

Abstract
Key points
Two‐pore channels (TPCs) were identified as a novel family of endolysosome‐targeted calcium release channels gated by nicotinic acid adenine dinucleotide phosphate, as also as intracellular Na+ channels able to control endolysosomal fusion, a key process in autophagic flux.
Autophagy, an evolutionarily ancient response to cellular stress, has been implicated in the pathogenesis of a wide range of cardiovascular pathologies, including heart failure.
We report direct evidence indicating that TPCs are involved in regulating autophagy in cardiomyocytes, and that TPC knockout mice show alterations in the cardiac lysosomal system. TPC downregulation implies a decrease in the viability of cardiomyocytes under starvation conditions. In cardiac tissues from both humans and rats, TPC transcripts and protein levels were higher in females than in males, and correlated negatively with markers of autophagy.
We conclude that the endolysosomal channels TPC1 and TPC2 are essential for appropriate basal and induced autophagic flux in cardiomyocytes, and also that they are differentially expressed in male and female hearts.
Abstract
Autophagy participates in physiological and pathological remodelling of the heart. The endolysosomal two‐pore channels (TPCs), TPC1 and TPC2, have been implicated in the regulation of autophagy. The present study aimed to investigate the role of TPC1 and TPC2 in basal and induced cardiac autophagic activity. In cultured cardiomyocytes, starvation induced a significant increase in TPC1 and TPC2 transcripts and protein levels that paralleled the increase in autophagy identified by increased LC3‐II and decreased p62 levels. Small interfering RNA depletion of TPC2 alone or together with TPC1 increased both LC3II and p62 levels under basal conditions and in response to serum starvation, suggesting that, under conditions of severe energy depletion (serum plus glucose starvation), changes in the autophagic flux (as assessed by use of bafilomycin A1) occurred either when TPC1 or TPC2 were downregulated. The knockdown of TPCs diminished cardiomyocyte viability under starvation and simulated ischaemia. Electron micrographs of hearts from TPC1/2 double knockout mice showed that cardiomyocytes contained large numbers of immature lysosomes with diameters significantly smaller than those of wild‐type mice. In cardiac tissues from humans and rats, TPC1 and TPC2 transcripts and protein levels were higher in females than in males. Furthermore, transcript levels of TPCs correlated negatively with p62 levels in heart tissues. TPC1 and TPC2 are essential for appropriate basal and induced autophagic flux in cardiomyocytes (i.e. there is a negative effect on cell viability under stress conditions in their absence) and they are differentially expressed in male and female human and murine hearts, where they correlate with markers of autophagy.
Key points
Two‐pore channels (TPCs) were identified as a novel family of endolysosome‐targeted calcium release channels gated by nicotinic acid adenine dinucleotide phosphate, as also as intracellular Na+ channels able to control endolysosomal fusion, a key process in autophagic flux.
Autophagy, an evolutionarily ancient response to cellular stress, has been implicated in the pathogenesis of a wide range of cardiovascular pathologies, including heart failure.
We report direct evidence indicating that TPCs are involved in regulating autophagy in cardiomyocytes, and that TPC knockout mice show alterations in the cardiac lysosomal system. TPC downregulation implies a decrease in the viability of cardiomyocytes under starvation conditions. In cardiac tissues from both humans and rats, TPC transcripts and protein levels were higher in females than in males, and correlated negatively with markers of autophagy.
We conclude that the endolysosomal channels TPC1 and TPC2 are essential for appropriate basal and induced autophagic flux in cardiomyocytes, and also that they are differentially expressed in male and female hearts.
Abbreviations
- DMEM
Dulbecco's modified Eagle's medium
- FBS
fetal bovine serum
- MTT
3‐(4,5 dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide
- NAADP
nicotinic acid adenine dinucleotide phosphate
- NCS
newborn calf serum
- siRNA
small interfering RNA
- TPC
two‐pore channel
Introduction
Two‐pore channels (TPCs) were initially identified as a novel family of endolysosome‐targeted calcium release channels gated by nicotinic acid adenine dinucleotide phosphate (NAADP) (Calcraft et al. 2009), and subsequently as intracellular Na+ channels able to control endolysosomal fusion (Wang et al. 2012). Three subtypes have been characterized: TPC1 and TPC3 locate to endosomes and TPC2 locates to lysosomes (Zhu et al. 2010). Cang et al. (2013) showed that TPCs can couple the metabolic state of the cell to endolysosomal function and it has been suggested that TPCs may regulate autophagic processes in various tissues (Pereira et al. 2011; Neely Kayala et al. 2012; Lu et al. 2013; Lin et al. 2015). Autophagy, an evolutionarily ancient response to cellular stress and starvation, has been implicated in the pathogenesis of a wide range of cardiovascular pathologies, including heart failure (Lavandero et al. 2013). In heart failure, changes in autophagy are associated with metabolic remodelling (Lavandero et al. 2013). We have previously reported that, in the failing human left ventricular myocardium, changes in metabolism and cell viability correlate with changes in the expression of the TPC genes TPCN1 and TPCN2 (García‐Rúa et al. 2012). We now report direct evidence indicating that TPCs are involved in regulating cardiac autophagy. In the present study, we demonstrate that, in cultured cardiomyocytes, starvation‐induced progressive autophagy (identified as an increase in LC3 II/GAPDH and a decrease in p62 protein levels) was accompanied by a significant increase in TPC1 and TPC2 levels. Furthermore, the silencing of TPC1 or TPC2, or both, caused an increase in basal LC3II/GAPDH levels in cardiomyocytes, as well as a significant accumulation of p62, under both basal and starvation conditions. Moreover, we observed that the cardiac tissue of TPC1/2 knockout mice analysed by electronic microscopy demonstrated a greatly increased number of primary (non‐mature) lysosomes with a diameter significantly lower than those in wild‐type mice. Finally, we show that, in women and female rat cardiac tissue, TPC1 and TPC2 levels are higher than in men and male rats, respectively, and that they correlate negatively with cardiac basal p62 levels. Taken together, these results demonstrate the crucial role of TPCs in cardiomyocyte autophagy regulation.
Methods
All reagents were obtained from Sigma‐Aldrich (St Louis, MO, USA) unless otherwise stated.
Ethical approval
The study protocol was approved by the Galician Clinical Research Ethics Committee (2007/304). All acquisitions of human tissues were performed after obtaining written informed consent and in accordance with the Declaration of Helsinki 2008, and also, when pertinent, the European Convention of Human Rights and Biomedicine (ETS 164) and the UK Human Tissue Act 2004, after approval by local medical/health research ethics committees. All animals were maintained and killed using protocols approved by the Animal Care Committee of the University of Santiago de Compostela in accordance with European Union Directive 2010/63.
Human tissue samples
Right atrial appendages were obtained from 299 patients (199 men and 100 women) undergoing valve surgery or coronary artery bypass grafting in the Department of Cardiology of the University Clinical Hospital, Santiago de Compostela, Spain. Patient characteristics are listed in Table 1.
Table 1.
Demographic and clinical characteristics of cardiac surgery patients donating the cardiac tissue used in the present study
| Men (n = 199) | Women (n = 100) | |
|---|---|---|
| Age (years) | 69.9 | 70.7 |
| Body mass index (kg m–2) | 28.8 | 29.6 |
| Hypertension | 142 (71.4) | 76 (76) |
| Type 2 diabetes mellitus | 58 (29.1) | 21 (21) |
| Hyperlipidaemia | 105 (52.8) | 52 (52) |
| Laboratory findings | ||
| Triglycerides (mg dL–1) | 112.4 | 124.7 |
| Cholesterol (mg dL–1) | 164.9 | 187.3 |
| Glucose (mg dL–1) | 115.5 | 117.3 |
| Drugs | ||
| Antiplatelets | 84(40.2) | 31 (31) |
| Anticoagulants | 39 (19.2) | 22 (22) |
| ACE inhibitors | 55 (27.6) | 20 (20) |
| ARBs | 46 (23.1) | 25 (25) |
| β‐blockers | 71 (35.7) | 35 (35) |
| CCBs | 43 (21.6) | 12 (12) |
| Diuretics | 65 (32.7) | 46 (46) |
| Oral antidiabetics | 31 (15.6) | 17 (17) |
| Insulin | 16 (8) | 7 (7) |
Data are shown as the mean or n (%). ACE, angiotensin‐converting enzyme; ARB, angiotensin II receptor blocker; CCB, calcium channel blocker.
Animals
Male (n=16) and female (n=16), 8‐week‐old Sprague–Dawley rats were killed by decapitation. Hearts were collected and quickly frozen at −80°C. Neonatal (1‐ to 3‐day‐old) Sprague–Dawley rats were killed by cervical dislocation, and their hearts were used to establish primary cardiomyocyte cultures.
Wild‐type C57Bl/6;129 mice (n = 3) and TPC1/2 double knockout (KO) (Lear et al. 2015) mice (n = 3) of the same background strain, derived from dihybrid crosses between TPC1−/− mice (Ruas et al. 2014) and TPC2−/− mice (Calcraft et al. 2009), were supplied by the Department of Pharmacology, Oxford University (Oxford, UK). TPC1/2 KO mice were compared with wild‐type mice of the same age (9 months) and sex (male).
Cell cultures
Cardiomyocytes from human atrial appendages, neonatal rat hearts, and HL‐1 adult mouse atrial cardiomyocytes (a gift from Dr W. C. Claycomb, Louisiana State University Medical Centre, New Orleans, LA, USA) were all cultured as described previously (González‐Juanatey et al. 2003). Briefly, HL‐1 cardiomyocytes were cultured on fibronectin‐coated plates with Claycomb medium supplemented with fetal bovine serum (FBS), penicillin/streptomycin and l‐glutamine. To establish primary cultures of rat neonatal cardiomyocytes, the hearts of 1‐day‐old Sprague–Dawley rats were digested with collagenase and pancreatin at 37°C in three 30 min digestion cycles, cells were centrifuged from the pooled supernatants, and fibroblasts were removed by differential seeding. The cardiomyocytes were seeded at a density of 35,000 cm–2 on gelatin‐coated plates containing Dulbecco's modified Eagle's medium (DMEM)/M199 medium (Lonza, Basel, Switzerland) supplemented with FBS (Biochrom Gmbh, Darmstadt, Germany), newborn calf serum (NCS), l‐glutamine and penicillin/streptomycin. To establish primary cultures of human cardiomyocytes, right atrial appendage pieces excised for catheter passage during surgery requiring cardiopulmonary bypass were cut into small pieces and digested at 37°C in three 5 min cycles with PBS containing 0.25% trypsin, 0.15% collagenase and 0.02% glucose, after which the cells were centrifuged from the pooled supernatants for 5 min at 580 g and cultured in Iscove's modified Dulbecco's medium supplemented with 10% NCS, l‐glutamine and penicillin/streptomycin.
Electron microscopy
Electron microscopy was performed on three left cardiac ventricles from wild‐type mice and three from TPC1/2 KO mice. Samples were fixed and postfixed in 2.5% glutaraldehyde and osmium tetroxide in sodium cacodylate buffer and 1% OsO4 in the same buffer, and then included in Spurr's epoxy resin. Ultrathin sections were stained with uranyl acetate and lead citrate. Morphometric characteristics were determined using an MOP 20 image analyser (Carl Zeiss, Oberkochen, Germany) with constant magnification for each sample. Area, lysosome area and the number of lysosomes per cell were determined in 63 cells.
Small interfering RNA (siRNA) knockdown
Neonatal rat cardiomyocytes were cultured as described above. Twenty‐four hours after plating, cardiomyocytes were incubated with siRNA negative control (SIC 001; Sigma‐Aldrich) and siRNAs targeting TPC1 [SASI_Rn01_00107855 (sequence: GAUGGAACCUGUUCGAUUU) and SASI_Rn01_00107855_AS (sequence: AAAUCGAACAGGUUCCAUC); Sigma‐Aldrich] and TPC2 [ON‐TARGET plus rat Tpcn2 (sequence CCGAGAACUUCCUGCGAGU); GE Healthcare, Little Chalfont, UK], all used in accordance with the manufacturer's instructions. In brief, siRNAs were reconstituted in 50 μm stock solution. Next, 200 nm TPC1 and 100 nm TPC2 were mixed with RNAiMax transfectant (Thermo Fisher Scientific, Waltham, MA, USA) in Opti‐MEM medium (Thermo Fisher Scientific). Cardiomyocytes were incubated with the RNAiMax for 6 h, followed by the addition of culture medium containing 10% serum. Then, 48 h after the siRNA incubation, the cardiomyocytes were subjected to (a) serum starvation or (b) serum starvation plus deprivation of glucose (nutrient deprivation; DMEM medium without glucose; Sigma‐Aldrich). Finally, 100 nm bafilomycin A1 was added for the last 2 h as indicated.
Simulated ischaemia in cultured cells
Forty‐eight hours after siRNA incubation, ischaemia (defined as shortage of nutrients and oxygen simultaneously) was imposed in neonatal rat cardiomyocytes by a buffer exchange to ischaemia‐mimetic solution (in mm: 10 deoxyglucose, 139 NaCl, 12 KCl, 0.5 MgCl2, 1.3 CaCl2, 20 lactic acid and 5 Hepes, pH 6.2) and placing the culture plates in a humidified gas anoxia chamber equilibrated with 94.9% N2, 5% CO2 and ≤0.1% O2 for 6 h (Xie et al. 2014; Xu et al. 2015). Controls incubated in normoxic culture medium with 10% FBS were prepared in parallel.
Adenoviral infection of cultured cardiomyocytes
Neonatal rat cardiomyocytes were plated in coverslips. Twenty‐four hours after siRNA knockdown, cells were infected with adenovirus expressing GFP‐LC3 (multiplicity of infection 10), kindly provided by Beverly Rothermel and Joseph Hill (Zhu et al. 2007) (University of Texas Southwestern Medical Centre, Dallas, TX, USA). After 24 h, cardiomyocytes were subjected to serum starvation, and then fixed with 4% paraformaldehyde and examined by confocal fluorescence microscopy. Finally, 100 nm bafilomycin A1 was added for the last 2 h as indicated.
Quantitative RT‐PCR
RNA was extracted using a NucleoSpin kit in accordance with the manufacturer's instructions (Macherey‐Nagel, Duren, Germany). RNA quality and quantity were determined using a NanoDrop (Thermo Fischer Scientific) spectrophotometer.
Reverse‐transcription for TPCN1 and TPCN2 was performed with 1000 ng of RNA using Transcriptor First Strand cDNA Synthesis kits (Roche, Upper Bavaria, Germany). Quantitative PCR was performed using 1 μl of cDNA per well and Roche TaqMan‐MGB master mix, probes and pre‐optimized primers for the predesigned primers: rat TPCN1: 68 bp, Assay Id 50591 (Ref 05583055001), reference position 222, RefSeq NM_139332.3; rat TPCN2: 71 bp, Assay Id 505914 (Ref 05583055001), reference position 150, RefSeq NM_001107566; rat 18S: 78 bp, Assay Id 502300 (Ref 05532957001), reference position 592, RefSeq XO1117; mouse TPCN1: 70 bp, Assay Id 317112 (Ref 05583055001), reference position 2530, RefSeq NM_145853; mouse TPCN2: 78 bp, Assay Id 317114 (Ref 05583055001), reference position 1361, RefSeq NM_146206; and mouse 18S: 106 bp, Assay Id 307906 (Ref 05532957001), reference position 950 RefSeq NR_003278. Amplification conditions were: 95ºC for 10 min, followed by 40 cycles of 95ºC for 15 s and 60ºC for 60 s.
We also performed quantitative RT‐PCR using Brilliant III SYBR Green QRT‐PCR Master Mix Kit (Agilent Technologies, Santa Clara, CA, USA) with 50 ng of RNA and the specific primers: human TPCN1: TGTGGAGTACTTGTCTTCCG (forward) and CAGCACGTTGCGGTAGCG (reverse), 175 bp, RefSeq Accession NM_001143819.2; human TPCN2 GCTGACCACTTACCGCAG (forward) and GAAGCTCAAAGTCCGTTGGC (reverse), 190 bp, RefSeq Accession NM_139075.3; and human GAPDH: GATCATCAGCAATGCCTCCT (forward) and ATGGCATGGACTGTGGTCAT (reverse), 108 bp, RefSeq Accession NM_001256799.2. Reverse transcription conditions were: 50°C for 10 min and 95°C for 3 min. Quantitative PCR conditions were: 40 cycles of 95°C for 20 s, 56°C for 20 s, and then 95º for 1 min, 55ºC for 30 s and 95º for 30 s. The PCR efficiency was determined to be 90–110%. Results were analysed using MxPro, version 4 (Stratagene, La Jolla, CA, USA).
Western blotting
Cultured cardiomyocytes (35,000 cells cm–2), HL‐1 cells (3 × 105 in P6 wells) or heart tissues were lysed and subjected to SDS‐PAGE/western blotting as described previously (Lear et al. 2010). Briefly, cells were lysed with Triton X‐100 (1% in a buffer consisting of 50 mm Tris‐HCl, 150 mm NaCl, 5 mm EDTA, 1 mm phenylmethylsulphonylfluoride, 10 μg ml−1 leupeptin, 10 μg ml−1 aprotinin, 10 μg ml−1 trypsin inhibitor and 1 mm NaVO4). Samples were separated by 8%, 10% or 12% SDS‐PAGE (depending on the molecular weight of the protein) and transferred to polyvinyl difluoride membranes (GE Healthcare), which were incubated for 1 h in blocking solution (50 mm Tris‐HCl, 200 mm NaCl, 0.1% Tween 20 and 5% skimmed milk power) unless the manufacturer's instructions for the antibody indicated otherwise. The primary antibodies used were: TPC1 (dilution 1:500; Santa Cruz Biotechnology, Dallas, TX, USA), TPC2 (dilution 1:200; Santa Cruz Biotechnology), Thr172‐phospho‐AMPK (dilution 1:1000; Cell Signaling Technology, Danvers, MA, USA), AMPK (dilution 1:1000; Cell Signaling Technology), Ser2448‐phospho‐mTOR (dilution 1:1000; Santa Cruz Biotechnology), mTOR (dilution 1:1000; Santa Cruz Biotechnology), LC3B (dilution 1:1000; Cell Signaling Technology), p62 (dilution 1:1000; Cell Signaling Technology), LAMP‐1 (dilution 1:700, Abcam, Cambridge, UK), Rab7 (dilution 1:1000, Cell Signaling Technology), GAPDH (dilution 1:1000; Sigma‐Aldrich) and β‐actin (dilution 1:1000; Thermo Fisher Scientific). Membranes were incubated for 1 h in horseradish‐peroxidase‐conjugated anti‐IgG secondary antibody (Santa Cruz Biotechnology) and visualized using an Immobilon HRP chemiluminescence detection system (Millipore, Billerica, MA, USA).
MTT
Cell viability was assessed by assaying mitochondrial metabolic activity with 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT).
Statistical analysis
All experimental data were obtained from at least three independent experiments. Comparisons between groups of data were performed with the appropriate test, depending on the normality of the data. We used the Mann–Whitney U test with non‐Gaussian data. All the correlation coefficients are reported as Spearman ρ. All analyses were performed using SPSS, version 15.0 (SPSS Inc., Chicago, IL, USA) or Prism 5 (GraphPad Software Inc., San Diego, CA, USA).
Results
TPC1 and TPC2 levels increase during starvation‐induced autophagy in cardiomyocytes
Serum starvation of neonatal rat cardiomyocytes for 12/24 h induced significant increases in TPC1 and TPC2 mRNA (Fig. 1 A) and protein (Fig. 1 B and C). In the case of TPC2, the increase in mRNA levels was even more evident when the cells were submitted to nutrient deprivation conditions comprising serum starvation and deprivation of glucose (Fig. 1 A). The protein results were enchoed in HL‐1 cardiomyocytes (Fig. 1 D and E) and the mRNA results in cultured human atrial cardiomyocytes (Fig. 1 F). These increases occurred concomitant with changes indicative of activated autophagy: AMPK, phosphoAMPK and LC3II rose, whereas mTOR phosphorylation and p62 fell (Fig. 2).
Figure 1. TPCs levels increase under starvation in cardiomyocytes .

RT‐PCR (A) and a western blot (B and C) show increases of TPC1 and TPC2 after 12/24 h of serum starvation or serum plus glucose starvation in neonatal rat cardiomyocytes (n = 6). A western blot (D and E) confirmed the increase of TPCs after serum starvation or after serum plus glucose starvation in HL‐1 cells (n = 7). RT‐PCR (F) confirmed increases of TPCs transcripts after 24 h of serum starvation in human cardiomyocytes (n = 4). Bar graphs show the mean ± SEM. Statistical significance (Mann–Whitney U test): *P ≤ 0.05; **P ≤ 0.01.
Figure 2. Effect of starvation on pAMPK, AMPK, mTOR, pmTOR, LC3 and p62 in cardiomyocytes .
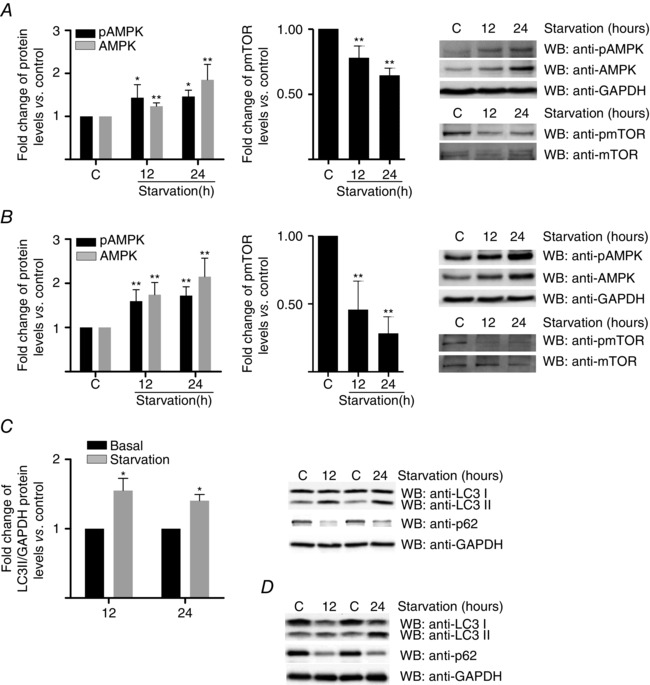
A, western blot corresponding to neonatal rat cardiomyocytes lysates immunostained with pAMPK, AMPK, pmTOR and mTOR (n = 5). B, representative western blot of HL‐1 lysates immunostained with pAMPK, AMPK, mTOR and pmTOR (n = 5). C, western blot corresponding to neonatal rat cardiomyocytes lysates immunostained with LC3 and p62. D, western blot corresponding to HL‐1 lysates immunostained with LC3 and p62 (n = 5). Bar graphs show the mean ± SEM. Statistical significance (Mann–Whitney U test): *P ≤ 0.05; **P ≤ 0.01.
TPC1 and TPC2 knockdown alters the progression of autophagy under basal and starvation‐induced conditions in neonatal rat cardiomyocytes
Silencing of TPC1 or TPC2, or both, using siRNAs did not modify cell viability under basal nutrient‐rich conditions, as measured by the MTT assay (data not shown). In all experiments, the knockdown was ≥60% (Fig. 3). Silencing TPC1 consistently reduced TPC2 mRNA (by 26.4 ± 18.2%) and vice versa (by 10.2 ± 9.9%). Knockdown of TPC2, either alone or in conjunction with TPC1, significantly increased GFP‐LC3 puncta and the LC3II/GAPDH ratio under nutrient‐rich conditions (Figs 4 A to C and 5 A), whereas TPC1 knockdown alone showed a tendency for an increase (Figs 4 C and 5 A). In cardiomyocytes under serum starvation for 24 h, the depletion of TPC2 did not increase the number of GFP‐LC3‐positive puncta further (Fig. 4 A and B), nor the processing of LC3I to LC3II compared to cardiomyocytes transfected with siRNA negative control (Figs 4 C and 5 A), although there appeared to be a trend in that direction (Fig. 4 C). However, under conditions of nutrient (glucose plus serum) deprivation (i.e. the experimental condition under which autophagy in cardiomyocytes is strongly induced as a result of severe energy depletion and is significantly elevated compared to under serum starvation; Matsui et al. 2007), TPC2 knockdown significantly increased the LC3II/GAPDH ratio compared to cardiomyocytes transfected with siRNA negative control (TPC1 silencing also inducing a bias in that direction) (Fig. 5 A), suggesting an alteration in the control of cardiomyocyte stress‐induced autophagy provoked by the absence of TPC2.
Figure 3. Levels of expression of TPCs after knockdown .
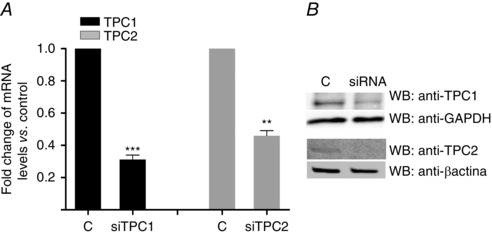
Confirmation of knockdown of TPC1 and TPC2 by using RT‐PCR (A) and western blot (B) in neonatal rat cardiomyocytes (n = 6). Bar graphs show the mean ± SEM. Statistical significance (Mann–Whitney U test): **P ≤ 0.01; ***P ≤ 0.001.
Figure 4. TPC2 and TPC1/2 downregulation increases LC3II in neonatal rat cardiomyocytes .
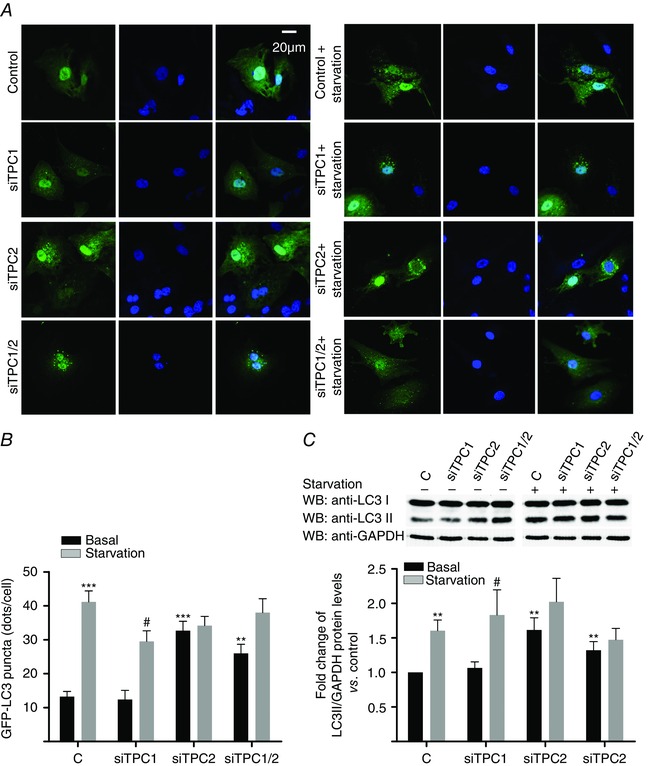
A and B, immunocytochemistry/confocal analysis showed that downregulation of TPC2 and TPC1/2 (but not of TPC1) increases basal GFP‐LC3 (n = 4). C, immunoblotting confirmed these results for LC3II/GAPDH ratio (n = 5). Bar graphs show the mean ± SEM. Statistical significance (Mann–Whitney U test): *P ≤ 0.05 vs. basal control; **P ≤ 0.01 vs. basal control; ***P ≤ 0.001 vs. basal control; # P ≤ 0.05 vs. non‐starved siTPC1.
Figure 5. TPC downregulation affects autophagic flux in neonatal rat cardiomyocytes .
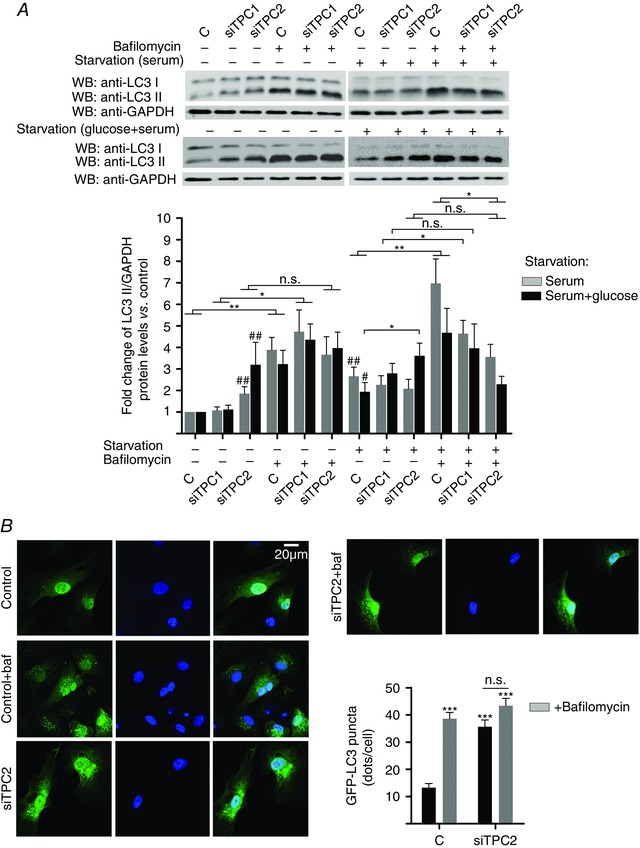
A, downregulation of TPC2 decreases autophagic flux (assessed by comparison of LC3II/GAPDH ratios between bafilomycin A1‐treated and bafilomycin A1‐non treated groups in cells in which TPC1 or TPC2 was silenced) in neonatal rat cardiomyocytes both under basal and serum starvation (24 h) conditions, whereas downregulation of TPC1 shows a tendency in the same direction (n=6). Under severe energy deprivation (serum plus glucose starvation for 16 h) conditions, either TPC1 or TPC2 downregulation causes alterations in autophagic flux (n = 7). B, immunocytochemistry/confocal analysis confirmed the above results for basal conditions (n = 4). Bar graphs show means ± SEM. Statistical significance (Mann–Whitney U test): *P ≤ 0.05; **P ≤ 0.01; # P ≤ 0.05 vs. basal control; ## P ≤ 0.01 vs. basal control. n.s., not significant.
Because the accumulation of autophagosomes or the measurement of LC3II may represent either the increased generation of autophagosomes and/or a block in autophagosomal maturation, we measured autophagic flux monitoring LC3 turnover, which is based on the observation that LC3II is degraded in autolysosomes: if cells are treated with lysosomotropic reagents that inhibit acidification inside the lysosome or inhibit autophagosome–lysosome fusion, the degradation of LC3II is blocked, resulting in the accumulation of LC3II (Mizushima et al. 2010). In the presence of such inhibitors, the accumulation of LC3II would be indicative of efficient autophagic flux, whereas the failure of LC3II to increase in the presence of such inhibitors would indicate a defect or delay in the process (Mizushima et al. 2010). Accordingly, differences in the amount of LC3II between samples in the presence and absence of lysosomal inhibitors represent the amount of LC3 that is delivered to lysosomes for degradation. We used a lysosomal inhibitor, bafilomycin A1, which is a specific inhibitor of the vacuolar type H+‐ATPase (V‐ATPase) (Klionsky et al. 2008), aiming to assess whether the knockdown of TPC1 and TPC2 could inhibit autophagic flux in neonatal rat cardiomyocytes. Under basal nutrient‐rich conditions, there were significant differences in the amount of LC3II between samples in the presence and absence of bafilomycin A1 in cardiomyocytes transfected with siRNA negative control, as well as in cardiomyocytes in which TPC1 was silenced (Fig. 5 A). However, in cardiomyocytes in which TPC2 was silenced, the amount of LC3II was not significantly different in the presence and absence of bafilomycin A1 (Fig. 5 A), suggesting that basal autophagic flux was already impeded by TPC2 knockdown (Fig. 5 A). Confirming these results, treatment of TPC2 knockdown cardiomyocytes with bafilomycin A1 did not increase the number of GFP‐LC3 puncta further under basal conditions (Fig. 5 B). Similar autophagic flux alterations were observed in cardiomyocytes in which TPC2 was silenced after they had been under serum starvation for 24 h (Fig. 5 A). Moreover, in cells subjected to severe energy depletion conditions (serum plus glucose starvation for 16 h), the silencing of TPC2 further strongly increased the LC3II/GAPDH ratio compared to cardiomyocytes transfected with siRNA negative control, whereas TPC1 knockdown again showed a clear tendency for an increase (Fig. 5 A). Importantly, either the absence of TPC1 or the absence of TPC2 caused alterations in autophagic flux when cardiomyocytes were subjected to severe energy depletion (Fig. 5 A), as demonstrated by the lack of a statistically significant difference in the LC3II/GAPDH ratio between bafilomycin A1‐treated and bafilomycin A1‐non‐treated groups for cells in which TPC1 or TPC2 was silenced (Fig. 5 A). Note that, under starvation conditions, we found a statistically significant decrease in the LC3II/GAPDH ratio in TPC2‐silenced cardiomyocytes treated with bafilomycin A1 compared to cardiomyocytes transfected with siRNA negative control treated with bafilomycin A1, and this was even more pronounced under severe energy depletion conditions (Fig. 5 A).
In line with these findings, and further confirming the alterations in autophagic flux induced by the absence of TPCs in cardiomyocytes, the knockdown of TPC2, with or without TPC1, significantly increased the basal levels of p62, a protein whose accumulation is generally considered to be a good indicator of defects in the turnover of poly‐ubiquitinated protein aggregates in autophagy (Barth et al. 2010) (Fig. 6 A), whereas knockdown of either TPC1 or TPC2 increased p62 levels under starvation conditions (Fig. 6 B and C).
Figure 6. TPC downregulation affects autophagy progression in neonatal rat cardiomyocytes .
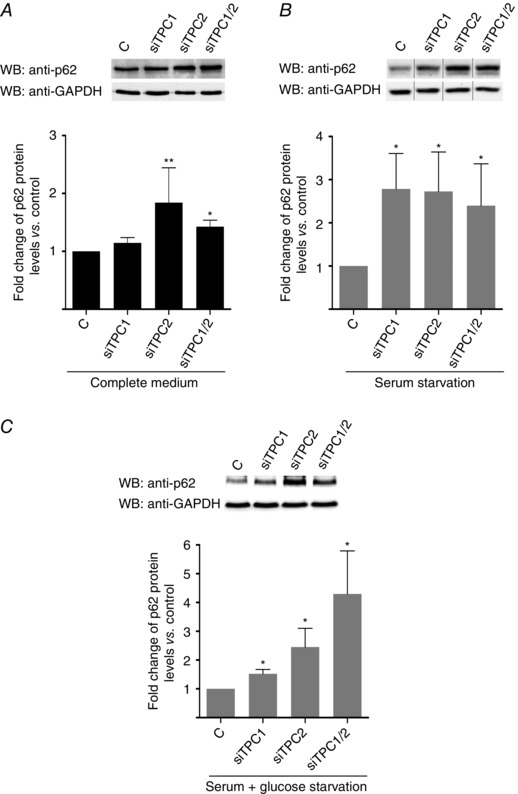
Downregulation of TPC2 and TPC1/2 induces accumulation of p62 under basal and starvation conditions (A to C; n = 5). Downregulation of TPC1 induces accumulation of p62 only after starvation (B and C; n = 5). Bar graphs show the mean ± SEM. Statistical significance (Mann–Whitney U test): *P ≤ 0.05; **P ≤ 0.01.
In addition, in TPC1/2 double knockout mice, the number of lysosomes in cardiac tissue was greater, and their mean diameter smaller, compared to wild‐type mice (Fig. 7).
Figure 7. Electron micrographs of cardiac tissue from TPC1/2 KO and wild‐type mice .

A, electron micrographs show increase in lysosome number and decrease in lysosome diameters in TPC1/2 KO vs. wild‐type mice. B, distribution of number vs. diameter of lysosomes (left) and statistical analysis of lysosome number (right) in TPC1/2 KO vs. wild‐type mice (n = 63 cardiomyocytes). Statistical significance (Mann–Whitney U test): *P ≤ 0.05.
The knockdown of TPCs decreases the viability of cardiomyocytes under ischaemia and starvation
To assess whether the absence of TPCs could affect cardiomyocyte viability under conditions of ischaemia or serum starvation (both of them comprising conditions for which an autophagic response has been reported in these cells; Lee et al. 2012; Shi et al. 2014), neonatal rat cultured cardiomyocytes, in which TPC1 or TPC2, or both, were knocked down using siRNAs, were subjected to in vitro simulated ischaemia for 6 h or to serum deprivation for 24 h. In vitro simulated ischaemia for 6 h severely decreased the viability (assessed by MTT) of cardiomyocytes transfected with siRNA negative control by 68.9 ± 2% vs. normoxic complete culture medium (P ≤ 0.001, n = 6), whereas serum deprivation for 24 h induced a moderate decrease in cardiomyocyte viability of 29.7 ± 5.7% vs. complete culture medium (P ≤ 0.01, n = 6), and all data were in agreement with previous studies (Kuzman et al. 2005; Xie et al. 2014; Xu et al. 2015). The silencing of TPC1 or TPC2, or both, did not affect the viability of neonatal rat cardiomyocytes after 6 h of normoxic nutrient‐rich conditions (data not shown); however, when cardiomyocytes were cultured under conditions of simulated ischaemia for 6 h, the silencing of TPCs significantly decreased cell viability by 20.2 ± 5.1% for TPC1, by 25.1 ± 6.2% for TPC2 and by 33.6 ± 4.7% for both (P ≤ 0.01, n = 8) vs. controls (cardiomyocytes subjected to simulated ischaemia and transfected with a siRNA negative control with no homology to known gene sequences) (Fig. 8 A). In cardiomyocytes subjected to serum deprivation for 24 h, the knockdown of TPC2, with or without TPC1, also decreased additionally cell viability by 10.5 ± 2.6% and 12.1 ± 2.2% (P ≤ 0.01, n = 8), respectively, vs. controls (cardiomyocytes subjected to serum deprivation and transfected with a siRNA negative control with no homology to known gene sequences) (Fig. 8 B).
Figure 8. The knockdown of TPCs decreases the viability of cardiomyocytes under ischaemia and starvation .
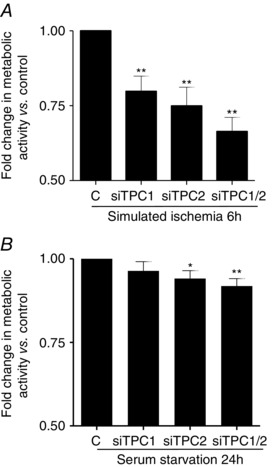
A, statistical analysis of MTT assays (n = 8) showing that, when cardiomyocytes were cultured under conditions of simulated ischaemia for 6 h, the silencing of TPCs significantly decreased cell viability vs. controls (cardiomyocytes subjected to simulated ischaemia and transfected with a siRNA negative control with no homology to known gene sequences). B, statistical analysis of MTT assays (n = 8) confirming that, in cardiomyocytes subjected to serum deprivation for 24 h, knockdown of TPC2, with or without TPC1, also decreased cell viability vs. controls. Statistical significance (Mann–Whitney U test): *P ≤ 0.05; **P ≤ 0.01.
The sex of an animal influences TPC1 and TPC2 levels and markers of autophagy in human and rat cardiac tissues
Because autophagy may impact cardiovascular diseases and exhibit a sex bias (Oliván et al. 2014), we considered whether TPCs and autophagy markers were differentially expressed in male and female hearts. We found that TPC1 and TPC2 mRNA and protein levels were significantly higher in the hearts of women and female rats than in those of men and male rats (Figs 9 A and B and 10 A and B). No other demographic, anthropometric, biochemical or pharmacological parameters in the patients included in the study correlated with TPCs levels. Interestingly, LC3II, LC3I and p62 levels were higher in the hearts of men and male rats than in those of women and female rats, respectively, although the LC3II/LC3I ratio was not significantly different (Figs 9 C and 10 C). Both in rat and human hearts, there was a striking negative correlation between p62 and transcript abundance of either TPC1 or TPC2 (Figs 9 D and 10 D). However, we could not find any significant influence of sex in the cardiac protein levels of the Rab GTP‐ase Rab7 and the lysosomal associated membrane protein LAMP‐1 (biomarker of lysosomes) (Figs 9 E and 10 E), which are essential proteins in the maturation of phagosomes (Harrison et al. 2003), and in the fusion of mature autophagosomes with lysosomes (Eskelinen, 2006; Campesi et al. 2013), respectively.
Figure 9. Sex‐dependence of cardiac TPCs levels and autophagy markers in cardiac surgery patients .
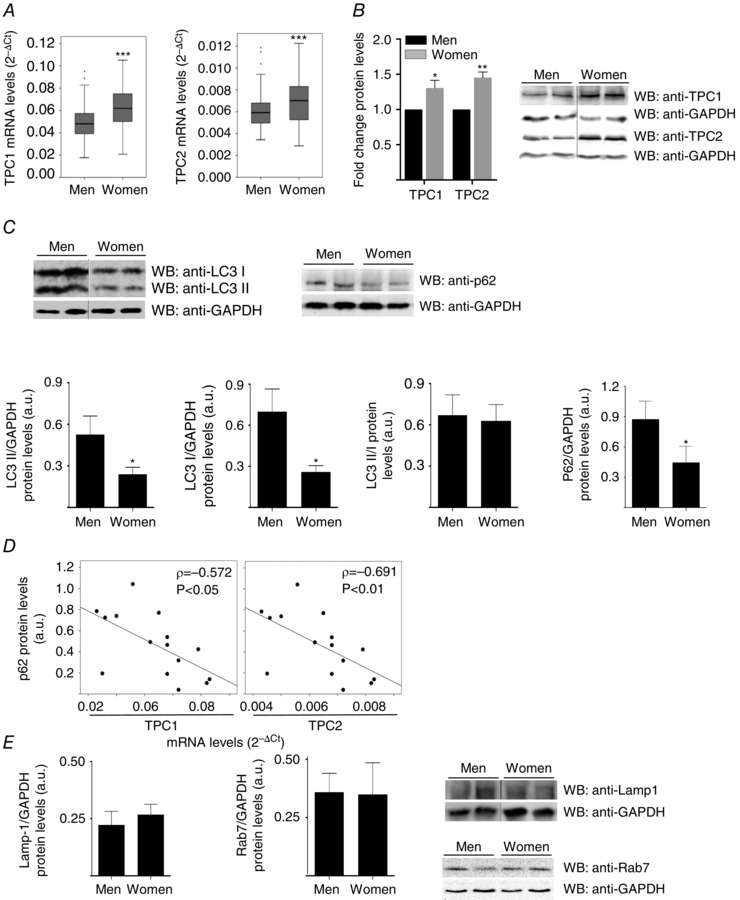
TPC1 and TPC2 transcripts are increased in women vs. men (A: women: n = 100; men: n = 199) hearts. Western blot confirmed increases of TPCs protein levels (B: women: n = 11; men: n = 11) in women hearts. C, human cardiac protein levels of LC3I (women: n = 14; men: n = 16), LC3II (women: n = 14; men: n = 16) and p62 (women: n = 10, men: n = 9), but not the LC3II/LC3I ratio (women: n = 14, men: n = 16), are lower in women than in men. D, negative correlation between p62 and TPCs transcripts in human hearts (n = 15). E, absence of a significant influence of sex in the cardiac protein levels of the Rab GTP‐ase Rab7 (women: n = 8; men: n = 8) and of the lysosomal associated membrane protein LAMP‐1 (women: n = 8; men: n = 8). Bar graphs show the mean ± SEM or median with interquartile range. The correlation coefficients were measured as the Spearman ρ. Statistical significance (Mann–Whitney U test): *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001. ρ, Spearman's rho.
Figure 10. Sex‐dependence of cardiac TPCs levels and autophagy markers in rats .
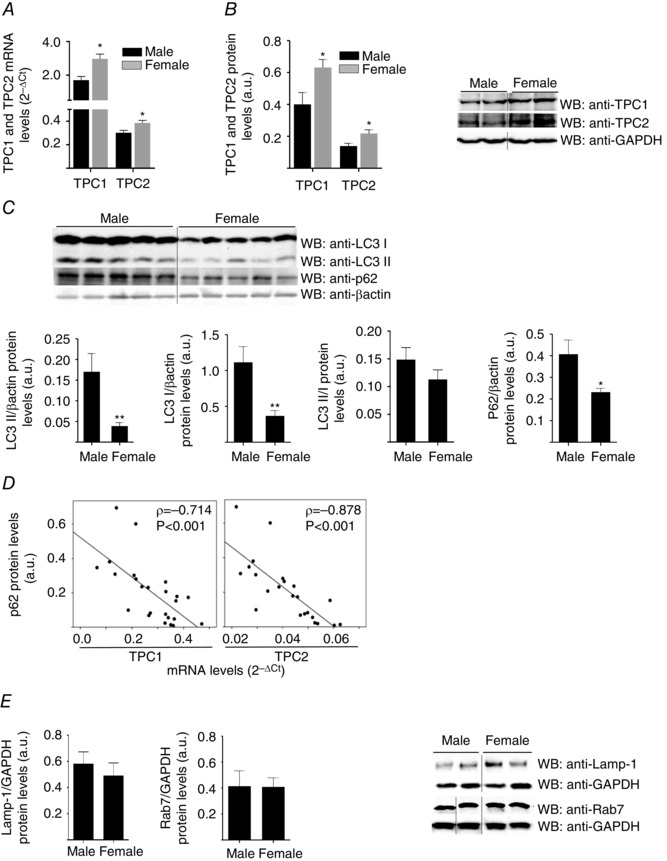
TPC1 and TPC2 transcripts are increased in females vs. males in rat (A: female: n = 9; male n = 9) hearts. Western blot confirmed increases of TPCs protein levels in rat (B: female: n = 7; male: 7) hearts. C, rat cardiac protein levels of LC3I, LC3II and p62, but not the LC3II/LC3I ratio, are lower in females (n = 7) than in males (n = 7). D, negative correlation between p62 and TPCs transcripts in rat hearts (n = 23). E, absence of a significant influence of sex in the cardiac protein levels of the Rab GTP‐ase Rab7 and of the lysosomal associated membrane protein LAMP‐1 (female: n = 10; male: n = 10). Bar graphs show the mean ± SEM or median with interquartile range. The correlation coefficients were measured as Spearman ρ. Statistical significance (Mann–Whitney U test): *P ≤ 0.05; **P ≤ 0.01; ρ, Spearman's rho.
Discussion
The results of the present study show that, in cultured cardiomyocytes, starvation induces a significant increase in TPC1 and TPC2 transcripts and protein levels that paralleled the increase in autophagic flux. In addition, siRNA depletion of TPC2 alone or together with TPC1 increases both LC3II and p62 levels under basal conditions and in response to serum starvation, suggesting a change in the autophagic process that, under conditions of severe energy depletion, is observed either when TPC1 or TPC2 are downregulated. Electron micrographs of cardiac tissue from TPC1/2 double knockout mice show that cardiomyocytes contain large numbers of immature lysosomes with diameters significantly smaller than those of wild‐type mice. Furthermore, the knockdown of TPCs decreases the viability of cardiomyocytes under ischaemia and starvation. Interestingly, in cardiac tissue from both humans and rats, TPC1 and TPC2 transcripts and protein levels are higher in females than in males, and transcript levels of both TPC1 and TPC2 correlate negatively with p62 levels in heart tissues.
Autophagy is an intracellular catabolic process in which damaged proteins and organelles are sequestered in autophagosomes and are degraded after fusion of the latter with endosomes and lysosomes, freeing amino acids and fatty acids for re‐use (Lavandero et al. 2013). Recent evidence reveals that cells constantly monitor lysosomal function and possess the ability to increase the number and activity of lysosomes in response to their energetic or degradative needs (Puertollano, 2014), upregulating the expression of critical regulators of the autophagic process, including several proteins implicated in the formation of autophagosomes as well as proteins required for the fusion between autophagosomes and lysosomes (Puertollano, 2014). With regard to this, two‐pore channels have been recently defined as intracellular Na+ ‐selective channels able to control endolysosomal fusion, and the result of some studies have suggested a potential regulatory role for these proteins in autophagic processes (Pereira et al. 2011; Neely Kayala et al. 2012; Lu et al. 2013; Lin et al. 2015). Interestingly, Cang et al. (2013) have suggested that TPCs could couple the cell's metabolic state to endolysosomal function: under nutrient‐ and ATP‐rich conditions, mTORC1 could bind and inhibit the activity of an endolysosomal ATP‐sensitive Na+ channel formed by the two‐pore channels TPC1 and TPC2 but, when the levels of ATP are reduced, mTORC1 redistributes to the cytosol, allowing the opening of the channel and the release of Na+ and other ions from the lumen of the lysosome to the cytosol. This depolarization of the lysosomal membrane may potentially affect many different lysosomal parameters such as fusion of lysosomes with autophagosomes or transport of nutrients. In summary, TPCs could emerge as key regulators of nutrient signalling and energy homeostasis. Furthermore, TPCs have also been identified as a family of endolysosomal NAADP‐mediated Ca2+ release channels that could control the intraluminal pH of acidic stores, and it was suggested that TPC2 might be involved in regulating the optimal functional conditions for lysosomal enzymes (Zhu et al. 2010).
In the present study, we show that, in response to starvation of either human or murine cardiomyocytes, there is an increase in the abundance of TPC proteins most probably driven by an increase in gene expression. This occurs in parallel with well‐known early steps in autophagy induction (AMPK activation, mTOR dephosphorylation, increased conjugation of LC3 to phosphatidylethanolamine to form LC3II, recruitment of LC3II to autophagosomal membranes) and a subsequent decrease in p62. Protein p62 is involved in the assembly of autophagosome cargoes (Jiang & Mizushima, 2014). Under basal conditions, p62 levels are themselves strictly regulated by autophagy, the impairment of which results in the massive accumulation of this protein: when autophagy is upregulated during starvation, p62 levels fall rapidly (Jiang & Mizushima, 2014).
Upregulation of TPCs in concert with autophagic activity could reflect co‐ordinate regulation or direct involvement of these molecules in the autophagic process. Indeed, we found that, under nutrient‐rich conditions, silencing of TPC2, with or without TPC1 knockdown, significantly increased LC3II and p62 (both of which degraded after fusion of autophagosomes to lysosomes; Jiang & Mizushima, 2014) and formation of GFP‐LC3 puncta, suggesting that TPC2 carries out a function important for maintaining normal autophagic function even in basal states. Silencing TPC1 or TPC2, or both, caused a significant accumulation of p62 under starvation conditions, which is a sign of impaired autophagic flux (Jiang & Mizushima, 2014) (as confirmed by the experiments using the lysosomal inhibitor bafilomycin to monitor LC3 turnover), suggesting that, under induced conditions, each protein has a unique role, and is also consistent with increased expression of both under inducing conditions.
The differential effects of TPC1 and TPC2 knockdown are probably related to differences in their sites of action. TPC1 is localized to mature endosomes, whereas TPC2 is associated with lysosomes. Our finding that starvation‐induced autophagy is dependent on both is in accordance with growing evidence indicating that autophagosomes fuse with both late and early endosomes (Szatmári et al. 2014); however, our results also suggest that, under basal conditions, autophagy can proceed without endosomal fusion or via a route involving only early endosomes.
Cardiomyocyte lysosomes of double TPC1/2 knockout mice were greater in number but smaller, resembling immature lysosomes (Guha & Padh, 2008) and consistent with a defect in vesicle fusion. In agreement with these results, Neely Kayala et al. (2012) showed that, in presenilin‐null fibroblastsm, altered TPC expression is associated with an increased number of lysosomes but a reduction in autophagy function.
To determine whether the absence of TPCs could affect cardiomyocyte viability under conditions of ischaemia or serum starvation (both of them comprising conditions for which an autophagic response has been reported in these cells; Lee et al. 2012; Shi et al. 2014), neonatal rat cultured cardiomyocytes in which TPC1 or TPC2, or both, were knocked down using siRNAs, were subjected to in vitro simulated ischaemia for 6 h (a condition that severely affects cardiomyocyte viability; Xie et al. 2014; Xu et al. 2015) or to serum deprivation for 24 h (a condition that affects moderately cardiomyocyte viability; Kuzman et al. 2005). The silencing of TPC1 or TPC2, or both, did not modify the viability of neonatal rat cardiomyocytes in normoxic nutrient‐rich conditions; however, when cardiomyocytes were cultured under conditions of simulated ischaemia or serum starvation, the silencing of TPCs significantly decreased cell viability. In recent years, several studies have reported that autophagy plays a fundamentally beneficial role during energy deprivation and ischaemia in the heart, defending it against metabolic stress and exerting cytoprotective effects (Sciarretta et al. 2014; Nishida et al. 2015; Wei et al. 2015), although it has also been suggested that excessive autophagy at cardiac level could be harmful in several pathological circumstances (Godar et al. 2015; Lavandero et al. 2015; Munasinghe et al. 2016). In our work, deregulated autophagy (consequence of TPCs silencing) implies a decrease in the viability of cardiomyocytes under starvation conditions, reinforcing the concept that the cardioprotection of autophagy is context‐dependent (Xu et al. 2015).
Our finding that basal TPC1 and TPC2 levels are higher in females than in males is the first report of a sex‐dependent difference in cardiac TPCs expression, and is in keeping with the observed sex differences in p62. However, we could not identify any differences between sexes in the cardiac levels of other two proteins that regulate the point where autophagosomes and endosomes fuse with lysosomes to permit the final degradation of their contents in the autophagy process, and thus are critical for the autophagy flux: the Rab GTP‐ase Rab7 (whose recruitment to phagosomes was found to precede and be essential for their fusion with late endosomes and/or lysosomes; Hyttinen et al. 2013) and the lysosomal marker LAMP‐1 (in agreement with the results from Campesi et al. 2013). Sex‐dependence of autophagy has previously been observed in vitro (Oliván et al. 2014), although the nature of the sex effect can differ depending upon cell type, tissue and organism (Koenig et al. 2014). At the cardiac level, the existence of sex differences in autophagy‐related gene expression was proposed and it was demonstrated that glycogen autophagy is more sensitive to metabolic stress in the hearts of female mice (Reichelt et al. 2013, Koenig et al. 2014 ). The results of the present study suggest a higher constitutive cardiomyocyte autophagy in females. It is generally recognized that women typically develop heart disease later than men and have a better prognosis (Lista et al. 2011). Whether autophagy could play a role in the sex differences detected in heart disease is still largely unknown; however, several studies have suggested the possible implication of hormones (especially androgens and oestrogens) in the modulation of autophagy at the cellular level, and it has been already demonstrated that female vascular smooth muscle cells exhibit a higher propensity to undergo autophagy and survival under unfavourable environmental conditions than those from men (Lista et al. 2011). Because the upregulation of autophagy in a diseased heart could be an adaptive protective response against haemodynamic or neurohormonal stresses, further work on the role of TPCs in those differences, as well as their implications for between‐sex differences in cardiac physiology and disease, is now warranted.
Additional information
Competing interests
The authors declare that they have no competing interests.
Funding
This work was supported by Carlos III Health Institute (FIS11/00497, Spain) and National Institutes of Health (R01 HL097768 USA). Diego Rodríguez‐Penas is funded by the International PhD School, Santiago de Compostela University; Vanessa García‐Rúa is funded by the University Professional Development Program (FPU) of the Spanish Ministry of Education; Sandra Feijóo‐Bandín is funded by the IDICHUS Research Foundation of Santiago de Compostela University Clinical Hospital; Ana Mosquera‐Leal is funded by the Health Research Fund of the Carlos III Health Institute; and Francisca Lago is funded by SERGAS.
Author contributions
VG‐R, VP, BAR and FL were responsible for the study conception and design. VG‐R, SF‐B, DR‐P, AM‐L and OG were responsible for the collection and assembly of data. VG‐R, SF‐B, DR‐P, AM‐L, AB, OG, VP, JH, BAR and FL were responsible for data analysis and interpretation. VG‐R, SF‐B, DR‐P, AM‐L, EA‐A, AB, LS, PL, JP, MP, ER‐L, MR, VP, JH, BAR, JG‐J and FL were responsible for writing the manuscript. VG‐R, SF‐B, DR‐P, AM‐L, EA‐A, AB, LS, PL, JP, MP, ER‐L, MR, OG, VP, JH, BAR, JG‐J and FL were responsible for the approval of the manuscript. EA‐A, OG, JH, BAR, JG‐J and FL were responsible for financial support. EA‐A, JH, BAR, JG‐J and FL were responsible for administrative support. LS, PL, JP, MP, ER‐L, MR and JG‐J were responsible for the provision of study materials and patients. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
References
- Barth S, Glick D & Macleod KF (2010). Autophagy: assays and artifacts. J Pathol 221, 117–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calcraft PJ et al (2009). NAADP mobilizes calcium from acidic organelles through two‐pore channels. Nature 459, 596–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campesi I, Straface E, Occhioni S, Montella A & Franconi F (2013). Protein oxidation seems to be linked to constitutive autophagy: a sex study. Life Sci 93, 145–152. [DOI] [PubMed] [Google Scholar]
- Cang C, Zhou Y, Navarro B, Seo Y‐J, Aranda K, Shi L, Battaglia‐Hsu S, Nissim I, Clapham DE & Ren D (2013). mTOR regulates lysosomal ATP‐sensitive two‐pore Na(+) channels to adapt to metabolic state. Cell 152, 778–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskelinen EL (2006). Roles of LAMP‐1 and LAMP‐2 in lysosome biogenesis and autophagy. Mol Aspects Med 27, 495–502. [DOI] [PubMed] [Google Scholar]
- García‐Rúa V, Otero MF, Lear PV, Rodríguez‐Penas D, Feijóo‐Bandín S, Noguera‐Moreno T, Calaza M, Álvarez‐Barredo M, Mosquera‐Leal A, Parrington J, Brugada J, Portolés M, Rivera M, González‐Juanatey JR & Lago F (2012). Increased expression of fatty‐acid and calcium metabolism genes in failing human heart. PLoS ONE 7, e37505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godar RJ, Ma X, Liu H, Murphy JT, Weinheimer CJ, Kovacs A, Crosby SD, Saftig P & Diwan A (2015). Repetitive stimulation of autophagy‐lysosome machinery by intermittent fasting preconditions the myocardium to ischemia‐reperfusion injury. Autophagy 11, 1537–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González‐Juanatey JR, Iglesias MJ, Alcaide C, Piñeiro R & Lago F (2003). Doxazosin induces apoptosis in cardiomyocytes cultured in vitro by a mechanism that is independent of alpha1‐adrenergic blockade. Circulation 107, 127–131. [DOI] [PubMed] [Google Scholar]
- Guha S & Padh H (2008). Cathepsins: fundamental effectors of endolysosomal proteolysis. Indian J Biochem Biophys 45, 75–90. [PubMed] [Google Scholar]
- Harrison RE, Bucci C, Vieira OV, Schroer TA & Grinstein S (2003). Phagosomes fuse with late endosomes and/or lysosomes by extension of membrane protrusions along microtubules: role of Rab7 and RILP. Mol Cell Biol 23, 6494–6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyttinen JM, Niittykoski M, Salminen A & Kaarniranta K (2013). Maturation of autophagosomes and endosomes: a key role for Rab7. Biochim Biophys Acta 1833, 503–510. [DOI] [PubMed] [Google Scholar]
- Jiang P & Mizushima N (2014). LC3‐ and p62‐based biochemical methods for the analysis of autophagy progression in mammalian cells. Methods 75, 13–18. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Elazar Z, Seglen PO & Rubinsztein DC (2008). Does bafilomycin A1 block the fusion of autophagosomes with lysosomes? Autophagy 4, 849–850. [DOI] [PubMed] [Google Scholar]
- Koenig A, Sateriale A, Budd RC, Huber SA & Buskiewicz IA (2014). The role of sex differences in autophagy in the heart during coxsackievirus B3‐induced myocarditis. J Cardiovasc Transl Res 7, 182–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzman JA, Gerdes AM, Kobayashi S & Liang Q (2005). Thyroid hormone activates Akt and prevents serum starvation‐induced cell death in neonatal rat cardiomyocytes. J Mol Cell Cardiol 39, 841–844. [DOI] [PubMed] [Google Scholar]
- Lavandero S, Troncoso R, Rothermel BA, Martinet W, Sadoshima J & Hill JA (2013). Cardiovascular autophagy: concepts, controversies, and perspectives. Autophagy 9, 1455–1466. [DOI] [PubMed] [Google Scholar]
- Lavandero S, Chiong M, Rothermel BA & Hill JA (2015). Autophagy in cardiovascular biology. J Clin Invest 125, 55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lear P V, González‐Touceda D, Porteiro Couto B, Viaño P, Guymer V, Remzova E, Tunn R, Chalasani A, García‐Caballero T, Hargreaves IP, Tynan PW, Christian HC, Nogueiras R, Parrington J & Diéguez C (2015). Absence of intracellular ion channels TPC1 and TPC2 leads to mature‐onset obesity in male mice, due to impaired lipid availability for thermogenesis in brown adipose tissue. Endocrinology 156, 975–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lear P V, Iglesias MJ, Feijóo‐Bandín S, Rodríguez‐Penas D, Mosquera‐Leal A, García‐Rúa V, Gualillo O, Ghè C, Arnoletti E, Muccioli G, Diéguez C, González‐Juanatey JR & Lago F (2010). Des‐acyl ghrelin has specific binding sites and different metabolic effects from ghrelin in cardiomyocytes. Endocrinology 151, 3286–3298. [DOI] [PubMed] [Google Scholar]
- Lee Y, Lee HY & Gustafsson AB (2012). Regulation of autophagy by metabolic and stress signaling pathways in the heart. J Cardiovasc Pharmacol 60, 118–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin P‐H, Duann P, Komazaki S, Park KH, Li H, Sun M, Sermersheim M, Gumpper K, Parrington J, Galione A, Evans AM, Zhu MX & Ma J (2015). Lysosomal two‐pore channel subtype 2 (TPC2) regulates skeletal muscle autophagic signaling. J Biol Chem 290, 3377–3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lista P, Straface E, Brunelleschi S, Franconi F & Malorni W (2011). On the role of autophagy in human diseases: a gender perspective. J Cell Mol Med 15, 1443‐1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Hao B‐X, Graeff R, Wong CWM, Wu W‐T & Yue J (2013). Two pore channel 2 (TPC2) inhibits autophagosomal‐lysosomal fusion by alkalinizing lysosomal pH. J Biol Chem 288, 24247–24263. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B & Sadoshima J (2007). Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP‐activated protein kinase and Beclin 1 in mediating autophagy. Circ Res 100, 914–922. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T & Levine B (2010). Methods in mammalian autophagy research. Cell 140, 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munasinghe PE, Riu F, Dixit P, Edamatsu M, Saxena P, Hamer NS, Galvin IF, Bunton RW, Lequeux S, Jones G, Lamberts RR, Emanueli C, Madeddu P & Katare R (2016). Type‐2 diabetes increases autophagy in the human heart through promotion of Beclin‐1 mediated pathway. Int J Cardiol 202, 13‐20. [DOI] [PubMed] [Google Scholar]
- Neely Kayala KM, Dickinson GD, Minassian A, Walls KC, Green KN & Laferla FM (2012). Presenilin‐null cells have altered two‐pore calcium channel expression and lysosomal calcium: implications for lysosomal function. Brain Res 1489, 8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida K, Taneike M & Otsu K (2015). The role of autophagic degradation in the heart. J Mol Cell Cardiol 78, 73–79. [DOI] [PubMed] [Google Scholar]
- Oliván S, Calvo AC, Manzano R, Zaragoza P & Osta R (2014). Sex differences in constitutive autophagy. Biomed Res Int 2014, 652817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira GJS, Hirata H, Fimia GM, do Carmo LG, Bincoletto C, Han SW, Stilhano RS, Ureshino RP, Bloor‐Young D, Churchill G, Piacentini M, Patel S & Smaili SS (2011). Nicotinic acid adenine dinucleotide phosphate (NAADP) regulates autophagy in cultured astrocytes. J Biol Chem 286, 27875–27881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puertollano R (2014). mTOR and lysosome regulation. F1000Prime Rep 6, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichelt ME, Mellor KM, Curl CL, Stapleton D & Delbridge LMD (2013). Myocardial glycophagy – a specific glycogen handling response to metabolic stress is accentuated in the female heart. J Mol Cell Cardiol 65, 67–75. [DOI] [PubMed] [Google Scholar]
- Ruas M, Chuang K‐T, Davis LC, Al‐Douri A, Tynan PW, Tunn R, Teboul L, Galione A & Parrington J (2014). TPC1 has two variant isoforms, and their removal has different effects on endo‐lysosomal functions compared to loss of TPC2. Mol Cell Biol 34, 3981–3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciarretta S, Yee D, Shenoy V, Nagarajan N & Sadoshima J (2014). The importance of autophagy in cardioprotection. High Blood Press Cardiovasc Prev 21, 21–28. [DOI] [PubMed] [Google Scholar]
- Shi C, Wu J, Fu M, Zhang B, Wang J, Yang X & Chi Y (2014). Ambra1 modulates starvation‐induced autophagy through AMPK signaling pathway in cardiomyocytes. Biochem Biophys Res Commun 452, 308–314. [DOI] [PubMed] [Google Scholar]
- Szatmári Z, Kis V, Lippai M, Hegedus K, T Faragó, Lorincz P, Tanaka T, Juhász G & Sass M (2014). Rab11 facilitates cross‐talk between autophagy and endosomal pathway through regulation of Hook localization. Mol Biol Cell 25, 522–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhang X, Dong X‐P, Samie M, Li X, Cheng X, Goschka A, Shen D, Zhou Y, Harlow J, Zhu MX, Clapham DE, Ren D & Xu H (2012). TPC proteins are phosphoinositide‐activated sodium‐selective ion channels in endosomes and lysosomes. Cell 151, 372–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei C, Gao J, Li M, Li H, Wang Y, Li H & Xu C (2015). Dopamine D2 receptors contribute to cardioprotection of ischemic post‐conditioning via activating autophagy in isolated rat hearts. Int J Cardiol 203, 837–839. [DOI] [PubMed] [Google Scholar]
- Xie M, Kong Y, Tan W, May H, Battiprolu PK, Pedrozo Z, Wang ZV, Morales C, Luo X, Cho G, Jiang N, Jessen ME, Warner JJ, Lavandero S, Gillette TG, Turer AT, & Hill JA (2014). Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation 129, 1139–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Li X, Lu Y, Shen L, Zhang J, Cao S, Huang X, Bin J & Liao Y (2015). Pharmacological modulation of autophagy to protect cardiomyocytes according to the time windows of ischaemia/reperfusion. Br J Pharmacol 172, 3072–3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA & Hill JA (2007). Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest 117, 1782–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu MX, Ma J, Parrington J, Calcraft PJ, Galione A & Evans a M (2010). Calcium signaling via two‐pore channels: local or global, that is the question. Am J Physiol Cell Physiol 298, C430–C441. [DOI] [PMC free article] [PubMed] [Google Scholar]