

Abstract
Studies over the past four decades have established that Ca2+ is a critical factor in control of salivary gland function and have led to identification of the critical components of this process. The major ion transport mechanisms and ion channels that are involved in fluid secretion have also been established. The key event in activation of fluid secretion is an increase in [Ca2+]i triggered by inositol 1,4,5‐trisphosphate (IP3)‐induced release of Ca2+ from ER via the IP3 receptor (IP3R). IP3Rs determine the site of initiation and the pattern of the [Ca2+]i signal in the cell. However, Ca2+ entry into the cell is required to sustain the elevation of [Ca2+]i and fluid secretion and is mediated by the store‐operated Ca2+ entry (SOCE) mechanism. Orai1, TRPC1, TRPC3 and STIM1 have been identified as critical components of SOCE in these cells. Cells finely tune the generation and amplification of [Ca2+]i signals for regulation of cell function. An important emerging area is the concept that unregulated [Ca2+]i signals in cells can directly cause cell damage, dysfunction and disease. Alternatively, aberrant [Ca2+]i signals can also amplify and increase the rates of cell damage. Such defects in Ca2+ signalling have been described in salivary glands in conjunction with radiation‐induced loss of salivary gland function as well as in the salivary defects associated with the autoimmune exocrinopathy Sjögren's syndrome. Such defects have been associated with altered function or expression of key Ca2+ signalling components, such as STIM proteins and TRP channels. These studies offer new avenues for examining the mechanisms underlying the disease and development of novel clinical targets and therapeutic strategies.
Abbreviations
- [Ca2+]i
cytosolic [Ca2+]
- CaSR
calcium‐sensing receptor
- ER
endoplasmic reticulum
- IP3
inositol 1,4,5‐trisphosphate
- IP3R
inositol trisphosphate receptor
- IR
irradiation
- NFAT
nuclear factor of activated T cells
- PIP2
phosphatidylinositol 4,5‐bisphosphate
- PM
plasma membrane
- pSS
primary Sjögren's syndrome
- ROS
reactive oxygen species
- SOCE
store‐operated calcium entry
- STIM1
stromal interacting molecule
- TRP
transient receptor potential
- TRPC
transient receptor potential canonical
Introduction
Salivary glands secrete fluid and proteins in response to specific neurotransmitter‐generated intracellular signals in acinar cells, which are the primary site of both types of secretion. Stimulation of β‐adrenergic receptors leads to 3′,5′‐cyclic adenosine monophosphate (cyclic AMP) generation, which activates exocytosis and protein secretion. In contrast, Ca2+ signals are induced in response to stimulation of plasma membrane (PM) receptors coupled to phospholipase C (PLC) activation and phosphatidylinositol 4,5‐bisphosphate (PIP2) hydrolysis, such as muscarinic cholinergic and α1‐adrenergic receptors, and result in activation of fluid secretion. The key trigger for stimulation of fluid secretion is an increase in cytosolic [Ca2+] ([Ca2+]i), which regulates ion channel activities that generate the appropriate osmotic gradient required to drive fluid secretion across the apical membrane (Melvin et al. 2005; Ambudkar, 2011, 2012). Physiologically, increase in [Ca2+]i in salivary acinar cells is initiated in response to inositol 1,4,5, trisphosphate (IP3)‐mediated release of Ca2+ from the endoplasmic reticulum (ER) Ca2+ store(s) via the inositol trisphosphate receptor (IP3R), a well characterized intracellular Ca2+‐release channel. The major subtypes of IP3Rs found in salivary gland cells are IP3R2 and ‐3, both of which are concentrated in the apical region of the acinar cells (Yule, 2001; Mikoshiba et al. 2008; Petersen & Tepikin, 2008). The apically localized IP3Rs account for the initial increase in [Ca2+]i detected in this region following stimulation. [Ca2+]i then increases in a wave‐like pattern from the apical to the basolateral areas of the cell (Yule, 2001; Melvin et al. 2005). This global increase in [Ca2+]i is essential for the coordinated regulation of various ion channels and transporters located in the apical and basolateral membrane regions of the cell (Melvin et al. 2005; Ambudkar, 2011, 2012).
The critical contribution of extracellular Ca2+ and Ca2+ entry in the regulation of fluid secretion was established more than a decade ago (Putney, 1986; Lee et al. 1997; Melvin et al. 2005; Ambudkar, 2011, 2012, 2014). IP3‐mediated Ca2+ release via IP3R, and resulting depletion of ER Ca2+, is the main triggering event in the activation of Ca2+ entry, which is mediated by the store‐operated Ca2+ entry (SOCE) mechanism (Putney, 1990). SOCE is achieved by the gating of plasma membrane Ca2+ channels, e.g. Orai1 and TRPC1, by STIM1, a Ca2+ binding protein that functions as the ER [Ca2+] sensor (Liou et al. 2005; Zhang et al. 2005; Zeng et al. 2008; Yuan et al. 2009; Hogan et al. 2010; Cheng et al. 2013; Prakriya, 2013). TRPC1 is an essential channel for salivary gland function, and lack of the channel results in significant loss of fluid secretion and SOCE (Liu et al. 2000, 2007; Hong et al. 2011). While Orai1 has been extensively studied, its exact role in salivary gland function is not yet known. One possible function of Orai1 in the gland could be to regulate TRPC1 function since studies with salivary gland cell lines have demonstrated that TRPC1 function is completely dependent on Orai1 (Cheng et al. 2008, 2011; Lee et al. 2010; Choi et al. 2014).
[Ca2+]i signals are a double‐edged sword: while [Ca2+]i increases are essential for the regulation of cell function, maintenance of Ca2+ homeostasis both in resting and stimulated cells is absolutely critical for cell survival. Unregulated increases in cytosolic Ca2+, due to release from either ER or other intracellular stores, or due to Ca2+ entry, can be extremely deleterious to cells (Berridge, 2012). Deficits in, or aberrant, Ca2+ signalling have been associated with cellular and functional damage in a number of cell types, including neuronal cells and lymphocytes. Two major conditions result in loss of salivary gland function and tissue damage, although loss of fluid secretion does not always correlate with loss of glandular tissue. The first condition is radiation‐induced xerostomia, or dry mouth condition, which occurs in patients who undergo radiation therapy for head‐and‐neck cancers that results in irreversible loss of salivary fluid secretion. This condition has been reproduced in several animal models, such as mouse, rats, mini‐pigs and non‐human primates. Interestingly, irradiation (IR) induces considerable loss of saliva flow in the absence of extensive tissue damage or loss of acinar cells. Fibrosis and loss of tissue occur subsequent to the functional loss, and the onset and severity of this phase of cellular damage differs among the various species. Irrespective of the progression of fibrosis, loss of salivary fluid secretion leads to xerostomia and associated complications such as difficulty swallowing, rampant dental caries, oral mucosal lesions and fungal infections that together severely affect the quality of life for the patients (Vissink et al. 2003; Delli et al. 2014). Thus, the mechanism involved in IR‐induced loss of salivary gland function is a subject of great interest in the field, with clinical studies being directed towards assessing therapies targeted to recovery of cell function, prevention of functional loss, or regrowth of salivary glands.
A second condition associated with progressive loss of salivary gland function is primary Sjögren's syndrome (pSS), a chronic autoimmune disease involving lymphocytic infiltration and loss of secretory function in salivary and lacrimal glands (Delaleu et al. 2005). The pathogenesis of this disease has not yet been elucidated although viral, hormonal, environmental and neuronal factors have been implicated (Hansen et al. 2005; Nikolov & Illei, 2009; Mavragani & Moutsopoulos, 2014). A major question which remains to be addressed is the poor correlation between salivary flow and extent of inflammation or tissue damage (Fox & Stern, 2002; Nikolov & Illei, 2009). It has been reported that a large number of patients have low levels of inflammation within their salivary glands and yet display substantial loss of function. The molecular alterations underlying this secretory defect are not yet known.
In this review we will summarize the current knowledge regarding the role of Ca2+ signalling in salivary gland function, with discussion of animal models that display defects in fluid secretion. Further, we will discuss some current studies that describe the involvement of Ca2+ signalling in salivary gland dysfunction.
Ca2+ signalling in salivary gland function
Generation of cytosolic Ca2+ signals
In resting cells [Ca2+]i is maintained at around 50–100 nm, which is lower than the threshold required to activate the ion flux systems that drive fluid secretion (Melvin et al. 2005; Yang et al. 2008; Ambudkar, 2014). Following stimulation, the increase in [Ca2+]i activates the K+ and Cl− channels as well as the Na+–K+–2Cl− cotransporter and Na+/H+ and Cl−/HCO3 − exchangers (Manganel & Turner, 1990; Nguyen et al. 2004). Together, the concerted activities of these ion‐flux mechanisms result in transepithelial flux of Cl−. Importantly, the rise in [Ca2+]i also triggers recruitment of aquaporin AQP5 water channels into the apical plasma membrane, thus substantially increasing fluid secretion (Ishikawa et al. 1998). The key molecular determinants of [Ca2+]i signal generation include the G‐protein‐coupled receptor‐signalling complex and the IP3Rs (Ambudkar, 2012, 2014). Muscarinic–cholinergic receptors (M1/M3) are major receptors involved in the regulation of salivary gland fluid secretion (Proctor, 2006; Proctor & Carpenter, 2007) as shown by the almost complete loss of fluid secretion in mice lacking both receptors. In contrast, M3−/− mice have a secretory defect only at low pilocarpine levels and exhibit problems with chewing and ingesting dry food (Gautam et al. 2004; Nakamura et al. 2004). Consistent with this, carbachol‐induced [Ca2+]i increase was attenuated in submandibular gland cells from mice lacking M3 receptors and absent in those lacking both M1 and M3 receptors.
One of the best characterized Ca2+ channels is the IP3R in the ER (Mikoshiba, 2007; Mikoshiba et al. 2008). IP3R is regulated by both IP3 and Ca2+. Ca2+ regulation of IP3R function displays a bell‐shaped curve with stimulation at low and inhibition at high [Ca2+]. Furthermore, an increase in IP3 levels increases the [Ca2+] sensitivity of IP3R. This exquisite regulation of IP3R by IP3 and [Ca2+]i ensures that the channel is relatively more active when [Ca2+]i is low and less active when [Ca2+]i is high, thus protecting ER Ca2+ stores and regulating [Ca2+]i within the physiological range required for the cell function (Mikoshiba, 2007). This feed‐forward and feedback regulation of IP3R can also explain the spread of the Ca2+ wave from the apical to the basal membrane in salivary gland cells (Lee et al. 1997; Yule, 2001; Melvin et al. 2005). Two major isoforms, IP3R2 and IP3R3, are found in salivary gland acinar cells (Yule, 2001; Melvin et al. 2005; Mikoshiba et al. 2008), and mice lacking either subtype display decreases in [Ca2+]i mobilization, while those lacking both receptors demonstrate complete loss of agonist‐stimulated intracellular Ca2+ release as well as fluid secretion (Futatsugi et al. 2005). More importantly, these data suggest that in the absence of IP3Rs there is no significant level of Ca2+ entry that can support elevation of [Ca2+]i or fluid secretion. Thus, the Ca2+ entry mechanisms activated directly by PIP2 hydrolysis appear to be minimal in these cells. Several cellular factors and proteins modulate IP3R function including ATP, cAMP, RACK1 and the IP3R‐binding protein known as inositol 1,4,5‐trisphosphate receptor‐binding protein (IRBIT) (Ahuja et al. 2014) and contribute to modification of [Ca2+]i increase as well as secretion (Ambudkar, 2011; Ando et al. 2014). cAMP‐dependent phosphorylation of the IP3R increases the sensitivity of the receptors for [IP3]. One underlying mechanism that has been revealed is that cAMP‐dependent phosphorylation of IP3R induces release of IRBIT from IP3R, which not only increases IP3R function but also allows IRBIT‐dependent activation of ion transport in the plasma membrane. Thus the cross talk between cAMP and IP3R has important physiological implications as typically both cAMP and Ca2+ signalling can be simultaneously activated in acinar cells (Park et al. 2013; Ahuja et al. 2014).
Mechanisms and physiological relevance of store‐operated Ca2+ influx
As noted above early studies established that Ca2+ influx is the primary determinant of sustained fluid secretion from salivary acinar cells (Ambudkar, 2014). It is now widely accepted that the primary mode of Ca2+ entry in acinar cells that is required for fluid secretion is mediated by SOCE. Physiologically SOCE can be activated by any stimulus that leads to depletion of ER Ca2+, typically via IP3‐induced activation of IP3R (Putney, 1997; Parekh & Putney, 2005). SOCE is a ubiquitous mode of Ca2+ influx that in addition to regulation of salivary gland fluid secretion is required for key physiological functions in other cell types: protein secretion in pancreatic acinar cells, platelet aggregation, endothelial cell permeability and migration, cell proliferation, T‐lymphocyte activation and mast cell degranulation among others (Parekh & Putney, 2005; Ong et al. 2014).
The main molecular components involved in SOCE in salivary gland acinar cells have now been identified. Members of the transient receptor potential canonical (TRPC) family, TRPC1 and TRPC3, contribute to SOCE in dispersed acinar cell preparations as well as salivary gland cell lines (Ambudkar, 2014; Hong et al. 2014; Sun et al. 2015). These two TRPC channels generate relatively non‐selective, Ca2+‐permeable channels that are activated downstream from receptor‐stimulated PIP2 hydrolysis. Mice lacking TRPC1 or TRPC3 show reduced SOCE and fluid secretion (Liu et al. 2007; Kim et al. 2009). Interestingly, the contribution of TRPC3 to SOCE is dependent on the presence of TRPC1 as TRPC1−/− mice do not display TRPC3‐dependent SOCE (Lee et al. 2014). Thus, it has been proposed that either the channels are assembled as a store‐operated heteromeric channel or that TRPC1 is required for store‐dependent regulation of TRPC3. Indeed, TRPC3–TRPC1 interaction is necessary for STIM1 regulation of the channels in salivary gland ductal cells. The two TRPC channels coimmunoprecipitate following cell stimulation together with STIM1. Loss of TRPC1 eliminates the association of STIM1 with TRPC3 (Yuan et al. 2007; Lee et al. 2014). The role of TRPC3 in SOCE‐independent regulation of salivary gland function has not yet been identified.
STIM1 and Orai have been identified as essential components of SOCE (Hogan et al. 2010; Cheng et al. 2013; Prakriya, 2013). STIM1, an ER Ca2+‐binding protein, responds to reductions in ER [Ca2+] and undergoes substantial conformational changes that result in multimerization of the protein, extension of its C‐terminal domain, and translocation to the periphery of the cells where it clusters at specific ER–PM junctions. At these locations, STIM1 interacts with and activates Orai1 and TRPC1 (Wu et al. 2006; Liou et al. 2007; Luik et al. 2008; Muik et al. 2011; Zhou et al. 2013). While the SOAR domain of STIM1 activates Orai1, the C‐terminal polybasic residues 639KK640 of STIM1 are involved in activation of TRPC1 (Zeng et al. 2008; Yuan et al. 2009). Orai1 has been conclusively established as the main pore‐forming component of store‐operated calcium release activated Ca2+ (CRAC) channels, although its role in salivary gland function is yet to be determined (Prakriya et al. 2006, 2013). Orai1 function has been studied in two other exocrine glands, lacrimal and pancreatic. Orai1−/− mice display loss of lacrimal gland function and reduced SOCE in lacrimal gland acinar cells, another exocrine gland cell type (Xing et al. 2014). Further, knockdown of Orai1 in isolated pancreatic ductal cells also resulted in loss of SOCE and Ca2+‐activated ion channel activity similar to that seen in TRPC1−/− cells (Hong et al. 2011). This suggests that TRPC1 is unlikely to function in the absence of Orai1 and, further, Orai1 cannot compensate for the lack of TRPC1.
Detailed studies on the role of TRPC1, Orai1 and STIM proteins have been carried out in salivary gland cells lines, e.g. human salivary gland adenocarcinoma HSG cells. Knockdown of either Orai1 or STIM1 leads to a complete loss of SOCE while knockdown of TRPC1 induces a > 50% decrease in SOCE (Ong et al. 2007 a; Cheng et al. 2008, 2011). These findings suggest that although TRPC1 is gated by STIM1, its function, and activation subsequent to ER‐Ca2+ store depletion, is dependent on Orai1. The critical mechanism underlying the functional interaction between TRPC1 and Orai1 was shown by a study (Cheng et al. 2011) demonstrating that Orai1‐mediated Ca2+ entry triggers recruitment of TRPC1 to the plasma membrane. Thus, TRPC1 and Orai1 form separate channels that are activated by STIM1 following neurotransmitter simulation of salivary gland cells and contribute to the [Ca2+]i increase seen in stimulated cells. Orai1 is the first channel to be activated while recruitment and activation of TRPC1 leads to amplification and modulation of [Ca2+]i due to Orai1 function.
STIM2 is a second ER‐localized Ca2+‐sensor protein that has been associated with SOCE and Ca2+ signalling, and it shares considerably sequence homology with STIM1. However, the Ca2+ sensitivity and activation kinetics of STIM2 differ from those of STIM1 (Stathopulos & Ikura, 2013). STIM2 has a relatively low affinity for Ca2+ and thus can detect small depletions of ER [Ca2+]. STIM1 on the other hand, with a higher Ca2+‐affinity, responds only to substantial ER Ca2+ depletion. Like STIM1, STIM2 clusters and translocates to form puncta in ER–PM junctions, where it clusters with Orai1 and STIM1 (Brandman et al. 2007; Darbellay et al. 2010; Wang et al. 2014; Ong & Ambudkar, 2015). However, STIM2 is a poor activator of Orai1 and SOCE. The distinct responses of STIM1 and STIM2 to ER [Ca2+] are physiologically relevant since in the levels of stimulus exerted on cells in vivo tend to be low and induce less depletion of ER Ca2+ (Ong et al. 2007 b). Evidence for a novel role of STIM2 in SOCE has been provided by a recent study that shows that STIM2 promotes STIM1 clustering in ER–PM junctions under conditions when there is less depletion of ER Ca2+ (Ong et al. 2015). This enhances assembly of the Orai1–STIM1 complex and activation of SOCE despite inability of STIM1 to detect the small change in ER [Ca2+]. Knockdown of STIM2 in cell lines induced about 10‐fold increase in the agonist concentration required for half‐maximal activation of SOCE. Importantly, targeted knockout of STIM2 within salivary glands of STIM2fl/fl mice resulted in a reduction of fluid secretion at relatively low stimulus intensities, and acinar cells isolated from the glands display reduced [Ca2+]i increases following stimulation with muscarinic receptor agonists. Together the findings suggest that STIM2 recruits STIM1 and facilitates formation of STIM1 puncta in ER–PM junctions at low stimulus intensities. This increases the agonist sensitivity of assembly of the STIM1–Orai1 channel complex and SOCE activation. (Ong et al. 2015). These findings demonstrate a critical role for STIM2 in regulating SOCE‐mediated Ca2+ signalling and cell function. However, other studies indicate that involvement of STIM2 in SOCE might vary (Bird et al. 2009; Kar et al. 2012). Bird et al. have previously suggested that the oscillatory pattern of SOCE is primarily dependent on the mobilization of STIM1, not STIM2. Another study by Kar et al. showed that stimulation of cysteinyl leukotriene type I receptor in mast cells leads to mobilization of STIM1, while that of the high‐affinity IgE receptor (FCεRI) involves mobilization of both STIM2 and STIM1.
Ca2+ signals generated due to Ca2+ entry via TRPC1 or Orai1 are decoded by cells for the regulation of distinct Ca2+‐dependent functions. In cells lines, TRPC1‐mediated Ca2+ regulates nuclear factor‐κB (NFκB), KCa and transmembrane member 16A (TMEM16A) activities. Studies with TRPC1−/− mice also suggest that the residual Orai1 in TRPC1−/− salivary gland acinar cells cannot sustain the activation of the ion channel mechanisms required for salivary secretion; e.g. KCa (Liu et al. 2007; Cheng et al. 2011). On the other hand, Orai1 function is sufficient for supporting nuclear factor of activated T cells (NFAT) activation, which is unaffected by knockdown of TRPC1, in salivary gland cell lines. Interestingly, the channels generate distinct patterns of Ca2+ signals in salivary gland cells lines, which might underlie the differential regulation of cell function (Ong et al. 2012). The exact nature of the Ca2+ signals generated by these two channels in salivary gland acinar cells and the Ca2+ sensors and effector proteins in the vicinity of the channels that determine the specificity of functional regulation remain to be elucidated (Fig. 1).
Figure 1. Schematic representation of Ca2+‐dependent regulation of fluid secretion in salivary gland acinar cells .
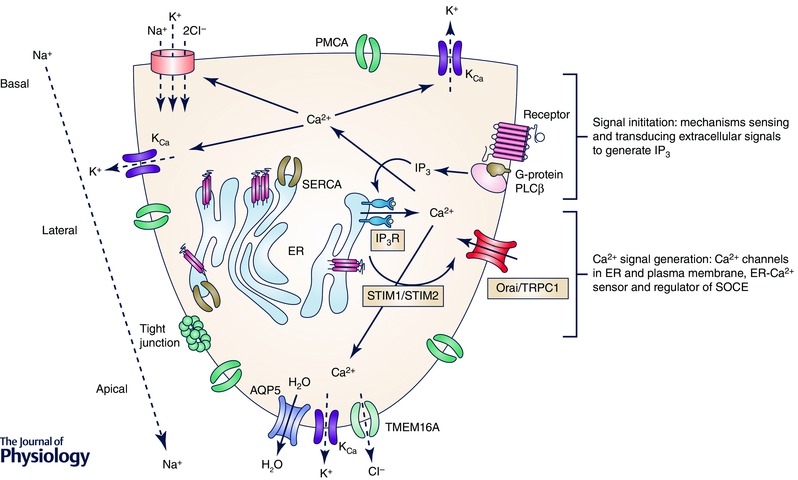
Key molecular components involved in initiation and generation of [Ca2+]i signals are indicated in the figure, as are ion channels and transporters regulated by [Ca2+]i elevation.
Aberrant [Ca2+]i signals and salivary gland dysfunction
Reactive oxygen species and Ca2+ in irradiation‐induced loss of salivary gland function
A debilitating side‐effect of IR in head and neck cancer patients is xerostomia, or dry mouth, caused by irreversible loss of salivary gland function due to bystander effects of the treatment. The exact mechanisms underlying the persistent loss of salivary gland function are not yet fully understood, although it has been suggested that IR might damage progenitor cells within the adult salivary gland leading to compromised regeneration of acinar cells (Stephens et al. 1991; Vissink et al. 1992; Konings et al. 2005; Asari et al. 2009). However, significant decrease in salivary fluid secretion is detected before onset of overt damage, or loss, of the gland. In mice glandular loss and fibrosis are not seen until 4 months after radiation while loss of function is almost immediate and persists even after the reactive oxygen species (ROS) has been cleared from the system. Importantly, there are no adequate therapies to protect against, or reverse, IR‐induced salivary hypofunction.
Radiation can induce DNA damage and membrane damage by generating intracellular ROS that can lead to long‐term irreversible salivary gland dysfunction. However, a recent study has implicated the involvement of Ca2+ signalling in radiation‐induced loss of salivary fluid secretion. While some calcium channels, including some transient receptor potential (TRP) channel members, are modulated by ROS due to protein modification, transient receptor potential melastatin‐like 2 (TRPM2), a Ca2+‐permeable non‐selective cation channel, is activated by metabolites generated as a direct consequence of ROS production in the cell (Di et al. 2012). H2O2 and other agents or conditions that produce ROS (Sumoza‐Toledo & Penner, 2011) trigger activation of poly‐ADP ribose polymerase (PARP) resulting in generation of adenosine diphosphate ribose (ADPR), which is the intracellular ligand that binds to and gates TRPM2. Thus, TRPM2 serves as an endogenous ROS sensor. A recent study showed that TRPM2 is present in salivary gland cell lines and acinar cells and is activated in response to IR, leading to enhancement of Ca2+ entry in salivary gland cells (Liu et al. 2013). In a variety of cell types, Ca2+ entry via TRPM2 has been associated with increased cell death and apoptosis. Further, TRPM2 activity also leads to cytokine production in lymphocytes and other haematopoetic cells (Takahashi et al. 2011). While the consequence of TRPM2‐mediated Ca2+ entry in acinar cells is still not known, unlike the persistent loss of saliva flow following IR of TRPM2+/+ mice, TRPM2−/− mice show a transient loss of salivary gland function which recovers to > 60% of the normal saliva flow within a month after treatment (Liu et al. 2013). Notably, treatment of WT mice with the PARP inhibitor 3AB or ROS scavenger Tempol induced significant protection of salivary gland function after IR. Together, these findings provide evidence that TRPM2 contributes to irreversible IR‐induced salivary gland dysfunction. This study also showed that while acini from untreated mice undergo robust decrease in cell volume upon stimulation with agonist (a process associated with secretion of fluid from the cells), this volume decrease is attenuated in cells from irradiated mice. Importantly, TRPM2−/− acinar cells, which are protected from IR‐induced loss of the secretory function, do not display deficits in cell volume decrease. These findings suggest that IR results in an inherent defect at the level of acinar cell function that can account for the loss of fluid secretion. What is significant is that this defect is associated with the presence and activation of TRPM2. Future studies should be directed towards delineating the intracellular targets of TRPM2‐mediated Ca2+ influx that can account for loss of fluid secretion. Possible factors are activation or inactivation of transcription factors, disruption of cell metabolism, altered Ca2+ signalling or ion channel or transport functions required for saliva flow.
Maximal activation of TRPC3 induces acinar cell toxicity
Aberrant increase in [Ca2+]i in pancreatic acinar cells has long been considered to be the basis for pancreatitis. Recent studies have implicated TRPC3 and Orai1 activities in the onset and progression of acinar cell damage and disease (Kim et al. 2009; Kim et al. 2011; Gerasimenko et al. 2014). Conditions that abrogate channel function result in protection of tissue. While Ca2+ signalling does not directly regulate protein secretion in salivary gland acinar cells, unregulated or a very high level of Ca2+ influx that is maintained for long time periods could have several intracellular consequences including ROS generation, mitochondrial dysfunction and membrane damage. A recent study demonstrated that prolonged activation of salivary gland acinar cells with high levels of agonist results in cytotoxity that is associated with TRPC3‐mediated Ca2+ entry (Kim et al. 2011). Similar conditions trigger TRPC3‐mediated acinar cell damage in pancreas as well. It is unclear whether these are due to direct effects of Ca2+ on the secretory granules or other intracellular organelles, or as a result of activation of proteases. By monitoring the release of glucose 6‐phosphate dehydrogenase (G6PD) and in the presence and absence of extracellular Ca2+, Kim et al. reported massive cell damage in acinar cells stimulated with 1 mm carbachol for 30–40 min at 37°C in the presence of external Ca2+. Removal of external Ca2+, lack of TRPC3, or inhibition of TRPC3 prevented the cell damage. Most notably, cells lacking TRPC3 or pharmacological inhibition of TRPC3 in +/+ cells by the selective inhibitor pyrazole‐3 (Pyr3) similarly protected salivary glands and the pancreas from Ca2+‐mediated cell toxicity.
It was suggested that Ca2+ entry in salivary gland acinar cells increases autophagosome formation as well as ER stress, both of which were reduced by eliminating TRPC3 function. Cell damage also resulted in an increased cellular ceramide content and accumulation of the lipid in the plasma membrane, which was also attenuated by blocking TRPC3. These studies demonstrate that under some conditions, TRPC3 activity can lead to acinar cell damage. Previous studies have shown that TRPC3 can be modified by ROS. Thus, further studies will be required to determine whether TRPC3 channels undergo further modification in cells stimulated at very high, non‐physiologically relevant levels of agonist. Since TRPC3 and TRPC1 have been suggested to generate a heteromeric channel (Lee et al. 2014), it will also of interest to determine whether heteromeric TRPC3–TRPC1 or homomeric TRPC3 channels contribute to the cytotoxic effects seen in salivary gland acinar cells. Inhibition of Orai1 function provides significant protection against pancreatitis. It is not clear whether Orai1 also contributes to agonist‐induced cytotoxicity in salivary gland acinar cells. However, since store‐dependent activation of TRPC3–TRPC1 heteromeric channels is likely to be dependent on Orai1 in these two types of acinar cells (Ong et al. 2014), knockdown of Orai1 will also reduce TRPC1–TRPC3 function. Thus, there is fine regulation of various ion channel activities in the maintenance of cellular Ca2+ homeostasis, and either a gain or a loss of Ca2+ channel or transporter activity can result in substantial disruption of Ca2+ signalling and physiological function.
Role of Ca2+ signalling in ductal epithelium in exocrine dysfunction
Exocrine dysfunction and damage have been linked to ductal tissue in both salivary and pancreatic glands. The duct carries out critical secretory as well as absorptive functions (Larusch & Whitcomb, 2012). In addition, factors secreted by acinar cells trigger autocrine responses in ductal epithelium. Purinergic receptors (P2Y2, P2X4, and P2X7) are localized in the ductal luminal membrane and sense [Ca2+] and ATP in the secreted fluid, both of which are increased during cell injury. Activation of purinergic receptors by either of these factors triggers intracellular Ca2+ signals that lead to activation of the cystic fibrosis transmembrane conductance regulator (CFTR) and secretion of bicarbonate‐rich fluid. Such secretion serves to flush the contents, such as mucins, out of the duct, thus decreasing the chance of ductal obstruction. Another receptor is the calcium‐sensing receptor (CaSR), a G‐protein‐coupled receptor that senses [Ca2+] in the secreted fluid. It is coupled to activation of cyclic‐AMP, which also leads to HCO3 − secretion via increasing CFTR activity. When [Ca2+] in pancreatic juice increases, such as following acinar cell injury, there is risk of trypsinogen activation in the ductal epithelium, which could increase the rate and severity of pancreatitis. However, activation of CaSR protects ductal physiology by activating ductal secretion (Ravi Kanth & Nageshwar Reddy, 2014) Although salivary glands do not exhibit a phenomenon similar to pancreatitis, many key components of ductal epithelium are similar, e.g. CaSR, CFTR and purinergic receptors (Bandyopadhyay et al. 2012; Hong et al. 2014; Jung & Lee, 2014) Thus, it is reasonable to hypothesize that elevations of ATP and [Ca2+] in secretory fluid could similarly affect ductal secretion in salivary glands by activating purinergic receptors and CaSR. This would relieve any mucin accumulation in the ducts, protecting against sialoadenitis, which will be extremely beneficial for the maintenance of salivary gland function and integrity.
Ca2+ signalling defects in Sjögren's syndrome
Primary Sjögren's syndrome (pSS) is a chronic autoimmune disease that results in lymphocytic infiltration and loss of secretory function in salivary and lacrimal glands (Nikolov & Illei, 2009). The pathogenesis of this disease has not yet been elucidated and neither has the basis for the loss of salivary fluid secretion been identified. While extensive lymphocytic infiltration is always associated with destruction of the gland within the area of the infiltrate, salivary gland dysfunction is seen in patients with little or no overt inflammation (Shen et al. 2013; Xuan et al. 2013). Thus, the hallmark of the disease, xerostomia, is poorly correlated with the level of lymphocytic infiltration. There are two areas within the gland that have been studied in attempts to understand the loss of salivary gland function. The first is the area within and around the lymphocytic infiltrate. The infiltrate itself is made up of multiple cell types including T and B lymphocytes. Most studies have focused on the T‐lymphocytes, although more is known about peripheral lymphocytes in the patients rather than the cells that populate the gland during the disease. It has been suggested that the infiltrating lymphocytes secrete cytokines and other factors that can alter the milieu and affect the epithelial cells in the vicinity. Some early studies therefore assessed the effects of Sjögren's syndrome‐associated cytokines such as interferon‐γ (IFNγ) and tumour necrosis factor‐α (TNFα) on cell function. However, no clear mechanism has emerged to account for the loss of function. The second, and more physiologically important, area is the relatively large amount of apparently morphologically intact salivary gland tissue in glands from SS patients. The extent of functional loss in the patients suggests that this relatively intact tissue has aberrant function.
Disruption of SOCE has major immunological consequences. Interestingly, loss of Orai1 function due to mutation results in the severe combined immune deficiency (SCID) phenotype, while patients with STIM1 mutations display autoimmunity and lymphoproliferation (Feske et al. 2015). T lymphocyte‐targeted deletion of STIM1 and STIM2 in mice resulted in loss of SOCE and SOCE‐dependent cytokine production in T cells and a decrease in functional regulatory T cells (Tregs) (Oh‐Hora et al. 2008). The mice displayed signs of autoimmunity, including dermatitis, blepharitis and lymphoproliferation, with lymphocytic infiltration in epithelial tissues, such as liver and lungs. Importantly, these mice developed spontaneous, rapid and progressive submandibular gland inflammation (Cheng et al. 2012). Within three months the glands in the mice resembled those of pSS patients, with severe salivary gland inflammation and damage. Evidence for development of SS in the mice was shown by the detection of major hallmarks of pSS: inflammation of salivary glands, loss of stimulated fluid secretion and elevated pSS‐specific autoantibodies. This study further assessed peripheral blood mononuclear cells (PBMCs) from pSS patients. Remarkably, PBMCs from pSS patients showed deficits in levels of STIM1 and STIM2 proteins, as well as diminished SOCE. Together these data show an association between STIM1 and STIM2 deficiencies in T cells, and the consequent aberrations in T cell function, with onset and progression of salivary gland exocrinopathy in Sjögren's syndrome. The exact mechanism(s) that cause these defects in lymphocytes or trigger their infiltration into salivary glands is not known.
In addition to the possible role of purinergic receptors in salivary gland dysfunction discussed above, the P2X7 purinergic receptor (P2X7R) has been reported to have an essential role in the innate immune response. It is involved in the modulation of several critical cellular functions, such as growth and replication, as well as activation of the NLRP3 inflammasome that results in release of IL‐1β and IL‐18. Notably, serum and salivary glands of patients with pSS show increases in levels of IL‐1β and IL‐18 that have been proposed to exert proinflammatory effects. In salivary glands, periductal CD68+ macrophages and ductal epithelial cells represent the major sources of IL‐18. A recent study has shown that P2X7R is overexpressed in glands of patients with pSS. Importantly, this has been linked to the activation of the inflammasome complex and increased release of IL‐18, suggesting a causative link between P2X7R, NLRP3 inflammasome activation and sialadenitis. Interestingly, inflammasome activation has also been described during acute pancreatitis, with the P2X7R upstream of the inflammasome and necessary for the development of pancreatic injury. In addition, P2X7R stimulation in salivary acinar cells has several deleterious consequences, including depolarization of the mitochondrial membrane and production of reactive oxygen species and cleavage and release of α‐fodrin. It has been suggested that P2X7R is involved in mechanisms that initiate or maintain salivary gland inflammation in pSS. These observations in pSS provide a rationale for novel P2X7R‐targeted therapies for this disease. (Woods et al. 2012; Baldini et al. 2013)
Recently, there has been more focus on the epithelial cell involvement in SS and it has been hypothesized that salivary gland acinar and ductal cells have functional defects that might account for the secretory dysfunction. Further, it has also been proposed that these cells synthesize and secrete cytokines that might contribute to disease progression. Some studies have shown that AQP5 localization is disrupted, which could account for the functional defect (Delporte, 2009; Soyfoo et al. 2012). Other studies suggested that pSS patients have autoantibodies to muscarinic receptors, which might dampen endogenous neurotransmitter stimulated salivary gland secretion (Nikolov & Illei, 2009). Data are now emerging from studies that directly examine acinar cell function in salivary gland biopsies obtained from patients that provide definitive evidence that neurotransmitter‐stimulated Ca2+ signalling and cell volume regulation are aberrant in acinar cells from pSS patients. A recent study links the decrease in Ca2+ signalling in acinar cells to loss of IP3R2 and IP3R3, two critical IP3R subtypes in this gland (Teos et al. 2015). This represents an important advancement in our understanding of the mechanism underlying the loss of saliva flow in pSS. However, further studies are required to establish the exact mechanism that initiates loss of the IP3Rs as well triggers P2X7–inflammasome activation. Such knowledge is likely to provide novel targets for therapy.
Conclusions
[Ca2+]i signals are a double‐edged sword: while [Ca2+]i increases are essential in the regulation of cell function, maintenance of Ca2+ homeostasis in both resting and stimulated cells is absolutely critical for cell survival. The key trigger for stimulation of fluid secretion is an increase in [Ca2+]i that occurs in response to stimulation of cells by neurotransmitters and that leads to regulation of ion channel activities and generation of the appropriate osmotic gradient required to drive fluid secretion. Key steps that determine the increase in cytosolic [Ca2+] are IP3‐mediated release of Ca2+ from the ER via the IP3R, and Ca2+ entry via plasma membrane SOCE‐channels (Putney, 1990). Critical components of SOCE in acinar cells include Orai1, STIM1, STIM2 and TRPC1. Thus, the major mechanisms and key players associated with regulation of Ca2+ signals in salivary gland function have now been elucidated. However, further studies are required to determine their assembly in the cells and delineate the Ca2+ sensors and other regulatory components involved in decoding the Ca2+ changes for regulation of specific cell functions.
Deficits and aberrant Ca2+ signalling have been associated with salivary gland dysfunction that results in xerostomia, or dry mouth condition, in patients. Consequences of this include difficulty swallowing, rampant dental caries, oral mucosal lesions, and fungal infections that together severely affect the quality of life for the patients (Fig. 2). The first is an irreversible loss of salivary gland function due to by‐stander effects of head and neck irradiation. This is a subject of great interest in the field with clinical studies being directed towards assessing therapies targeted to recovery of cell function, prevention of functional loss, or regrowth of salivary glands. Novel targets such as TRPM2 or mitochondrial or cytosolic ROS modulators could provide effective strategies for treatment. However, further studies are required to fully understand the mechanism underlying the loss of fluid secretion in the irradiated gland. The second is the progressive loss of salivary gland function associated with primary Sjögren's syndrome, a chronic autoimmune disease. Notably, there is poor correlation between salivary flow and extent of inflammation or tissue damage with a large number of patients displaying low levels of inflammation within their salivary glands with substantial loss of function. The molecular alterations underlying this secretory defect are not yet known. Emerging studies implicate Ca2+ signalling and functional defects in both lymphocytes as well as salivary gland acinar cells from patients diagnosed with this disease. Further studies are required to validate these initial observations and identify possible novel components that can be used to improve diagnosis or treatment for this disease.
Figure 2. Ca2+ signalling in salivary gland dysfunction .
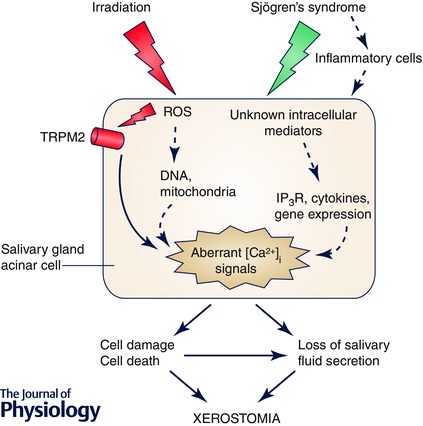
Two conditions, IR and Sjögren's syndrome, induce salivary gland dysfunction that leads to xerostomia. TRPM2 has been identified as a target for IR‐generated ROS in cells, and loss of TRPM2 function protects against IR‐induced defect in salivary gland acinar cell function. IR‐induced salivary gland dysfunction can include as yet unconfirmed effects on mitochondria and DNA (dashed arrows). Sjögren's syndrome also results in loss of salivary fluid secretion and xerostomia. While the condition causes inflammation in the gland, the intracellular mediators of the disease are not known. Decreases in IP3R function, cytokine generation and changes in gene expression could contribute to initiation and the progress of the disease.
Additional information
Competing interests
The author declares that there are no conflict of interests.
Funding
I.A. is funded by NIDCR‐DIR (Z01‐DE00438‐31).
Acknowledgements
I would like to thank Dr Hwei Ling Ong for her help in the preparation of this manuscript.
Biography
Indu Ambudkar received her PhD degree from Madurai Kamaraj University in India. She joined University of Maryland, School of Medicine, as a post‐doctoral researcher in 1980 and then moved to the National Institute of Dental and Craniofacial Research in 1985, where she is now Chief of the Molecular Physiology and Therapeutics Branch and the Secretory Physiology Section. Research in her laboratory is focused on Ca2+ entry mechanisms in salivary gland cells as well as the role of Ca2+ signalling in exocrine gland physiology and disease.

This review was presented at the symposium “New Insights into Ca2+ Stores and Intracellular Ca2+ Channels”, which took place at the Gordon Research Conference on Calcium Signalling ‐ Molecular and Cellular Mechanisms in Health and Disease in Maine, USA, 7–12 June, 2015.
References
- Ahuja M, Jha A, Maleth J, Park S & Muallem S (2014). cAMP and Ca2+ signaling in secretory epithelia: crosstalk and synergism. Cell Calcium 55, 385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambudkar IS (2011). Dissection of calcium signaling events in exocrine secretion. Neurochem Res 36, 1212–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambudkar IS (2012). Polarization of calcium signaling and fluid secretion in salivary gland cells. Curr Med Chem 19, 5774–5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambudkar IS (2014). Ca2+ signaling and regulation of fluid secretion in salivary gland acinar cells. Cell Calcium 55, 297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando H, Kawaai K & Mikoshiba K (2014). IRBIT: a regulator of ion channels and ion transporters. Biochim Biophys Acta 1843, 2195–2204. [DOI] [PubMed] [Google Scholar]
- Asari T, Maruyama K & Kusama H (2009). Salivation triggered by pilocarpine involves aquaporin‐5 in normal rats but not in irradiated rats. Clin Exp Pharmacol Physiol 36, 531–538. [DOI] [PubMed] [Google Scholar]
- Baldini C, Rossi C, Ferro F, Santini E, Seccia V, Donati V & Solini A (2013). The P2X7 receptor‐inflammasome complex has a role in modulating the inflammatory response in primary Sjogren's syndrome. J Intern Med 274, 480–489. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay BC, Swaim WD, Sarkar A, Liu X & Ambudkar IS (2012). Extracellular Ca2+ sensing in salivary ductal cells. J Biol Chem 287, 30305–30316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ (2012). Calcium signalling remodelling and disease. Biochem Soc Trans 40, 297–309. [DOI] [PubMed] [Google Scholar]
- Bird GS, Hwang SY, Smyth JT, Fukushima M, Boyles RR & Putney JW Jr (2009). STIM1 is a calcium sensor specialized for digital signaling. Curr Biol 19, 1724–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandman O, Liou J, Park WS & Meyer T (2007). STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell 131, 1327–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng KT, Alevizos I, Liu X, Swaim WD, Yin H, Feske S, Oh‐hora M & Ambudkar IS (2012). STIM1 and STIM2 protein deficiency in T lymphocytes underlies development of the exocrine gland autoimmune disease, Sjogren's syndrome. Proc Natl Acad Sci USA 109, 14544–14549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng KT, Liu X, Ong HL & Ambudkar IS (2008). Functional requirement for Orai1 in store‐operated TRPC1‐STIM1 channels. J Biol Chem 283, 12935–12940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng KT, Liu X, Ong HL, Swaim W & Ambudkar IS (2011). Local Ca2+ entry via Orai1 regulates plasma membrane recruitment of TRPC1 and controls cytosolic Ca2+ signals required for specific cell functions. PLoS Biol 9, e1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng KT, Ong HL, Liu X & Ambudkar IS (2013). Contribution and regulation of TRPC channels in store‐operated Ca2+ entry In Store‐Operated Calcium Channels, ed. Prakriya M, pp. 149–179. Elsevier, Amsterdam, The Netherlands. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S, Maleth J, Jha A, Lee KP, Kim MS, So I, Ahuja M & Muallem S (2014). The TRPCs‐STIM1‐Orai interaction. Handb Exp Pharmacol 223, 1035–1054. [DOI] [PubMed] [Google Scholar]
- Darbellay B, Arnaudeau S, Ceroni D, Bader CR, Konig S & Bernheim L (2010). Human muscle economy myoblast differentiation and excitation‐contraction coupling use the same molecular partners, STIM1 and STIM2. J Biol Chem 285, 22437–22447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaleu N, Jonsson R & Koller MM (2005). Sjogren's syndrome. Eur J Oral Sci 113, 101–113. [DOI] [PubMed] [Google Scholar]
- Delli K, Spijkervet FK, Kroese FG, Bootsma H & Vissink A (2014). Xerostomia. Monogr Oral Sci 24, 109–125. [DOI] [PubMed] [Google Scholar]
- Delporte C (2009). Aquaporins in secretory glands and their role in Sjogren's syndrome. Handb Exp Pharmacol 190, 185–201. [DOI] [PubMed] [Google Scholar]
- Di A, Gao XP, Qian F, Kawamura T, Han J, Hecquet C, Ye RD, Vogel SM & Malik AB (2012). The redox‐sensitive cation channel TRPM2 modulates phagocyte ROS production and inflammation. Nat Immunol 13, 29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feske S, Wulff H & Skolnik EY (2015). Ion channels in innate and adaptive immunity. Annu Rev Immunol 33, 291–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox RI & Stern M (2002). Sjogren's syndrome: mechanisms of pathogenesis involve interaction of immune and neurosecretory systems. Scand J Rheumatol Suppl 116, 3–13. [PubMed] [Google Scholar]
- Futatsugi A, Nakamura T, Yamada MK, Ebisui E, Nakamura K, Uchida K, Kitaguchi T, Takahashi‐Iwanaga H, Noda T, Aruga J & Mikoshiba K (2005). IP3 receptor types 2 and 3 mediate exocrine secretion underlying energy metabolism. Science 309, 2232–2234. [DOI] [PubMed] [Google Scholar]
- Gautam D, Heard TS, Cui Y, Miller G, Bloodworth L & Wess J (2004). Cholinergic stimulation of salivary secretion studied with M1 and M3 muscarinic receptor single‐ and double‐knockout mice. Mol Pharmacol 66, 260–267. [DOI] [PubMed] [Google Scholar]
- Gerasimenko JV, Gerasimenko OV & Petersen OH (2014). The role of Ca2+ in the pathophysiology of pancreatitis. J Physiol 592, 269–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen A, Lipsky PE & Dorner T (2005). Immunopathogenesis of primary Sjogren's syndrome: implications for disease management and therapy. Curr Opin Rheumatol 17, 558–565. [DOI] [PubMed] [Google Scholar]
- Hogan PG, Lewis RS & Rao A (2010). Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu Rev Immunol 28, 491–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong JH, Li Q, Kim MS, Shin DM, Feske S, Birnbaumer L, Cheng KT, Ambudkar IS & Muallem S (2011). Polarized but differential localization and recruitment of STIM1, Orai1 and TRPC channels in secretory cells. Traffic 12, 232–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong JH, Park S, Shcheynikov N & Muallem S (2014). Mechanism and synergism in epithelial fluid and electrolyte secretion. Pflugers Arch 466, 1487–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa Y, Eguchi T, Skowronski MT & Ishida H (1998). Acetylcholine acts on M3 muscarinic receptors and induces the translocation of aquaporin5 water channel via cytosolic Ca2+ elevation in rat parotid glands. Biochem Biophys Res Commun 245, 835–840. [DOI] [PubMed] [Google Scholar]
- Jung J & Lee MG (2014). Role of calcium signaling in epithelial bicarbonate secretion. Cell Calcium 55, 376–384. [DOI] [PubMed] [Google Scholar]
- Kar P, Bakowski D, Di Capite J, Nelson C & Parekh AB (2012). Different agonists recruit different stromal interaction molecule proteins to support cytoplasmic Ca2+ oscillations and gene expression. Proc Natl Acad Sci U S A 109, 6969–6974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Hong JH, Li Q, Shin DM, Abramowitz J, Birnbaumer L & Muallem S (2009). Deletion of TRPC3 in mice reduces store‐operated Ca2+ influx and the severity of acute pancreatitis. Gastroenterology 137, 1509–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Lee KP, Yang D, Shin DM, Abramowitz J, Kiyonaka S, Birnbaumer L, Mori Y & Muallem S (2011). Genetic and pharmacologic inhibition of the Ca2+ influx channel TRPC3 protects secretory epithelia from Ca2+‐dependent toxicity. Gastroenterology 140, 2107–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konings AW, Coppes RP & Vissink A (2005). On the mechanism of salivary gland radiosensitivity. Int J Radiat Oncol Biol Phys 62, 1187–1194. [DOI] [PubMed] [Google Scholar]
- Larusch J & Whitcomb DC (2012). Genetics of pancreatitis with a focus on the pancreatic ducts. Minerva Gastroenterol Dietol 58, 299–308. [PMC free article] [PubMed] [Google Scholar]
- Lee KP, Choi S, Hong JH, Ahuja M, Graham S, Ma R, So I, Shin DM, Muallem S & Yuan JP (2014). Molecular determinants mediating gating of transient receptor potential canonical (TRPC) channels by stromal interaction molecule 1 (STIM1). J Biol Chem 289, 6372–6382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KP, Yuan JP, Hong JH, So I, Worley PF & Muallem S (2010). An endoplasmic reticulum/plasma membrane junction: STIM1/Orai1/TRPCs. FEBS Lett 584, 2022–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MG, Xu X, Zeng W, Diaz J, Wojcikiewicz RJ, Kuo TH, Wuytack F, Racymaekers L & Muallem S (1997). Polarized expression of Ca2+ channels in pancreatic and salivary gland cells. Correlation with initiation and propagation of [Ca2+]i waves. J Biol Chem 272, 15765–15770. [DOI] [PubMed] [Google Scholar]
- Liou J, Fivaz M, Inoue T & Meyer T (2007). Live‐cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc Natl Acad Sci USA 104, 9301–9306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE Jr & Meyer T (2005). STIM is a Ca2+ sensor essential for Ca2+‐store‐depletion‐triggered Ca2+ influx. Curr Biol 15, 1235–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Cheng KT, Bandyopadhyay BC, Pani B, Dietrich A, Paria BC, Swaim WD, Beech D, Yildrim E, Singh BB, Birnbaumer L & Ambudkar IS (2007). Attenuation of store‐operated Ca2+ current impairs salivary gland fluid secretion in TRPC1–/– mice. Proc Natl Acad Sci USA 104, 17542–17547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Cotrim A, Teos L, Zheng C, Swaim W, Mitchell J, Mori Y & Ambudkar I (2013). Loss of TRPM2 function protects against irradiation‐induced salivary gland dysfunction. Nat Commun 4, 1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Wang W, Singh BB, Lockwich T, Jadlowiec J, O'Connell B, Wellner R, Zhu MX & Ambudkar IS (2000). Trp1, a candidate protein for the store‐operated Ca2+ influx mechanism in salivary gland cells. J Biol Chem 275, 3403–3411. [DOI] [PubMed] [Google Scholar]
- Luik RM, Wang B, Prakriya M, Wu MM & Lewis RS (2008). Oligomerization of STIM1 couples ER calcium depletion to CRAC channel activation. Nature 454, 538–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manganel M & Turner RJ (1990). Agonist‐induced activation of Na+/H+ exchange in rat parotid acinar cells is dependent on calcium but not on protein kinase C. J Biol Chem 265, 4284‐4289. [PubMed] [Google Scholar]
- Mavragani CP & Moutsopoulos HM (2014). Sjogren's syndrome. Annu Rev Pathol 9, 273–285. [DOI] [PubMed] [Google Scholar]
- Melvin JE, Yule D, Shuttleworth T & Begenisich T (2005). Regulation of fluid and electrolyte secretion in salivary gland acinar cells. Annu Rev Physiol 67, 445–469. [DOI] [PubMed] [Google Scholar]
- Mikoshiba K (2007). The IP3 receptor/Ca2+ channel and its cellular function. Biochem Soc Symp 74, 9–22. [DOI] [PubMed] [Google Scholar]
- Mikoshiba K, Hisatsune C, Futatsugi A, Mizutani A, Nakamura T & Miyachi K (2008). The role of Ca2+ signaling in cell function with special reference to exocrine secretion. Cornea 27 Suppl 1, S3–8. [DOI] [PubMed] [Google Scholar]
- Muik M, Fahrner M, Schindl R, Stathopulos P, Frischauf I, Derler I, Plenk P, Lackner B, Groschner K, Ikura M & Romanin C (2011). STIM1 couples to ORAI1 via an intramolecular transition into an extended conformation. EMBO J 30, 1678–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Matsui M, Uchida K, Futatsugi A, Kusakawa S, Matsumoto N, Nakamura K, Manabe T, Taketo MM & Mikoshiba K (2004). M3 muscarinic acetylcholine receptor plays a critical role in parasympathetic control of salivation in mice. J Physiol 558, 561–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HV, Stuart‐Tilley A, Alper SL & Melvin JE (2004). Cl−/HCO3 − exchange is acetazolamide sensitive and activated by a muscarinic receptor‐induced [Ca2+]i increase in salivary acinar cells. Am J Physiol Gastrointest Liver Physiol 286, G312–320. [DOI] [PubMed] [Google Scholar]
- Nikolov NP & Illei GG (2009). Pathogenesis of Sjogren's syndrome. Curr Opin Rheumatol 21, 465–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh‐Hora M, Yamashita M, Hogan PG, Sharma S, Lamperti E, Chung W, Prakriya M, Feske S & Rao A (2008). Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nat Immunol 9, 432–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong HL & Ambudkar IS (2015). Molecular determinants of TRPC1 regulation within ER‐PM junctions. Cell Calcium 58, 376–386. [DOI] [PubMed] [Google Scholar]
- Ong HL, Cheng KT, Liu X, Bandyopadhyay BC, Paria BC, Soboloff J, Pani B, Gwack Y, Srikanth S, Singh BB, Gill DL & Ambudkar IS (2007. a). Dynamic assembly of TRPC1‐STIM1‐Orai1 ternary complex is involved in store‐operated calcium influx. Evidence for similarities in store‐operated and calcium release‐activated calcium channel components. J Biol Chem 282, 9105–9116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong HL, de Souza LB, Cheng KT & Ambudkar IS (2014). Physiological functions and regulation of TRPC channels. Handb Exp Pharmacol 223, 1005–1034. [DOI] [PubMed] [Google Scholar]
- Ong HL, de Souza LB, Zheng C, Cheng KT, Liu X, Goldsmith CM, Feske S & Ambudkar IS (2015). STIM2 enhances receptor‐stimulated Ca2+ signaling by promoting recruitment of STIM1 to the endoplasmic reticulum‐plasma membrane junctions. Sci Signal 8, ra3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong HL, Jang SI & Ambudkar IS (2012). Distinct contributions of Orai1 and TRPC1 to agonist‐induced [Ca2+]i signals determine specificity of Ca2+‐dependent gene expression. PLoS One 7, e47146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong HL, Liu X, Tsaneva‐Atanasova K, Singh BB, Bandyopadhyay BC, Swaim WD, Russell JT, Hegde RS, Sherman A & Ambudkar IS (2007. b). Relocalization of STIM1 for activation of store‐operated Ca2+ entry is determined by the depletion of subplasma membrane endoplasmic reticulum Ca2+ store. J Biol Chem 282, 12176–12185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB & Putney JW Jr (2005). Store‐operated calcium channels. Physiol Rev 85, 757–810. [DOI] [PubMed] [Google Scholar]
- Park S, Shcheynikov N, Hong JH, Zheng C, Suh SH, Kawaai K, Ando H, Mizutani A, Abe T, Kiyonari H, Seki G, Yule D, Mikoshiba K & Muallem S (2013). Irbit mediates synergy between Ca2+ and cAMP signaling pathways during epithelial transport in mice. Gastroenterology 145, 232–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen OH & Tepikin AV (2008). Polarized calcium signaling in exocrine gland cells. Annu Rev Physiol 70, 273–299. [DOI] [PubMed] [Google Scholar]
- Prakriya M (2013). Store‐operated Orai channels: structure and function. Curr Top Membr 71, 1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A & Hogan PG (2006). Orai1 is an essential pore subunit of the CRAC channel. Nature 443, 230–233. [DOI] [PubMed] [Google Scholar]
- Proctor GB ( 2006). Muscarinic receptors and salivary secretion. J Appl Physiol (1985) 100, 1103–1104. [DOI] [PubMed] [Google Scholar]
- Proctor GB & Carpenter GH (2007). Regulation of salivary gland function by autonomic nerves. Auton Neurosci 133, 3–18. [DOI] [PubMed] [Google Scholar]
- Putney JW Jr (1986). Identification of cellular activation mechanisms associated with salivary secretion. Annu Rev Physiol 48, 75–88. [DOI] [PubMed] [Google Scholar]
- Putney JW Jr (1990). Capacitative calcium entry revisited. Cell Calcium 11, 611–624. [DOI] [PubMed] [Google Scholar]
- Putney JW Jr (1997). Capacitative calcium entry In Capacitative Calcium Entry, ed. Putney JW, Jr, pp. 53–64, R. G. Landes Company, Austin, TX, USA. [Google Scholar]
- Ravi Kanth V & Nageshwar Reddy D (2014). Genetics of acute and chronic pancreatitis: An update. World J Gastrointest Pathophysiol 5, 427–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Suresh L, Malyavantham K, Kowal P, Xuan J, Lindemann MJ & Ambrus JL Jr (2013). Different stages of primary Sjogren's syndrome involving lymphotoxin and type 1 IFN. J Immunol 191, 608–613. [DOI] [PubMed] [Google Scholar]
- Soyfoo MS, Konno A, Bolaky N, Oak JS, Fruman D, Nicaise C, Takiguchi M & Delporte C (2012). Link between inflammation and aquaporin‐5 distribution in submandibular gland in Sjogren's syndrome? Oral Dis 18, 568–574. [DOI] [PubMed] [Google Scholar]
- Stathopulos PB & Ikura M (2013). Structural aspects of calcium‐release activated calcium channel function. Channels (Austin) 7, 344–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens LC, Schultheiss TE, Price RE, Ang KK & Peters LJ (1991). Radiation apoptosis of serous acinar cells of salivary and lacrimal glands. Cancer 67, 1539–1543. [DOI] [PubMed] [Google Scholar]
- Sumoza‐Toledo A & Penner R (2011). TRPM2: a multifunctional ion channel for calcium signalling. J Physiol 589, 1515–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Birnbaumer L & Singh BB (2015). TRPC1 regulates calcium‐activated chloride channels in salivary gland cells. J Cell Physiol 230, 2848–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N, Kozai D, Kobayashi R, Ebert M & Mori Y (2011). Roles of TRPM2 in oxidative stress. Cell Calcium 50, 279–287. [DOI] [PubMed] [Google Scholar]
- Teos LY, Zhang Y, Cotrim AP, Swaim W, Won JH, Ambrus J, Shen L, Bebris L, Grisius M, Jang SI, Yule DI, Ambudkar IS & Alevizos I (2015). IP3R deficit underlies loss of salivary fluid secretion in Sjogren's Syndrome. Sci Rep 5, 13953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vissink A, Down JD & Konings AW (1992). Contrasting dose‐rate effects of γ‐irradiation on rat salivary gland function. Int J Radiat Biol 61, 275–282. [DOI] [PubMed] [Google Scholar]
- Vissink A, Jansma J, Spijkervet FK, Burlage FR & Coppes RP (2003). Oral sequelae of head and neck radiotherapy. Crit Rev Oral Biol Med 14, 199–212. [DOI] [PubMed] [Google Scholar]
- Wang X, Wang Y, Zhou Y, Hendron E, Mancarella S, Andrake MD, Rothberg BS, Soboloff J & Gill DL (2014). Distinct Orai‐coupling domains in STIM1 and STIM2 define the Orai‐activating site. Nat Commun 5, 3183–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods LT, Camden JM, Batek JM, Petris MJ, Erb L & Weisman GA (2012). P2X7 receptor activation induces inflammatory responses in salivary gland epithelium. Am J Physiol Cell Physiol 303, C790–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MM, Buchanan J, Luik RM & Lewis RS (2006). Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J Cell Biol 174, 803–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing J, Petranka JG, Davis FM, Desai PN, Putney JW & Bird GS (2014). Role of Orai1 and store‐operated calcium entry in mouse lacrimal gland signalling and function. J Physiol 592, 927–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xuan J, Shen L, Malyavantham K, Pankewycz O, Ambrus JL Jr & Suresh L (2013). Temporal histological changes in lacrimal and major salivary glands in mouse models of Sjogren's syndrome. BMC Oral Health 13, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YD, Cho H, Koo JY, Tak MH, Cho Y, Shim WS, Park SP, Lee J, Lee B, Kim BM, Raouf R, Shin YK & Oh U (2008). TMEM16A confers receptor‐activated calcium‐dependent chloride conductance. Nature 455, 1210–1215. [DOI] [PubMed] [Google Scholar]
- Yuan JP, Zeng W, Dorwart MR, Choi YJ, Worley PF & Muallem S (2009). SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat Cell Biol 11, 337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan JP, Zeng W, Huang GN, Worley PF & Muallem S (2007). STIM1 heteromultimerizes TRPC channels to determine their function as store‐operated channels. Nat Cell Biol 9, 636–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yule DI (2001). Subtype‐specific regulation of inositol 1,4,5‐trisphosphate receptors: controlling calcium signals in time and space. J Gen Physiol 117, 431–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng W, Yuan JP, Kim MS, Choi YJ, Huang GN, Worley PF & Muallem S (2008). STIM1 gates TRPC channels, but not Orai1, by electrostatic interaction. Mol Cell 32, 439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA & Cahalan MD (2005). STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 437, 902–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Srinivasan P, Razavi S, Seymour S, Meraner P, Gudlur A, Stathopulos PB, Ikura M, Rao A & Hogan PG (2013). Initial activation of STIM1, the regulator of store‐operated calcium entry. Nat Struct Mol Biol 20, 973–981. [DOI] [PMC free article] [PubMed] [Google Scholar]