

Abstract
Glucose derived from extracellular sources serves as an energy source in virtually all eukaryotic cells, including skeletal muscle. Its contribution to energy turnover increases with exercise intensity up to moderately heavy workloads. However, at very high workloads, the contribution of extracellular glucose to energy turnover is negligible, despite the high rate of glucose transport. Reactive oxygen species (ROS) are involved in the stimulation of glucose transport in isolated skeletal muscle preparations during intense repeated contractions. Consistent with this observation, heavy exercise is associated with significant production of ROS. However, during more mild to moderate stimulation or exercise conditions (in vitro, in situ and in vivo) antioxidants do not affect glucose transport. It is noteworthy that the production of ROS is limited or not observed under these conditions and that the concentration of the antioxidant used was extremely low. The results to date suggest that ROS involvement in activation of glucose transport occurs primarily during intense short‐term exercise and that other mechanisms are involved during mild to moderate exercise. What remains puzzling is why ROS‐mediated activation of glucose transport would occur under conditions where glucose transport is highest and utilization (i.e. phosphorylation of glucose by hexokinase) is low. Possibly ROS production is involved in priming glucose transport during heavy exercise to accelerate glycogen biogenesis during the initial recovery period after exercise, as well as altering other aspects of intracellular metabolism.
![]()
Abbreviations
- AMPK
AMP‐dependent protein kinase
- BTS
N‐benzyl‐p‐toluene sulphonamide
- EDL
extensor digitorum longus
- NAC
N‐acetylcysteine
- ROS
reactive oxygen species
- SOD
superoxide dismutase
Introduction
More than a century ago, increased glucose uptake was observed in the equine masseter muscle during chewing (Chauveau & Kaufmann, 1887). Studies in the 1950 s confirmed that contractions increased muscle glucose uptake in rats and dogs (Goldstein et al. 1953; Huycke, 1955) and in the 1960 s and 1970 s measurements of muscle glucose uptake were performed in humans (Reichard et al. 1961; Sanders et al. 1964; Havel et al. 1967; Whichelow et al. 1968; Jorfeldt & Wahren, 1970; Wahren et al. 1971; Ahlborg et al. 1974). It is generally implied or implicitly stated that extracellular glucose is an important fuel for muscle function or performance, especially under conditions of prolonged submaximal exercise (Richter & Hargreaves, 2013). A link between reduced extracellular glucose (hypoglycaemia) and fatigue was identified in early (Levine et al. 1924; Christensen, 1939) and later studies as well (Coyle et al. 1983), although hypoglycaemia does not always lead to fatigue during prolonged submaximal exercise (Felig et al. 1982).
Glucose transport and carbohydrate utilization
The extent to which glucose transport contributes to carbohydrate utilization in muscle depends on the experimental conditions (e.g. intensity, duration and mode of exercise, muscle mass involved, glycogen and blood flow analyses, etc.). Almost 30 years ago, it was shown that during maximal leg cycling exercise (corresponding to ∼100% of maximal oxygen uptake (), leg glucose uptake increased approximately 50‐fold vs. rest, but virtually none of the glucose that entered the muscle was metabolized (metabolized and/or utilized denotes glucose phosphorylation by hexokinase followed by entry into glycolysis), presumably due to inhibition of hexokinase by glucose‐6‐P (Katz et al. 1986). Thus under conditions of intense short‐term exercise, despite maximal rates of glucose transport, the contribution of extracellular glucose to muscle carbohydrate utilization is negligible. Indeed during the initial phase of moderate cycling exercise (first 40 min at 75% of ), where high rates of glucose uptake are achieved, extra‐cellular glucose contributes to only about 5% of muscle carbohydrate utilization. And even during the latter half of the exercise (the last 35 min), extracellular glucose accounted for less than 20% of carbohydrate utilization. These estimates were based on direct measurements of muscle glucose uptake (leg blood flow measurements and arterial and femoral venous glucose concentrations) and muscle glycogen contents (Katz et al. 1991). The contribution of extracellular glucose to carbohydrate utilization increases as the glycogen store is depleted and this becomes especially clear when performing exercise with initial high or low muscle glycogen content (Gollnick et al. 1981). In summary, extracellular glucose contributes little to muscle carbohydrate utilization during moderate and heavy exercise. Therefore the use of carbohydrate supplements will be of limited benefit under many exercise conditions when glycogen stores are full or adequate (Coyle et al. 1986). Only when muscle glycogen stores reach low levels will carbohydrate supplements have positive results on muscle performance (Coyle et al. 1986).
Limitations of glucose metabolism
What limits glucose metabolism in skeletal muscle has been an issue of debate. Three specific sites or processes have been suggested: (1) glucose delivery, (2) glucose transport, and (3) glucose phosphorylation by hexokinase. Richter and Hargreaves have discussed this issue in depth and the reader is referred to their recent review for details (Richter & Hargreaves, 2013). Various experimental approaches have been employed, including alterations in the expression of enzymes or transport proteins (e.g. hexokinase, glycogen synthase, GLUT1 and GLUT4), or use of isotopic glucose analogues and calculations based on specific assumptions in mice, as well as increasing extracellular glucose concentrations, to assess what limits glucose metabolism. Depending on the manipulation and the conditions studied, it has been concluded that all three mechanisms can be limiting (Schultz et al. 1977; Furler et al. 1991; Ren et al. 1993; Manchester et al. 1996; Azpiazu et al. 2000; Wasserman, 2009; Richter & Hargreaves, 2013). Thus conclusions regarding the limiting step are likely to depend on the assumptions made and conditions of study. Noteworthy was the important observation that mice exhibit muscle glycogen concentrations that are only about 10% of those in humans (Coyle et al. 1986; Katz et al. 1991; Pederson et al. 2004; Zhang et al. 2008). Further, mice that lack glycogen synthase, and therefore lack glycogen as well, are able to run as well as wild‐type (WT) mice (Pederson et al. 2005). Thus caution should be used in extrapolating results from such models to intact humans. Briefly, it appears that during the onset of moderate exercise or during short‐term intense exercise in humans, glucose phosphorylation limits glucose metabolism. This conclusion is based on measurements of accumulation of free glucose in the muscle, and it appears that the free glucose derives from the extracellular space (from glucose transported into muscle) rather than release from intracellular glycogen (via debranching enzyme) (Katz et al. 1986, 1991; Sahlin et al. 1989).
Reactive oxygen species and glucose transport
Background information on free radical chemistry and the role of reactive oxygen species (ROS) in exercise‐induced oxidative stress are presented elsewhere (Powers & Jackson, 2008). That ROS are involved in activation of glucose transport in isolated muscle preparations has been reviewed earlier (Balon & Yerneni, 2001; Katz, 2007; Merry & McConell, 2009, 2012; Richter & Hargreaves, 2013). The present brief review will examine literature published since the initial observation that ROS is involved in the activation of glucose transport after intense short‐term exercise (Sandström et al. 2006). In the latter study, it was found that repeated tetanic contractions in isolated mouse extensor digitorum longus muscle (EDL; fast‐twitch, glycolytic) increased glucose transport about threefold and the general anti‐oxidant N‐acetylcysteine (NAC) blocked about half of the increase but did not affect force (Sandström et al. 2006). The effect of NAC appeared to be specific for exercise since NAC did not affect basal, hypoxia‐, or insulin‐mediated glucose transport. Although NAC has anti‐oxidant properties, it has additional biological effects, including binding to transition and heavy metal ions and provision of precursor for glutathione synthesis (Samuni et al. 2013). Therefore experiments were performed to examine if NAC was functioning as an antioxidant in the isolated EDL muscle. Repeated contractions resulted in increased intracellular ROS levels (using the fluorescent indicator, 5‐(and 6)‐chloromethyl‐2′,7′‐dichlorohydrofluorescein) and the ratio of oxidized to reduced glutathione (GSSG/GSH, reflecting increased ROS levels), and both of these increases were attenuated or blocked by NAC, demonstrating that NAC was functioning as an anti‐oxidant under the conditions studied (Sandström et al. 2006). To identify the species of ROS involved, an additional anti‐oxidant, ebselen was used (a selenoorganic compound that induces glutathione peroxidase‐like reduction of H2O2 in the presence of GSH) (Cotgreave et al. 1987). Ebselen also inhibited about half of contraction‐mediated glucose transport. This finding implicated H2O2 or its derivative(s) as the active ROS species. To further study the potential role of H2O2 as the activating agent, muscles from mice overexpressing mitochondrial Mn2+‐dependent superoxide dismutase (SOD; to enhance conversion of O· − to H2O2 in mitochondria) were studied. The strategy was to enhance conversion of superoxide (O· −), the parent free radical that is produced in mitochondria, to H2O2 and thereby increase contraction‐mediated glucose transport. Indeed, glucose transport after repeated contractions was increased by about 25% in EDL muscle overexpressing Mn2+‐dependent SOD compared with values from WT muscle (Sandström et al. 2006). In contrast, neither basal nor insulin‐mediated glucose transport was affected by SOD overexpression. Addition of H2O2 to the medium bathing resting muscle also increased glucose transport. Overall the results indicated a role for ROS in activation of glucose transport during intense repeated contractions in isolated mouse muscle and suggested that H2O2 or its derivative(s) was the activating ROS species.
Interestingly, glucose transport after repeated contractions was almost 45% higher in soleus muscles from mice overexpressing SOD vs. muscles from WT mice (Katz, 2007). This may derive from a higher mitochondrial O· − production in soleus owing to a higher mitochondrial volume and/or faster conversion of O· − to H2O2 because of higher activity of Mn2+‐dependent SOD in soleus vs. EDL muscle (Oh‐Ishi et al. 1995; Hollander et al. 1999). Thus one might expect that contraction‐mediated glucose transport should be higher in soleus (type I, more oxidative) than in EDL (type II, more glycolytic) muscle owing to a higher mitochondrial capacity, as well as higher expression of GLUT4 protein in type I vs. type II muscle (Richter & Hargreaves, 2013). Indeed during treadmill running in rodents this is the case. Glucose disposal during moderate treadmill running in rats is markedly higher in type I vs. type II muscle (James et al. 1985). However, glycogen breakdown is substantial in type I muscle but negligible in type II muscle under these conditions (James et al. 1985), suggesting that type II muscle was not significantly activated. In contrast, in vitro studies, where isolated muscles are activated under identical or similar conditions, show that contraction‐mediated activation of glucose transport is actually higher in EDL than in soleus muscle (Higaki et al. 2001; Johansson et al. 2007; Merry et al. 2010 b; Sylow et al. 2013), although sometimes the activation of glucose transport in soleus and EDL muscles can be similar (Jorgensen et al. 2004). This appears to be in contrast to the idea that endogenous ROS production is important for activation of glucose transport during exercise. It is noteworthy that type II muscle has lower levels of antioxidant buffering systems than oxidative muscle (Lawler et al. 1993; Leeuwenburgh et al. 1994; Hollander et al. 1999; Zhang et al. 2007; Anderson & Neufer, 2006). Consequently, EDL may be more sensitive to oxidants. Support for this idea comes from the observation that exposure of soleus and EDL (or epitrochlearis, also a type II muscle) muscles to exogenously administered H2O2 (or H2O2 produced by exogenously administered glucose oxidase) results in a greater degree of inactivation of aconitase (an enzyme of the tricarboxylic acid cycle in mitochondria), as well as increased phosphorylation of signalling molecules (e.g. insulin receptor, insulin receptor substrate‐1 and protein kinase B (Akt)) and activation of glucose transport in EDL/epitrochlearis muscle (Kim et al. 2006; Zhang et al. 2007; Jensen et al. 2008; Zhang et al. 2010). Interestingly, glycolytic muscle actually has a higher capacity than oxidative muscle to generate H2O2 in mitochondria (Anderson & Neufer, 2006). This should be considered together with the observation that the ROS‐dependent activation of glucose transport in EDL muscle is associated with activation of AMP‐dependent protein kinase (AMPK) (see below) (Sandström et al. 2006). Since activation of AMPK (α2 isoform) during repeated contractions is less in soleus than in EDL muscle (Jorgensen et al. 2004), this can also contribute to the lower degree of contraction‐mediated glucose transport in soleus muscle. Exogenous H2O2 was shown to selectively activate the α1 isoform of AMPK in epitrochlearis muscle (Toyoda et al. 2004), and contraction‐mediated activation of this isoform is similar in soleus and EDL muscle (Jorgensen et al. 2004). On the other hand, others have shown that H2O2 does not activate either isoform in isolated EDL muscle (Higaki et al. 2008), but activates both α1 and α2 isoforms in isolated soleus muscle (Jensen et al. 2008). In our hands exogenous H2O2 increases total AMPK activity in isolated EDL muscle threefold (Sandström et al. 2006). Such differential results make it difficult to reach a conclusive statement regarding the effects of H2O2 on AMPK. What appears clear is that contraction‐mediated glucose transport is less in isolated soleus than in EDL, possibly owing to a decreased sensitivity to endogenously produced ROS.
Contraction‐mediated glucose transport is believed to involve activation of AMPK, although under some conditions a dissociation between AMPK activation and glucose transport has been reported (for references see Sandström et al. 2006; Richter & Hargreaves, 2013). Initially it was found that intense repeated contractions induced about a 10‐fold increase in AMPK activity and phosphorylation in isolated EDL muscle. Pre‐incubation of muscle in NAC blocked about 50% of the increase in AMPK activation (similar to the effect of NAC on contraction‐mediated activation of glucose transport) (Sandström et al. 2006). Thus the results indicated that endogenously produced ROS resulted in activation of AMPK and this led to the acceleration of glucose transport during repeated contractions (Sandström et al. 2006).
Since the initial observation of ROS involvement in contraction‐mediated activation of glucose transport, it was shown that isolated EDL muscle from WT and adenylate kinase‐1 knockout mice exhibited a twofold increase in glucose transport after repeated contractions and that NAC blocked about 30% of the increase in both groups (Zhang et al. 2008). In another study, the role of cross‐bridge function in contraction‐mediated glucose transport was investigated. By blocking cross‐bridge activity with the myosin II ATPase inhibitor N‐benzyl‐p‐toluene sulphonamide (BTS), force production during repeated contractions was decreased to ∼5% of control (Sandström et al. 2007). Despite the almost complete abolition of force, glucose transport after repeated contractions was similar in both control and BTS groups (∼4‐fold that of basal). Importantly, the GSSG/GSH ratio was markedly increased, as was AMPK activity, in both groups. These results confirmed and extended the original observations, indicating that ROS was involved in the activation of AMPK and glucose transport during electrical stimulation of isolated glycolytic muscle (Sandström et al. 2007; Zhang et al. 2008). Further, the activation of glucose transport does not appear to depend on force generation, which is consistent with earlier work (Holloszy & Narahara, 1965) and discussed in more detail elsewhere (Sandström et al. 2007). Others subsequently used BTS, or BTS together with blebbistatin (non‐muscle myosin II inhibitor), to study the role of mechanical load on contraction‐mediated glucose transport (Blair et al. 2009; Jensen et al. 2014; Sylow et al. 2015). When Jensen et al. (2014) used BTS together with blebbistatin and our stimulation conditions on isolated mouse EDL muscle (Sandström et al. 2007), they too found that contraction‐mediated glucose transport was unaffected by the myosin ATPase inhibitors, although force was almost completely suppressed. However, with more mild stimulation protocols, the myosin ATPase inhibitors suppressed contraction‐mediated glucose transport (Jensen et al. 2014). Under other experimental conditions (including higher concentrations of BTS), the myosin ATPase inhibitors also partially blocked contraction‐mediated glucose transport and activation of signalling molecules (including AMPK) (Blair et al. 2009; Sylow et al. 2015). The reason for these apparently divergent results is not clear and therefore additional studies are warranted.
McConell and colleagues embarked on a series of experiments to investigate the role of ROS in contraction‐mediated glucose transport. First they confirmed that repeated contractions in isolated mouse EDL muscle, under conditions similar to those reported earlier (Sandström et al. 2006), resulted in activation of glucose transport that was blocked by ∼50% by NAC (20 mm) (Merry & McConell, 2009). Similar findings were observed in the isolated soleus muscle. However, in contrast to earlier findings (Sandström et al. 2006), contraction‐mediated activation of AMPK was not affected by NAC either in EDL or in soleus muscle (Merry & McConell, 2009). Further, repeated contractions of muscles isolated from mice expressing a kinase‐dead form of the α2 AMPK isoform (AMPK‐KD) resulted in normal activation of glucose transport and the activation was also blocked by NAC. However, there was no activation AMPK in AMPK‐KD muscles. These findings led the authors to conclude that ROS are involved in activation of glucose uptake during repeated contractions via a mechanism that is independent of AMPK (Merry & McConell, 2009).
In additional studies, McConnell et al. examined the effects of NAC infusion on glucose uptake in the rat hindlimb in situ during repeated contractions, as well as in humans during cycle ergometry (∼60% of ) (Merry et al. 2010 a,c). The authors studied light to moderate exercise to mimic physiological conditions. In both studies significant increases in glucose uptake or disposal were noted during exercise, but NAC infusion did not affect glucose metabolism during exercise in either study. In neither study was the GSSG/GSH ratio in muscle altered by exercise although S‐glutathiolynation of an unidentified protein(s) was increased and this increase was blocked by NAC (Merry et al. 2010 a,c). It was concluded that ROS were not significantly involved in activation of glucose transport during moderate exercise conditions (Merry et al. 2010 a,c). In these studies, the level of NAC was about 20 μm in rat plasma and about 200 μm in human plasma. This should be contrasted with a concentration of 20 mm in the medium bathing isolated muscles (Sandström et al. 2006; Zhang et al. 2008; Merry et al. 2010 b). In this context it was recently demonstrated that the effect of NAC on contractile function of isolated mouse EDL muscle is temperature and concentration dependent (Katz et al. 2014). Indeed 1 mm NAC was ineffective, whereas 10 mm NAC had a small but significant effect and 20 mm NAC had a robust effect on force generation (Katz et al. 2014). Thus, although the small degree of oxidative stress during moderate exercise (S‐glutathionlyation of unknown proteins) was blocked by NAC (Merry et al. 2010 a,c), it appears that neither substantial oxidative stress nor antioxidant buffer capacity was achieved in these studies (Merry et al. 2010 a,c). Of interest, exercise at 75% of results in a robust increase in leg glucose uptake (Katz et al. 1991) and at this exercise intensity significant increases in the muscle GSSG content are detected in human muscle (GSSG + GSH remain unchanged) (Zhang et al. 2007). However, it is not known if exogenous antioxidants affect glucose uptake under these conditions. Finally one should consider that experiments performed in one species under given conditions may not always be applicable to other conditions or species (e.g. mouse vs. human).
Summary and conclusions
From recent reviews it appears that ROS are not involved in activation of glucose transport during light to moderate exercise conditions, but may contribute during intense exercise (Merry & McConell, 2012; Richter & Hargreaves, 2013), as depicted in Fig. 1. In an earlier review, it was pointed out that the effect of ROS on glucose transport would be expected to be most significant during heavy exercise, where oxidative stress is expected to be highest (Katz, 2007). Indeed, the ROS effect may become most apparent at exercise intensities exceeding 75% of (Gohil et al. 1988; Sahlin et al. 1991; Wang et al. 2006; Zhang et al. 2007). The source of the ROS is not clear but it may derive from O· − produced in mitochondria (Katz, 2007), as supported by the experiments on muscle from mice overexpressing mitochondrial Mn2+‐dependent SOD (see above). But the question remains as to what the physiological significance is of ROS for muscle metabolism and function. During exercise at 100% of , where the effects of ROS are assumed to be near maximal and the highest rates of glucose transport are achieved, virtually none of the glucose is metabolized. The physiological benefit of ROS under these conditions is not clear. Possibly the activation of glucose transport under these conditions is not for glucose supply for energy metabolism during exercise but rather for rapid glycogenesis during the initial phase of recovery after exercise. Moreover, the effects of ROS may not be relegated solely to glucose transport but to other intracellular processes as well. The finding that NAC results in marked changes in glucose‐6‐P (enhanced) and malate (decreased) after just a few near maximal tetanic contractions in isolated muscle indicates that ROS is significantly involved in metabolic processes occurring in the cytosol and mitochondria during intense exercise (Katz et al. 2014), in addition to affecting contractile function (Powers & Jackson, 2008). Thus full elucidation of the role of ROS in control of glucose transport and other metabolic events in muscle during exercise awaits further research.
Figure 1. Scheme for key steps in activation of glucose transport during exercise in skeletal muscle .
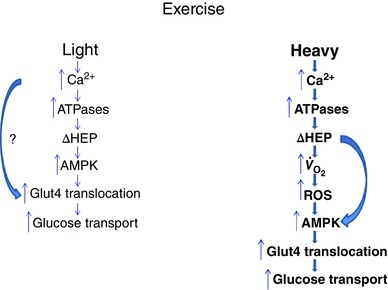
Light and Heavy denote exercise intensity. Bold lettering and bold arrows denote greater activities/levels (in Heavy). ΔHEP, change in levels of high energy phosphates (decreases in phosphocreatine and ATP and increases in ADP and AMP); AMPK, AMP‐activated protein kinase; , oxygen uptake. Curved arrow in Light denotes a Ca2+‐dependent enhancement of Glut4 translocation via an unknown mechanism(s) (denoted by ?) in addition to a low degree of activation of AMPK. Curved arrow in Heavy denotes AMP‐mediated activation of AMPK. The figure is partially derived from Katz (2007).
Additional information
Competing interests
None declared.
Biography
Abram Katz received his PhD in Medical Sciences from the Karolinska Institutet, Sweden. He is currently Professor at the Department of Physical Therapy, Ariel University, Israel. Previously he was on faculty at the Karolinska Institutet and the University of Illinois at Urbana‐Champaign, USA. His research focuses on the regulation of carbohydrate metabolism in skeletal muscle in the context of exercise, hormone action and disease.

References
- Ahlborg G, Felig P, Hagenfeldt L, Hendler R & Wahren J (1974). Substrate turnover during prolonged exercise in man. Splanchnic and leg metabolism of glucose, free fatty acids, and amino acids. J Clin Invest 53, 1080–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson EJ & Neufer PD (2006). Type II skeletal myofibres possess unique properties that potentiate mitochondrial H2O2 generation. Am J Physiol Cell Physiol 290, C844–C851. [DOI] [PubMed] [Google Scholar]
- Azpiazu I, Manchester J, Skurat AV, Roach PJ & Lawrence JC Jr (2000). Control of glycogen synthesis is shared between glucose transport and glycogen synthase in skeletal muscle fibres. Am J Physiol Endocrinol Metab 278, E234–E243. [DOI] [PubMed] [Google Scholar]
- Balon TW & Yerneni KK (2001). Redox regulation of skeletal muscle glucose transport. Med Sci Sports Exerc 33, 382–385. [DOI] [PubMed] [Google Scholar]
- Blair DR, Funai K, Schweitzer GG & Cartee GD (2009). A myosin II ATPase inhibitor reduces force production, glucose transport, and phosphorylation of AMPK and TBC1D1 in electrically stimulated rat skeletal muscle. Am J Physiol Endocrinol Metab 296, E993–E1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauveau MA & Kaufmann M (1887). Experiences pour la determination du coefficient de l'activite nutritive et respiratoire des muscles en repos et en travail. C R Acad Sci III 104, 1126–1132. [Google Scholar]
- Christensen EHHO (1939). Arbeitsfahigkeit und ernahrung. Skand Arch Physiol 81, 160–171. [Google Scholar]
- Cotgreave IA, Sandy MS, Berggren M, Moldeus PW & Smith MT (1987). N‐acetylcysteine and glutathione‐dependent protective effect of PZ51 (Ebselen) against diquat‐induced cytotoxicity in isolated hepatocytes. Biochem Pharmacol 36, 2899–2904. [DOI] [PubMed] [Google Scholar]
- Coyle EF, Coggan AR, Hemmert MK & Ivy JL (1986). Muscle glycogen utilization during prolonged strenuous exercise when fed carbohydrate. J Appl Physiol (1985) 61, 165–172. [DOI] [PubMed] [Google Scholar]
- Coyle EF, Hagberg JM, Hurley BF, Martin WH, Ehsani AA & Holloszy JO (1983). Carbohydrate feeding during prolonged strenuous exercise can delay fatigue. J Appl Physiol Respir Environ Exerc Physiol 55, 230–235. [DOI] [PubMed] [Google Scholar]
- Felig P, Cherif A, Minagawa A & Wahren J (1982). Hypoglycemia during prolonged exercise in normal men. N Engl J Med 306, 895–900. [DOI] [PubMed] [Google Scholar]
- Furler SM, Jenkins AB, Storlien LH & Kraegen EW (1991). In vivo location of the rate‐limiting step of hexose uptake in muscle and brain tissue of rats. Am J Physiol Endocrinol Metab 261, E337–E347. [DOI] [PubMed] [Google Scholar]
- Gohil K, Viguie C, Stanley WC, Brooks GA & Packer L (1988). Blood glutathione oxidation during human exercise. J Appl Physiol (1985) 64, 115–119. [DOI] [PubMed] [Google Scholar]
- Goldstein MS, Mullick V, Huddelstun B & Levine R (1953). Action of muscular work on transfer of sugars across cell barriers; comparison with action of insulin. Am J Physiol 173, 212–216. [DOI] [PubMed] [Google Scholar]
- Gollnick PD, Pernow B, Essen B, Jansson E & Saltin B (1981). Availability of glycogen and plasma FFA for substrate utilization in leg muscle of man during exercise. Clin Physiol 1, 27–42. [Google Scholar]
- Havel RJ, Pernow B & Jones NL (1967). Uptake and release of free fatty acids and other metabolites in the legs of exercising men. J Appl Physiol 23, 90–99. [DOI] [PubMed] [Google Scholar]
- Higaki Y, Hirshman MF, Fujii N & Goodyear LJ (2001). Nitric oxide increases glucose uptake through a mechanism that is distinct from the insulin and contraction pathways in rat skeletal muscle. Diabetes 50, 241–247. [DOI] [PubMed] [Google Scholar]
- Higaki Y, Mikami T, Fujii N, Hirshman MF, Koyama K, Seino T, Tanaka K & Goodyear LJ (2008). Oxidative stress stimulates skeletal muscle glucose uptake through a phosphatidylinositol 3‐kinase‐dependent pathway. Am J Physiol Endocrinol Metab 294, E889–E897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollander J, Fiebig R, Gore M, Bejma J, Ookawara T, Ohno H & Ji LL (1999). Superoxide dismutase gene expression in skeletal muscle: fibre‐specific adaptation to endurance training. Am J Physiol Regul Integr Comp Physiol 277, R856–R862. [DOI] [PubMed] [Google Scholar]
- Holloszy JO & Narahara HT (1965). Studies of tissue permeability. X. Changes in permeability to 3‐methylglucose associated with contraction of isolated frog muscle. J Biol Chem 240, 3493–3500. [PubMed] [Google Scholar]
- Huycke EKP (1955). Effects of insulin and muscular exercise upon the uptake of hexoses by muscle cells. Acta Physiol Scand 34, 231–249. [DOI] [PubMed] [Google Scholar]
- James DE, Kraegen EW & Chisholm DJ (1985). Muscle glucose metabolism in exercising rats: comparison with insulin stimulation. Am J Physiol Endocrinol Metab 248, E575–E580. [DOI] [PubMed] [Google Scholar]
- Jensen TE, Schjerling P, Viollet B, Wojtaszewski JF & Richter EA (2008). AMPK α1 activation is required for stimulation of glucose uptake by twitch contraction, but not by H2O2, in mouse skeletal muscle. PLoS One 3, e2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen TE, Sylow L, Rose AJ, Madsen AB, Angin Y, Maarbjerg SJ & Richter EA (2014). Contraction‐stimulated glucose transport in muscle is controlled by AMPK and mechanical stress but not sarcoplasmatic reticulum Ca2+ release. Mol Metab 3, 742–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson SM, Salehi A, Sandström ME, Westerblad H, Lundquist I, Carlsson PO, Fredholm BB & Katz A (2007). A1 receptor deficiency causes increased insulin and glucagon secretion in mice. Biochem Pharmacol 74, 1628–1635. [DOI] [PubMed] [Google Scholar]
- Jorfeldt L & Wahren J (1970). Human forearm muscle metabolism during exercise. V. Quantitative aspects of glucose uptake and lactate production during prolonged exercise. Scand J Clin Lab Invest 26, 73–81. [DOI] [PubMed] [Google Scholar]
- Jorgensen SB, Viollet B, Andreelli F, Frosig C, Birk JB, Schjerling P, Vaulont S, Richter EA & Wojtaszewski JF (2004). Knockout of the α2 but not α1 5′‐AMP‐activated protein kinase isoform abolishes 5‐aminoimidazole‐4‐carboxamide‐1‐β‐4‐ribofuranosidebut not contraction‐induced glucose uptake in skeletal muscle. J Biol Chem 279, 1070–1079. [DOI] [PubMed] [Google Scholar]
- Katz A (2007). Modulation of glucose transport in skeletal muscle by reactive oxygen species. J Appl Physiol 102, 1671–1676. [DOI] [PubMed] [Google Scholar]
- Katz A, Broberg S, Sahlin K & Wahren J (1986). Leg glucose uptake during maximal dynamic exercise in humans. Am J Physiol Endocrinol Metab 251, E65–E70. [DOI] [PubMed] [Google Scholar]
- Katz A, Hernandez A, Caballero DM, Briceno JF, Amezquita LV, Kosterina N, Bruton JD & Westerblad H (2014). Effects of N‐acetylcysteine on isolated mouse skeletal muscle: contractile properties, temperature dependence, and metabolism. Pflugers Arch 466, 577–585. [DOI] [PubMed] [Google Scholar]
- Katz A, Sahlin K & Broberg S (1991). Regulation of glucose utilization in human skeletal muscle during moderate dynamic exercise. Am J Physiol Endocrinol Metab 260, E411–E415. [DOI] [PubMed] [Google Scholar]
- Kim JS, Saengsirisuwan V, Sloniger JA, Teachey MK & Henriksen EJ (2006). Oxidant stress and skeletal muscle glucose transport: roles of insulin signalling and p38 MAPK. Free Radic Biol Med 41, 818–824. [DOI] [PubMed] [Google Scholar]
- Lawler JM, Powers SK, Visser T, Van Dijk H, Kordus MJ & Ji LL (1993). Acute exercise and skeletal muscle antioxidant and metabolic enzymes: effects of fibre type and age. Am J Physiol Regul Integr Comp Physiol 265, R1344–R1350. [DOI] [PubMed] [Google Scholar]
- Leeuwenburgh C, Fiebig R, Chandwaney R & Ji LL (1994). Aging and exercise training in skeletal muscle: responses of glutathione and antioxidant enzyme systems. Am J Physiol Regul Integr Comp Physiol 267, R439–R445. [DOI] [PubMed] [Google Scholar]
- Levine S, Gordon B & Derick C (1924). Some changes in the constituents of the blood following a marathon race. JAMA 82, 1778–1779. [Google Scholar]
- Manchester J, Skurat AV, Roach P, Hauschka SD & Lawrence JC Jr (1996). Increased glycogen accumulation in transgenic mice overexpressing glycogen synthase in skeletal muscle. Proc Natl Acad Sci USA 93, 10707–10711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merry TL, Dywer RM, Bradley EA, Rattigan S & McConell GK (2010. a). Local hindlimb antioxidant infusion does not affect muscle glucose uptake during in situ contractions in rat. J Appl Physiol (1985) 108, 1275–1283. [DOI] [PubMed] [Google Scholar]
- Merry TL & McConell GK (2009). Skeletal muscle glucose uptake during exercise: a focus on reactive oxygen species and nitric oxide signalling. IUBMB Life 61, 479–484. [DOI] [PubMed] [Google Scholar]
- Merry TL & McConell GK (2012). Do reactive oxygen species regulate skeletal muscle glucose uptake during contraction? Exerc Sport Sci Rev 40, 102–105. [DOI] [PubMed] [Google Scholar]
- Merry TL, Steinberg GR, Lynch GS & McConell GK (2010. b). Skeletal muscle glucose uptake during contraction is regulated by nitric oxide and ROS independently of AMPK. Am J Physiol Endocrinol Metab 298, E577–E585. [DOI] [PubMed] [Google Scholar]
- Merry TL, Wadley GD, Stathis CG, Garnham AP, Rattigan S, Hargreaves M & McConell GK (2010. c). N‐Acetylcysteine infusion does not affect glucose disposal during prolonged moderate‐intensity exercise in humans. J Physiol 588, 1623–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh‐Ishi S, Kizaki T, Yamashita H, Nagata N, Suzuki K, Taniguchi N & Ohno H (1995). Alterations of superoxide dismutase iso‐enzyme activity, content, and mRNA expression with aging in rat skeletal muscle. Mech Ageing Dev 84, 65–76. [DOI] [PubMed] [Google Scholar]
- Pederson BA, Chen H, Schroeder JM, Shou W, DePaoli‐Roach AA & Roach PJ (2004). Abnormal cardiac development in the absence of heart glycogen. Mol Cell Biol 24, 7179–7187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pederson BA, Cope CR, Schroeder JM, Smith MW, Irimia JM, Thurberg BL, DePaoli‐Roach AA & Roach PJ (2005). Exercise capacity of mice genetically lacking muscle glycogen synthase: in mice, muscle glycogen is not essential for exercise. J Biol Chem 280, 17260–17265. [DOI] [PubMed] [Google Scholar]
- Powers SK & Jackson MJ (2008). Exercise‐induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol Rev 88, 1243–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichard GA, Issekutz B Jr, Kimbel P, Putnam RC, Hochellna NJ & Weinhouse S (1961). Blood glucose metabolism in man during muscular work. J Appl Physiol 16, 1001–1005. [DOI] [PubMed] [Google Scholar]
- Ren JM, Marshall BA, Gulve EA, Gao J, Johnson DW, Holloszy JO & Mueckler M (1993). Evidence from transgenic mice that glucose transport is rate‐limiting for glycogen deposition and glycolysis in skeletal muscle. J Biol Chem 268, 16113–16115. [PubMed] [Google Scholar]
- Richter EA & Hargreaves M (2013). Exercise, GLUT4, and skeletal muscle glucose uptake. Physiol Rev 93, 993–1017. [DOI] [PubMed] [Google Scholar]
- Sahlin K, Broberg S & Katz A (1989). Glucose formation in human skeletal muscle. Influence of glycogen content. Biochem J 258, 911–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahlin K, Ekberg K & Cizinsky S (1991). Changes in plasma hypoxanthine and free radical markers during exercise in man. Acta Physiol Scand 142, 275–281. [DOI] [PubMed] [Google Scholar]
- Samuni Y, Goldstein S, Dean OM & Berk M (2013). The chemistry and biological activities of N‐acetylcysteine. Biochim Biophys Acta 1830, 4117–4129. [DOI] [PubMed] [Google Scholar]
- Sanders CA, Levinson GE, Abelmann WH & Freinkel N (1964). Effect of exercise on the peripheral utilization of glucose in man. N Engl J Med 271, 220–225. [DOI] [PubMed] [Google Scholar]
- Sandström ME, Zhang SJ, Silva JP, Reid MB, Westerblad H & Katz A (2006). Role of reactive oxygen species in contraction‐mediated glucose transport in skeletal muscle. J Physiol 575, 251–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandström ME, Zhang SJ, Westerblad H & Katz A (2007). Mechanical load plays little role in contraction‐mediated glucose transport in mouse skeletal muscle. J Physiol 579, 527–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz TA, Lewis SB, Westbie DK, Wallin JD & Gerich JE (1977). Glucose delivery: a modulator of glucose uptake in contracting skeletal muscle. Am J Physiol Endocrinol Metab 233, E514–E518. [DOI] [PubMed] [Google Scholar]
- Sylow L, Jensen TE, Kleinert M, Mouatt JR, Maarbjerg SJ, Jeppesen J, Prats C, Chiu TT, Boguslavsky S, Klip A, Schjerling P & Richter EA (2013). Rac1 is a novel regulator of contraction‐stimulated glucose uptake in skeletal muscle. Diabetes 62, 1139–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sylow L, Moller LL, Kleinert M, Richter EA & Jensen TE (2015). Stretch‐stimulated glucose transport in skeletal muscle is regulated by Rac1. J Physiol 593, 645–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoda T, Hayashi T, Miyamoto L, Yonemitsu S, Nakano M, Tanaka S, Ebihara K, Masuzaki H, Hosoda K, Inoue G, Otaka A, Sato K, Fushiki T & Nakao K (2004). Possible involvement of the α1 isoform of 5′AMP‐activated protein kinase in oxidative stress‐stimulated glucose transport in skeletal muscle. Am J Physiol Endocrinol Metab 287, E166–E173. [DOI] [PubMed] [Google Scholar]
- Wahren J, Felig P, Ahlborg G & Jorfeldt L (1971). Glucose metabolism during leg exercise in man. J Clin Invest 50, 2715–2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JS, Lee T & Chow SE (2006). Role of exercise intensities in oxidized low‐density lipoprotein‐mediated redox status of monocyte in men. J Appl Physiol (1985) 101, 740–744. [DOI] [PubMed] [Google Scholar]
- Wasserman DH (2009). Four grams of glucose. Am J Physiol Endocrinol Metab 296, E11–E21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whichelow MJ, Butterfield WJ, Abrams ME, Sterky G & Garratt CJ (1968). The effect of mild exercise on glucose uptake in human forearm tissues in the fasting state and after oral glucose administration. Metabolism 17, 84–95. [DOI] [PubMed] [Google Scholar]
- Zhang SJ, Sandström M, Ahlsen M, Ivarsson N, Zhu H, Ma J, Ren JM, Westerblad H & Katz A (2010). 2‐Methoxyoestradiol inhibits glucose transport in rodent skeletal muscle. Exp Physiol 95, 892–898. [DOI] [PubMed] [Google Scholar]
- Zhang SJ, Sandström ME, Aydin J, Westerblad H, Wieringa B & Katz A (2008). Activation of glucose transport and AMP‐activated protein kinase during muscle contraction in adenylate kinase‐1 knockout mice. Acta Physiol (Oxf) 192, 413–420. [DOI] [PubMed] [Google Scholar]
- Zhang SJ, Sandström ME, Lanner JT, Thorell A, Westerblad H & Katz A (2007). Activation of aconitase in mouse fast‐twitch skeletal muscle during contraction‐mediated oxidative stress. Am J Physiol Cell Physiol 293, C1154–C1159. [DOI] [PubMed] [Google Scholar]