however, minocycline affected Iba‐1 expression in the preBötC in both NX and SH‐treated rats (A). Inset: higher magnification of microglia within each representative treatment group. Low‐magnification images of Iba‐1 and NK‐1R (and merged images) are provided to illustrate orientation of the preBötC. Values are means ± SD and expressed as arbitrary units of fluorescence intensity within the AOI, or number of Iba‐1‐positive cells per AOI; P < 0.05.
Figure 8. Serotonergic expression in the nAmb and preBotC following SH and minocycline treatment .
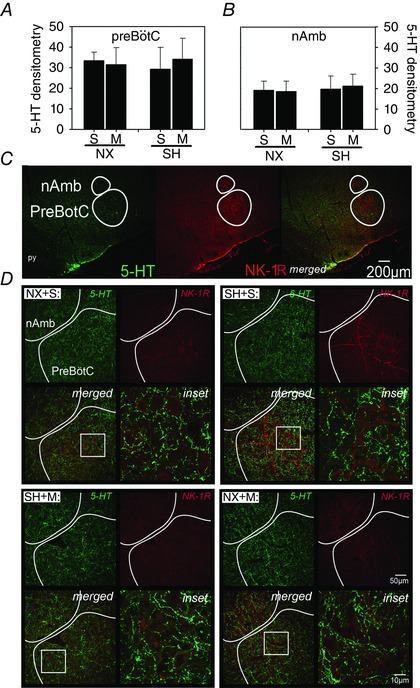
Changes in expression of 5‐HT in the (A) preBötC and (B) nAmb of P16 rats after being raised in normoxia (NX) or exposed to SH between P11 and P15, and treated with minocycline (M) or saline (S; control). D, representative images of both brain regions of rats from each treatment group. Note that double‐labelling of neurons with an antibody specific for the neurokinin‐1 receptor (NK‐R1) was used to anatomically identify the preBötC and nAmb (C). SH did not affect 5‐HT expression in any brain region. Inset: higher magnification of 5‐HT within each representative treatment group. Low‐magnification images of 5‐HT and NK‐1R (and merged images) are provided to illustrate orientation of the preBötC. Values are means ± SD and expressed as arbitrary units of mean fluorescence intensity within the AOI; P < 0.05.
Hypoglossal motonucleus and cuneate nucleus
Neither SH nor minocycline affected Iba‐1 densitometry or the number of microglia in either the XII (Fig. 9 A) or the CN (Fig. 9 C). Although surprisingly, minocycline increased the number of microglia in the XII compared to saline treatment in room air control rats (P = 0.02; Fig. 9 A). In contrast to the decreased 5‐HT immunoreactivity observed in the nTS and DMNV following SH (Fig. 5), it was increased in the XII (P = 0.006; Fig. 9 B); this effect was prevented by minocycline treatment (P = 0.01). 5‐HT expression was not affected by SH or minocycline in the CN (Fig. 9 D).
Figure 9. Microglia (Iba‐1) and serotonergic expression in the XII and CN following SH and minocycline treatment .
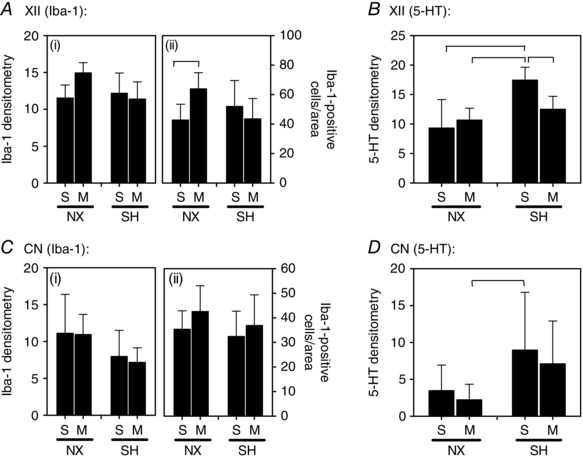
Changes in expression of (A and C) Iba‐1 and (B and D) 5‐HT in the XII nucleus and a non‐respiratory region, the CN of P16 rats after being raised in normoxia (NX) or sustained hypoxia (SH), and treated with minocycline (M) or saline (S; control). Note that SH did not affect Iba‐1 expression in either brain region compared to normoxia control rats; however, there was increased 5‐HT expression in the XII following SH, which was prevented by minocycline treatment (B). Values are means ± SD and expressed as arbitrary units of fluorescence intensity within the AOI, or number of Iba‐1‐positive cells per AOI; P < 0.05).
Discussion
In the current study, the attenuated HVR, increased respiratory variability (CV) and high incidence of mortality following neonatal SH exposure during the P11–15 period were consistent with our previous observations (Mayer et al. 2014). However, we were unable to confirm our previous finding in which SH attenuated the HCVR (Mayer et al. 2014). This may be because there was only a subtle reduction in the HCVR (Mayer et al. 2014) and thus it may be more variable in its reproducibility. We further showed that the deleterious effects of SH were associated with reduced 5‐HT immunoreactivity in the nTS and DMNV. As there was no effect of SH on 5‐HT expression in the other brainstem regions (with the exception of increased expression in the XII), the serotonergic system in the nTS and DMNV appears to be uniquely vulnerable to SH. The opposing effects of SH on 5‐HT in the XII motor nucleus (increased expression), a region containing neurons that innervate the tongue, compared to the nTS and DMNV (decreased expression) suggest the selective effects of SH may depend on the functional role of the specific brainstem region. 5‐HT expression in the nTS and DMNV represents projections from neurons located in the ROb, and the reduced expression following SH exposure, together with the unexpected mortality and impaired respiratory control during a critical window of development (Mayer et al. 2014), was consistent with the characteristics of SIDS (Kinney, 2009; Kinney et al. 2009; Duncan et al. 2010; Bejjani et al. 2013; Machaalani & Waters, 2014). One inconsistency, however, was that we did not observe any appreciable change in the number of 5‐HT neurons in the ROb, which seems to be higher in a subpopulation of SIDS infants (Duncan et al. 2010). One explanation for this discrepancy could be that the immunohistochemistry performed in the current study, which was intended to be used to show an association between brainstem neurochemistry and the attenuated HVR, was performed immediately after SH exposure, whereas the mortality did not occur until another 2–3 days later. The timing of our analysis of 5‐HT neurons in the ROb may be too early and therefore may not allow for direct comparisons with the data from SIDS infants, which for obvious reasons are performed post‐mortem. The increased 5‐HT neurons in the ROb has been proposed to be a compensatory response to the reduced expression in the brainstem regions where these neurons send their projections to cardio‐respiratory control regions (Duncan et al. 2010). An alternative explanation for the inconsistencies in ROb 5‐HT neuron counts could be related to the unusually high degree of variability in our normoxia control group (Fig. 6 D). Two of the seven rats exhibited 5‐HT counts that were more than 5‐fold higher than their littermates, which eliminated the statistical power to detect differences between groups. Admittedly not a preferable statistical approach, a re‐analysis after intentionally excluding these two rats revealed that there was a significant (3‐fold) increase in the number of ROb 5‐HT neurons following SH (data not shown). Despite the variability and although we did not detect an overall change in the number of 5‐HT neurons in the ROb, the disturbances in 5‐HT expression in the nTS (and DMNV) could explain the loss of the HVR following SH as it receives and integrates inputs arising from the carotid body (CB). In fact, the results from our rat model would indicate that reductions in 5‐HT expression in the cardio‐respiratory regions where the raphe neurons project to may be the relevant abnormality in SIDS that underlies the respiratory disturbances independently of any changes in the ROb neurons. Indeed, not all SIDS cases exhibit increased numbers of ROb 5‐HT neurons, which appears to be a consistent observation (Duncan et al. 2010). Disturbances in other brain regions such as the preBötC (or nAmb) could also explain such a profound disturbance in respiratory control, including the increased variability of breathing (CV) (Fig. 1), although we did not observe any appreciable changes in 5‐HT expression in this region.
We also demonstrated that the changes in 5‐HT expression in the nTS and the DMNV were correlated with an increased number of Iba‐1‐positive cells (i.e. microglia). The alterations in 5‐HT and microglia expression in both the nTS and the DMNV were prevented by minocycline treatment, suggesting that the vulnerability of the serotonergic system to SH in these distinct brain regions may be modulated by microglia activity. The effects of minocycline on the SH‐induced changes in nTS and DMNV neurochemistry were also associated with a reduction in respiratory variability and a moderate improvement of the attenuated HVR; a more robust effect of minocycline on the latter might have been expected with a higher dose. Changes in glial expression (increased gliosis) have also been documented in the DMNV and nTS in SIDS cases (Naeye, 1976; Takashima et al. 1978; Obonai et al. 1996; Sawaguchi et al. 2002). The increased gliosis (increased size and number of astrocytes) of the nTS and DMNV of SIDS cases was proposed to result from chronic hypoxia/ischaemia (Naeye, 1976). However, the 5‐HT abnormalities in SIDS is thought to have a fetal (Paterson et al. 2006) or even genetic origin (Weese‐Mayer et al. 2007). We provide evidence, however, that postnatal exposure to SH during a critical window of postnatal development is at least one risk factor that is sufficient to decrease 5‐HT levels in key cardio‐respiratory control regions.
Microglia are the resident immune cells and play an important role in CNS development, including synaptic pruning (Paolicelli et al. 2011) and, through interactions with neurons, shape the neural circuity of the maturing CNS (Wake et al. 2011). However, microglia transform from a ‘resting’ ramified morphology to an ‘activated’ amoeboid state as part of a pro‐inflammatory response to CNS injury (Streit et al. 2005; Graeber et al. 2011). Whether the microglia were ‘activated’ and became part of a pro‐inflammatory response, which also involves changes in chemokine and cytokine expression, is beyond the scope of this study. However, in the early neonatal period, microglia in the brain are represented by both ‘activated’ and ‘resting’ morphological phenotypes, suggesting a complex pro‐inflammatory and anti‐inflammatory environment in the developing CNS (Crain et al. 2013). Prevention of the respiratory and serotonergic disturbances and also the incidence of mortality by minocycline would imply the increased microglia expression contribute to a deleterious effect of SH. Thus, the increased Iba‐1 immunoreactivity observed in both the nTS and the DMNV may represent an aberrant microglial response leading to a lethal disturbance in brainstem neurochemistry and/or neuroarchitecture. However, the results of the current study do not allow us to determine whether the increased number of microglia following SH was the result of migration from other regions, or a change in the number of ramified microglia that transitioned into the amoeboid ‘activated’ state. Nevertheless, the ‘protective’ effects of minocycline against the changes in 5‐HT and microglia that occurred in the nTS and the DMNV would indicate a selective and preventable effect of SH on cardio‐respiratory control regions. Previous studies also showed that serotonergic deficits in the CNS were prevented by minocycline treatment in a neonatal rat model of hypoxia/ischaemia injury (Wixey et al. 2011 a). Whether the protective effects of minocycline are the result of a direct action on microglia, however, is unclear. Minocycline is a tetracycline antibiotic and inhibits microglia ‘activation’ by preventing nuclear translocation of NF‐kB (Pang et al. 2012; Kobayashi et al. 2013). It appears to have little if any effect on astrocytes (Kim & Suh, 2009), but side‐effects can include pitting and staining of teeth, and bone stunting (Chabner et al. 2011). Bone stunting could be consistent with the smaller size of normoxia rats treated with minocycline compared to saline controls (Table 1). The increased number of microglia in the nTS (Fig. 4 A) and Iba‐1 expression in the preBötC (Fig. 7 A) in normoxia control rats is additional evidence of potential non‐specific effects of minocycline, and its mechanism of action (despite being protective) should be interpreted cautiously.
We speculate that the vulnerability to SH during the P11–15 critical period may also be related to disturbances in other neurochemical factors. Substantial constitutive changes in brainstem neurochemistry (GABA, NMDA, glutamate, 5‐HT) occur abruptly and in some cases transiently (depending on the receptor and/or neurotransmitter) at ∼P12 in various brainstem regions including the nTS and the DMNV (Liu et al. 2002; Liu & Wong‐Riley, 2010 a,b,c). Such changes signify a critical period of development that appears necessary for ‘normal’ maturation of the brainstem neural circuitry. We chose an SH exposure period (between P11 and P15) with the intention that it encompassed the P12 critical period. Thus, the deleterious effects of SH may be explained by a microglia‐mediated disturbance in the trajectory of necessary developmental changes in constitutive 5‐HT expression and other neurochemical factors involved in maturation of cardio‐respiratory defence responses. SH exposure between P3 and P10 caused selective changes in enzymes that regulate glucose metabolism in the pons and medulla compared to other brain regions (Lai et al. 2003), and there are also unique changes in brainstem mitochondrial enzymes that take place between P11 and P13 (Liu & Wong‐Riley, 2003). We also showed that SH increased whole body metabolism in the P11–15 age group (Mayer et al. 2014), although whether there were changes in brain metabolism is yet to be determined. As activated microglia undergo metabolic reprogramming during an inflammatory response (Gimeno‐Bayon et al. 2014), further studies are underway in our laboratory to investigate whether the heightened vulnerability of the neonate around P11–15 is related to dysregulation of brain energy metabolism (Joseph et al. 2000) and aberrant inflammatory responses (Jurcovicova, 2014).
The distinct changes in microglia and 5‐HT neurochemistry that occurred in the nTS and the DMNV and not in the other brainstem regions could be a response mediated by modification of afferent inputs arising from their peripheral organs. For example, inflammation localised to the hind paw of 10‐day‐old rats increased the number of microglia in the corresponding spinal dorsal horn, which incidentally was also prevented by an intrathecal injection of minocycline (Vega‐Avelaira et al. 2013). SH has been considered pro‐inflammatory to the CB (Gauda et al. 2013) and blunted its sensitivity to acute hypoxia (Hanson et al. 1989). There is also evidence from several species that removal of CB afferents through surgical denervation can be lethal if it occurs during a critical period of development (Bureau et al. 1985 a,b; Haddad & Donnelly, 1988; Cote et al. 1996). Mortality resulted if denervation of the rat CB was performed at P7–8 compared to denervation in older animals (Serra et al. 2001). In piglets, CB denervation at P15 caused a higher mortality than at P10 and death occurred within 4–7 days (Donnelly & Haddad, 1990), which is similar to the timing (∼3 days) of mortality observed in the current study following SH exposure. CB denervation also disturbed the arousal response to acute hypoxia in sleeping dogs (Bowes et al. 1981 a,b) and surviving denervated piglets exhibited pronounced apnoea (Donnelly & Haddad, 1990); the latter would also be consistent with increased CV of breathing of rats following SH (Fig. 1). It is therefore reasonable that the protective effects of minocycline on the HVR were mediated by changes in breathing frequency, not tidal volume. This observation would be consistent with a potential effect of SH on CB function, which might also explain why the changes in microglia and 5‐HT were unique to the nTS (and DMNV) where the CB afferents terminate. Thus, minocycline might protect the deleterious effects of SH on the CB, rather than via a direct effect on the brainstem. Furthermore, morphological and neurochemical abnormalities in the CB have been implicated in SIDS (reviewed by Darnall, 2013; Porzionato et al. 2013), and may in part contribute to their respiratory disturbances (Valdes‐Dapena, 1980; Kahn et al. 1992, 2003; Slotkin et al. 1995; Gaultier, 2000). The changes in microglia and 5‐HT observed in the DMNV, however, are more difficult to explain, although nTS neurons do project to the DMNV suggesting it may be an indirect effect. A thorough discussion of how SH could affect the organs (e.g. heart and lung) that are innervated by this region is beyond the scope of this study. However, because the DMNV provides parasympathetic activity to the heart (lungs and other regions), a neurochemical disturbance in the DMNV could modify bradycardic responses to acute hypoxia (Dyavanapalli et al. 2014). Altered bradycardic and auto‐resuscitative capabilities have been shown in 5‐HT‐deficient mice (Cummings et al. 2011) and are consistent with the abnormalities associated with SIDS (Poets et al. 1999; Sridhar et al. 2003).
Summary
We showed the critical period of development (P11–15) during which the neonatal rat exhibits a heightened vulnerability to SH exposure is associated with increased microglia in distinct brainstem regions involved in cardio‐respiratory control. The increased microglia activity appears to modulate the constitutive expression of brainstem neurochemistry in association with a lethal disturbance in important defence responses to acute hypoxia, all of which can be pharmacologically preventable with an inhibitor of microglia. These observations bear a striking resemblance to the anatomical, neurochemical and functional abnormalities found in SIDS cases. Although these data demonstrate a protective effect of minocycline, its broad range of effects (e.g. as an antibiotic) independently of inhibiting activated microglia would preclude its prophylactic use to defend against a SIDS scenario. Instead, these data provide a possible explanation for the brainstem serotonergic abnormalities associated with SIDS and emphasises the importance of avoiding scenarios in which infants may be at increased risk of exposure to environmental hypoxia, or other conditions that may initiate microglial responses during postnatal periods of heightened brainstem vulnerability.
Additional information
Competing interests
The authors have no competing or conflicts of interests to disclose.
Author contributions
P.M.M.: conception, design, analysis, interpretation of data; drafting and revising content of the article; final approval. C.A.M.: analysis and interpretation of data; drafting and revising content of the article; final approval. D.G.L.: analysis of data; revising content of the article; final approval.
Funding
Funding was provided by the Department of Pediatrics, Case Western Reserve University/Rainbow Babies & Children's Hospital.
Acknowledgements
We would like to express our sincere gratitude to Steve Torontali for fabrication of the plethysmograph.
References
- Bejjani C, Machaalani R & Waters KA (2013). The dorsal motor nucleus of the vagus (DMNV) in sudden infant death syndrome (SIDS): pathways leading to apoptosis. Respir Physiol Neurobiol 185, 203–210. [DOI] [PubMed] [Google Scholar]
- Bowes G, Townsend ER, Bromley SM, Kozar LF & Phillipson EA (1981. a). Role of the carotid body and of afferent vagal stimuli in the arousal response to airway occlusion in sleeping dogs. Am Rev Respir Dis 123, 644–647. [DOI] [PubMed] [Google Scholar]
- Bowes G, Townsend ER, Kozar LF, Bromley SM & Phillipson EA (1981. b). Effect of carotid body denervation on arousal response to hypoxia in sleeping dogs. J Appl Physiol 51, 40–45. [DOI] [PubMed] [Google Scholar]
- Bureau MA, Lamarche J, Foulon P & Dalle D (1985. a). Postnatal maturation of respiration in intact and carotid body‐chemodenervated lambs. J Appl Physiol 59, 869–874. [DOI] [PubMed] [Google Scholar]
- Bureau MA, Lamarche J, Foulon P & Dalle D (1985. b). The ventilatory response to hypoxia in the newborn lamb after carotid body denervation. Respir Physiol 60, 109–119. [DOI] [PubMed] [Google Scholar]
- Chabner B, Brunton L & Knollman B (2011). Goodman and Gilman's The Pharmacological Basis of Therapeutics, 12th edn. McGraw‐Hill Education, New York. [Google Scholar]
- Cote A, Porras H & Meehan B (1996). Age‐dependent vulnerability to carotid chemodenervation in piglets. J Appl Physiol 80, 323–331. [DOI] [PubMed] [Google Scholar]
- Crain JM, Nikodemova M & Watters JJ (2013). Microglia express distinct M1 and M2 phenotypic markers in the postnatal and adult central nervous system in male and female mice. J Neurosci Res 91, 1143–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings KJ, Commons KG, Hewitt JC, Daubenspeck JA, Li A, Kinney HC & Nattie EE (2011). Failed heart rate recovery at a critical age in 5‐HT‐deficient mice exposed to episodic anoxia: implications for SIDS. J Appl Physiol (1985) 111, 825–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnall RA (2013). The carotid body and arousal in the fetus and neonate. Respir Physiol Neurobiol 185, 132–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly DF & Haddad GG (1990). Prolonged apnea and impaired survival in piglets after sinus and aortic nerve section. J Appl Physiol 68, 1048–1052. [DOI] [PubMed] [Google Scholar]
- Drorbaugh JE & Fenn WO (1955). A barometric method for measuring ventilation in newborn infants. Pediatrics 16, 81–87. [PubMed] [Google Scholar]
- Drummond GB (2009). Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol 587, 713–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan JR, Paterson DS, Hoffman JM, Mokler DJ, Borenstein NS, Belliveau RA, Krous HF, Haas EA, Stanley C, Nattie EE, Trachtenberg FL & Kinney HC (2010). Brainstem serotonergic deficiency in sudden infant death syndrome. JAMA 303, 430–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyavanapalli J, Jameson H, Dergacheva O, Jain V, Alhusayyen M & Mendelowitz D (2014). Chronic intermittent hypoxia‐hypercapnia blunts heart rate responses and alters neurotransmission to cardiac vagal neurons. J Physiol 592, 2799–2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley JC & Katz DM (1992). The central organization of carotid body afferent projections to the brainstem of the rat. Brain Res 572, 108–116. [DOI] [PubMed] [Google Scholar]
- Gauda EB, Shirahata M, Mason A, Pichard LE, Kostuk EW & Chavez‐Valdez R (2013). Inflammation in the carotid body during development and its contribution to apnea of prematurity. Respir Physiol Neurobiol 185, 120–131. [DOI] [PubMed] [Google Scholar]
- Gaultier C (2000). Development of the control of breathing: implications for sleep‐related breathing disorders in infants. Sleep 23(Suppl 4), S136–139. [PubMed] [Google Scholar]
- Gimeno‐Bayon J, Lopez‐Lopez A, Rodriguez MJ & Mahy N (2014). Glucose pathways adaptation supports acquisition of activated microglia phenotype. J Neurosci Res 92, 723–731. [DOI] [PubMed] [Google Scholar]
- Graeber MB, Li W & Rodriguez ML (2011). Role of microglia in CNS inflammation. FEBS Lett 585, 3798–3805. [DOI] [PubMed] [Google Scholar]
- Haddad GG & Donnelly DF (1988). The interaction of chemoreceptors and baroreceptors with the central nervous system. A critical role in early life. Ann N Y Acad Sci 533, 221–227. [DOI] [PubMed] [Google Scholar]
- Hanson MA, Kumar P & Williams BA (1989). The effect of chronic hypoxia upon the development of respiratory chemoreflexes in the newborn kitten. J Physiol 411, 563–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges MR & Richerson GB (2010. a). Medullary serotonin neurons and their roles in central respiratory chemoreception. Respir Physiol Neurobiol 173, 256–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges MR & Richerson GB (2010. b). The role of medullary serotonin (5‐HT) neurons in respiratory control: contributions to eupneic ventilation, CO2 chemoreception, and thermoregulation. J Appl Physiol 108, 1425–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges MR, Wehner M, Aungst J, Smith JC & Richerson GB (2009). Transgenic mice lacking serotonin neurons have severe apnea and high mortality during development. J Neurosci 29, 10341–10349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph V, Soliz J, Pequignot J, Sempore B, Cottet‐Emard JM, Dalmaz Y, Favier R, Spielvogel H & Pequignot JM (2000). Gender differentiation of the chemoreflex during growth at high altitude: functional and neurochemical studies. Am J Physiol Regul Integr Comp Physiol 278, R806‐816. [DOI] [PubMed] [Google Scholar]
- Jurcovicova J (2014). Glucose transport in brain ‐ effect of inflammation. Endocr Regul 48, 35–48. [DOI] [PubMed] [Google Scholar]
- Kahn A, Groswasser J, Franco P, Scaillet S, Sawaguchi T, Kelmanson I & Dan B (2003). Sudden infant deaths: stress, arousal and SIDS. Early Hum Dev 75 Suppl, S147–166. [DOI] [PubMed] [Google Scholar]
- Kahn A, Groswasser J, Rebuffat E, Sottiaux M, Blum D, Foerster M, Franco P, Bochner A, Alexander M, Bachy A, et al (1992). Sleep and cardiorespiratory characteristics of infant victims of sudden death: a prospective case‐control study. Sleep 15, 287–292. [DOI] [PubMed] [Google Scholar]
- Kim HS & Suh YH (2009). Minocycline and neurodegenerative diseases. Behav Brain Res 196, 168–179. [DOI] [PubMed] [Google Scholar]
- Kinney HC (2009). Brainstem mechanisms underlying the sudden infant death syndrome: evidence from human pathologic studies. Dev Psychobiol 51, 223–233. [DOI] [PubMed] [Google Scholar]
- Kinney HC, Richerson GB, Dymecki SM, Darnall RA & Nattie EE (2009). The brainstem and serotonin in the sudden infant death syndrome. Ann Rev Pathol 4, 517–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney HC & Thach BT (2009). The sudden infant death syndrome. N Engl J Med 361, 795–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Imagama S, Ohgomori T, Hirano K, Uchimura K, Sakamoto K, Hirakawa A, Takeuchi H, Suzumura A, Ishiguro N & Kadomatsu K (2013). Minocycline selectively inhibits M1 polarization of microglia. Cell Death Dis 4, e525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai JC, White BK, Buerstatte CR, Haddad GG, Novotny EJ, Jr & Behar KL (2003). Chronic hypoxia in development selectively alters the activities of key enzymes of glucose oxidative metabolism in brain regions. Neurochem Res 28, 933–940. [DOI] [PubMed] [Google Scholar]
- Li A, Zhou S & Nattie E (2006). Simultaneous inhibition of caudal medullary raphe and retrotrapezoid nucleus decreases breathing and the CO2 response in conscious rats. J Physiol 577, 307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q & Wong‐Riley MT (2002). Postnatal expression of neurotransmitters, receptors, and cytochrome oxidase in the rat pre‐Botzinger complex. J Appl Physiol 92, 923–934. [DOI] [PubMed] [Google Scholar]
- Liu Q & Wong‐Riley MT (2003). Postnatal changes in cytochrome oxidase expressions in brain stem nuclei of rats: implications for sensitive periods. J Appl Physiol (1985) 95, 2285–2291. [DOI] [PubMed] [Google Scholar]
- Liu Q & Wong‐Riley MT (2005). Postnatal developmental expressions of neurotransmitters and receptors in various brain stem nuclei of rats. J Appl Physiol 98, 1442–1457. [DOI] [PubMed] [Google Scholar]
- Liu Q & Wong‐Riley MT (2010. a). Postnatal development of N‐methyl‐d‐aspartate receptor subunits 2A, 2B, 2C, 2D, and 3B immunoreactivity in brain stem respiratory nuclei of the rat. Neuroscience 171, 637–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q & Wong‐Riley MT (2010. b). Postnatal changes in tryptophan hydroxylase and serotonin transporter immunoreactivity in multiple brainstem nuclei of the rat: implications for a sensitive period. J Comp Neurol 518, 1082–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q & Wong‐Riley MT (2010. c). Postnatal changes in the expressions of serotonin 1A, 1B, and 2A receptors in ten brain stem nuclei of the rat: implication for a sensitive period. Neuroscience 165, 61–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YY, Wong‐Riley MT, Liu JP, Jia Y, Liu HL, Jiao XY & Ju G (2002). GABAergic and glycinergic synapses onto neurokinin‐1 receptor‐immunoreactive neurons in the pre‐Botzinger complex of rats: light and electron microscopic studies. European J Neurosci 16, 1058–1066. [DOI] [PubMed] [Google Scholar]
- Machaalani R & Waters KA (2014). Neurochemical abnormalities in the brainstem of the sudden infant death syndrome (SIDS). Paediatr Respir Rev 15, 293–300. [DOI] [PubMed] [Google Scholar]
- Mayer CA, Ao J, Di Fiore JM, Martin RJ & MacFarlane PM (2013). Impaired hypoxic ventilatory response following neonatal sustained and subsequent chronic intermittent hypoxia in rats. Respir Physiol Neurobiol 187, 167–175. [DOI] [PubMed] [Google Scholar]
- Mayer CA, Di Fiore JM, Martin RJ & Macfarlane PM (2014). Vulnerability of neonatal respiratory neural control to sustained hypoxia during a uniquely sensitive window of development. J Appl Physiol (1985) 116, 514–521. [DOI] [PubMed] [Google Scholar]
- Naeye RL (1976). Brain‐stem and adrenal abnormalities in the sudden‐infant‐death syndrome. Am J Clin Pathol 66, 526–530. [DOI] [PubMed] [Google Scholar]
- Obonai T, Takashima S, Becker LE, Asanuma M, Mizuta R, Horie H & Tanaka J (1996). Relationship of substance P and gliosis in medulla oblongata in neonatal sudden infant death syndrome. Pediatr Neurol 15, 189–192. [DOI] [PubMed] [Google Scholar]
- Pang T, Wang J, Benicky J & Saavedra JM (2012). Minocycline ameliorates LPS‐induced inflammation in human monocytes by novel mechanisms including LOX‐1, Nur77 and LITAF inhibition. Biochim Biophys Acta 1820, 503–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L, Ragozzino D & Gross CT (2011). Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458. [DOI] [PubMed] [Google Scholar]
- Paterson DS, Hilaire G & Weese‐Mayer DE (2006). Medullary serotonin defects and respiratory dysfunction in sudden infant death syndrome. Respir Physiol Neurobiol 168, 133–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxino G & Watson C (1998). The rat brain in stereotaxic coordinates. Academic Press, London. [Google Scholar]
- Penatti EM, Berniker AV, Kereshi B, Cafaro C, Kelly ML, Niblock MM, Gao HG, Kinney HC, Li A & Nattie EE (2006). Ventilatory response to hypercapnia and hypoxia after extensive lesion of medullary serotonergic neurons in newborn conscious piglets. J Appl Physiol 101, 1177–1188. [DOI] [PubMed] [Google Scholar]
- Poets CF, Meny RG, Chobanian MR & Bonofiglo RE (1999). Gasping and other cardiorespiratory patterns during sudden infant deaths. Pediatr Res 45, 350–354. [DOI] [PubMed] [Google Scholar]
- Porzionato A, Macchi V, Stecco C & De Caro R (2013). The carotid body in sudden infant death syndrome. Respir Physiol Neurobiol 185, 194–201. [DOI] [PubMed] [Google Scholar]
- Richter DW, Schmidt‐Garcon P, Pierrefiche O, Bischoff AM & Lalley PM (1999). Neurotransmitters and neuromodulators controlling the hypoxic respiratory response in anaesthetized cats. J Physiol 514, 567–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rourke K, Mayer C & MacFarlane P (2014). Systemic LPS attenuates the acute hypoxic ventilatory response: implications for a uniquely sensitive window of neonatal development. FASEB J 28, Suppl 1177.4. [Google Scholar]
- Sawaguchi T, Franco P, Kato I, Shimizu S, Kadhim H, Groswasser J, Sottiaux M, Togari H, Kobayashi M, Takashima S, Nishida H, Sawaguchi A & Kahn A (2002). From epidemiology to physiology and pathology: apnea and arousal deficient theories in sudden infant death syndrome (SIDS)–with particular reference to hypoxic brainstem gliosis. Forensic Sci Int 130(Suppl), S21–29. [DOI] [PubMed] [Google Scholar]
- Serra A, Brozoski D, Hedin N, Franciosi R & Forster HV (2001). Mortality after carotid body denervation in rats. J Appl Physiol 91, 1298–1306. [DOI] [PubMed] [Google Scholar]
- Slotkin TA, Lappi SE, McCook EC, Lorber BA & Seidler FJ (1995). Loss of neonatal hypoxia tolerance after prenatal nicotine exposure: implications for sudden infant death syndrome. Brain Res Bull 38, 69–75. [DOI] [PubMed] [Google Scholar]
- Sridhar R, Thach BT, Kelly DH & Henslee JA (2003). Characterization of successful and failed autoresuscitation in human infants, including those dying of SIDS. Pediatr Pulmonol 36, 113–122. [DOI] [PubMed] [Google Scholar]
- Stornetta RL, Rosin DL, Wang H, Sevigny CP, Weston MC & Guyenet PG (2003). A group of glutamatergic interneurons expressing high levels of both neurokinin‐1 receptors and somatostatin identifies the region of the pre‐Botzinger complex. J Compar Neurol 455, 499–512. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Conde JR, Fendrick SE, Flanary BE & Mariani CL (2005). Role of microglia in the central nervous system's immune response. Neurol Res 27, 685–691. [DOI] [PubMed] [Google Scholar]
- Takashima S, Armstrong D, Becker L & Bryan C (1978). Cerebral hypoperfusion in the sudden infant death syndrome? Brainstem gliosis and vasculature. Ann Neurol 4, 257–262. [DOI] [PubMed] [Google Scholar]
- Valdes‐Dapena MA (1980). Sudden infant death syndrome: a review of the medical literature 1974–1979. Pediatrics 66, 597–614. [PubMed] [Google Scholar]
- Vega‐Avelaira D, Ballesteros JJ & Lopez‐Garcia JA (2013). Inflammation‐induced hyperalgesia and spinal microglia reactivity in neonatal rats. Eur J Pain 17, 1180–1188. [DOI] [PubMed] [Google Scholar]
- Wake H, Moorhouse AJ & Nabekura J (2011). Functions of microglia in the central nervous system–beyond the immune response. Neuron Glia Biol 7, 47–53. [DOI] [PubMed] [Google Scholar]
- Weese‐Mayer DE, Ackerman MJ, Marazita ML & Berry‐Kravis EM (2007). Sudden infant death syndrome: review of implicated genetic factors. Am J Med Genet A 143A, 771–788. [DOI] [PubMed] [Google Scholar]
- Wixey JA, Reinebrant HE & Buller KM (2011. a). Inhibition of neuroinflammation prevents injury to the serotonergic network after hypoxia‐ischemia in the immature rat brain. J Neuropathol Exp Neurol 70, 23–35. [DOI] [PubMed] [Google Scholar]
- Wixey JA, Reinebrant HE & Buller KM (2012). Post‐insult ibuprofen treatment attenuates damage to the serotonergic system after hypoxia‐ischemia in the immature rat brain. J Neuropathol Exp Neurol 71, 1137–1148. [DOI] [PubMed] [Google Scholar]
- Wixey JA, Reinebrant HE, Spencer SJ & Buller KM (2011. b). Efficacy of post‐insult minocycline administration to alter long‐term hypoxia‐ischemia‐induced damage to the serotonergic system in the immature rat brain. Neuroscience 182, 184–192. [DOI] [PubMed] [Google Scholar]
- Wong‐Riley MT & Liu Q (2005). Neurochemical development of brain stem nuclei involved in the control of respiration. Respir Physiol Neurobiol 149, 83–98. [DOI] [PubMed] [Google Scholar]