

Abstract
Vascular smooth muscle cell (VSMC) phenotypic conversion from a contractile to ‘synthetic’ state contributes to vascular pathologies including restenosis, atherosclerosis and vascular calcification. We have recently found that the secretion of exosomes is a feature of ‘synthetic’ VSMCs and that exosomes are novel players in vascular repair processes as well as pathological vascular thrombosis and calcification. Pro‐inflammatory cytokines and growth factors as well as mineral imbalance stimulate exosome secretion by VSMCs, most likely by the activation of sphingomyelin phosphodiesterase 3 (SMPD3) and cytoskeletal remodelling. Calcium stress induces dramatic changes in VSMC exosome composition and accumulation of phosphatidylserine (PS), annexin A6 and matrix metalloproteinase‐2, which converts exosomes into a nidus for calcification. In addition, by presenting PS, VSMC exosomes can also provide the catalytic surface for the activation of coagulation factors. Recent data showing that VSMC exosomes are loaded with proteins and miRNA regulating cell adhesion and migration highlight VSMC exosomes as potentially important communication messengers in vascular repair. Thus, the identification of signalling pathways regulating VSMC exosome secretion, including activation of SMPD3 and cytoskeletal rearrangements, opens up novel avenues for a deeper understanding of vascular remodelling processes.
Abbreviations
- ECM
extracellular matrix
- ESCRT
endosomal sorting complex required for transport
- EV
extracellular vesicle
- MGP
matrix Gla protein
- MV
matrix vesicle
- MVB
multivesicular body
- oxLDL
oxidized phospholipid
- PDGF
platelet‐derived growth factor
- PS
phosphatidyl serine
- SM
smooth muscle
- SMPD3
sphingomyelin phosphodiesterase 3
- TNFα
tumour necrosis factor‐α
- TF
tissue factor
- VSMC
vascular smooth muscle cell
Exosomes released from vascular smooth cells mediate calcification
Mature contractile vascular smooth muscle cells (VSMCs) are quiescent cells that play a key role in the conductive function of the vasculature by maintaining vessel wall integrity and regulating arterial tone. VSMC contractile function is mediated by a unique contractile apparatus consisting, in part, of smooth muscle (SM)‐specific marker proteins such as α‐SM actin, SM myosin heavy chain, h1‐calponin, SM22α and smoothelin. Pathological cues such as ageing, oxidative stress, inflammation and mechanical injury induce phenotypic modulation of VSMCs leading to cell proliferation, migration and extracellular matrix (ECM) remodelling (Chamley‐Campbell et al. 1979; Gomez & Owens, 2012). This phenotypic modulation contributes to a range of vascular pathologies including restenosis, atherosclerosis, thoracic aortic aneurysms and dissections, transplant vasculopathy, pulmonary hypertension and vascular calcification. Recent definitive in vivo lineage studies have established a dominant role for VSMC phenotypic modulation in atherosclerosis and vascular repair (Nemenoff et al. 2011; Robbins et al. 2013; Shankman et al. 2015). In addition, conditional knockout in postnatal mice of myocardin, the master regulator of SM‐specific gene expression, induces profound loss of vascular SM contractile markers and extensive matrix remodelling leading to arterial aneurysms, aortic dissections, and rupture and death (Wang et al. 2003; Huang et al. 2015). The phenotypic plasticity of VSMCs is associated with expression of multiple markers including those from macrophages, mesenchymal stem cells, myofibrobalsts and osteoblasts (Shanahan et al. 1994; Rong et al. 2003; Shankman et al. 2015) and this is especially apparent within the atherosclerotic plaque. Importantly, the functionality of newly acquired phenotypes is limited and often maladaptive; take for example osteogenic VSMCs, which mediate vascular calcification, a pathology that has been intensively studied (Shanahan et al. 1994; Shankman et al. 2015; Vengrenyuk et al. 2015).
Recent advances have shown that vascular calcification is a tightly regulated process resembling bone mineralization (Shanahan et al. 2011) driven by VSMC osteogenic conversion and commonly observed in the aged population and patients with chronic kidney disease and type 2 diabetes. Deposition of insoluble calcium phosphate crystals reduces vascular compliance, promotes inflammation and stimulates VSMC death resulting in plaque biomechanical instability (Nadra et al. 2005; Ewence et al. 2008; Sage et al. 2011; Hutcheson et al. 2014 b). A breakthrough in calcification studies came from the identification of small membrane‐enclosed extracellular vesicles (EVs) found within the matrix and secreted by VSMCs (Tanimura et al. 1983; Reynolds et al. 2004) as well as infiltrating macrophages in atherosclerotic plaques (New et al. 2013). Similar EVs called matrix vesicles (MVs) were previously detected in the bone growth plate where they were postulated to form the nidus for mineralization (Anderson, 1995). In physiological conditions, MVs secreted by VSMCs are loaded with calcification inhibitors, such an endogenously expressed matrix Gla protein (MGP) and circulating fetuin‐A (α2‐Heremens–Schmid glycoprotein) (Reynolds et al. 2005). However, a prolonged mineral imbalance and/or inflammation induces depletion of MGP and fetuin‐A in MV and enrichment with a protein–lipid complex consisting of phosphatidyl serine (PS) and annexin A6, which converts MV to the nidus for calcification by providing mineral nucleation sites (Reynolds et al. 2005; Bobryshev et al. 2007; Shroff et al. 2008; Kapustin et al. 2011). We and others were able to observe calcifying MVs in the vasculature in vitro and in vivo, though it was not clear whether these were derived from apoptotic cells or formed in an intracellular compartment and budded from live cells. It is generally accepted that MVs secreted by bone‐derived cells are budded from the plasma membrane, suggesting similar mechanisms could be implicated in MV production by VSMCs (Anderson, 1995).
To identify the origin of calcifying VSMC MVs we used the potent calcification inhibitor fetuin‐A as a tracer. Fetuin‐A is not expressed by VSMCs yet is efficiently loaded into MVs (Reynolds et al. 2005). We found that alexa488‐labelled fetuin‐A is rapidly taken up by human VSMCs and delivered to early and late endosomal compartments (Kapustin et al. 2015). From here a subset of late endosomal compartment, multivesicular bodies (MVBs), is involved in the production of small, ∼100 nm, extracellular vesicles or exosomes which are generated by the inverted budding of the MVB limiting membrane into the MVB lumen. MVBs are transported to the cell periphery where the fusion of the MVB limiting membrane and plasma membrane releases intraluminal exosomes into the extracellular matrix (Harding et al. 1983; Raposo & Stoorvogel, 2013). We observed colocalization of fetuin‐A positive intracellular organelles and a MVB marker in VSMCs indicating that fetuin‐A is delivered to MVBs and recycled via the exosomal pathway. Further studies identified an enrichment of exosomal protein markers, tetraspanins CD9 and CD63, as well as the presence of the endosomal sorting complex required for transport (ESCRT) machinery, Tsg101, in VSMC MVs, isolated by differential ultracentrifugation. A comparison of the proteomic composition revealed high levels of similarity between VSMC‐derived MVs and exosomes secreted by other cells. The size and morphological appearance of VSMC‐derived vesicles detected with nanoparticle tracking analysis and electron microscopy also were indicative of exosome characteristics (Kapustin et al. 2015). Sphingomyelin phosphodiesterase 3 (SMPD3) has been implicated in exosome biogenesis so we next tested whether it played a role in VSMC calcification. We found that inhibition of SMPD3 blocks exosome secretion and VSMC calcification (Trajkovic et al. 2008; Kapustin et al. 2015). Moreover, elevated extracellular calcium, a known cause of calcification, induces SMPD3 expression and exosome production (Kapustin et al. 2015). Interestingly, these data are in agreement with the phenotype of fragilitas ossium (fro/fro) mice with a SMPD3 gene deletion that inactivates enzyme activity. These mice are characterized by an osteogenesis imperfecta phenotype with significant impairment in growth plate structure, short‐limbed dwarfism and undermineralised bones and teeth (Aubin et al. 2005; Khavandgar et al. 2011). Importantly, loss of SMPD3 activity in osteoblasts also dramatically reduced their mineralization capacity in vitro confirming the local role of SMDP3 during bone development (Khavandgar et al. 2011). However, further work is required to determine whether this is via a similar MV/exosome pathway as shown for VSMCs. The exact intracellular origin of macrophage‐derived EVs that contribute to vascular calcification also remains unknown.
At the molecular level calcium phosphate crystal formation is triggered in osteoblast‐derived MVs by nucleation sites consisting of PS and annexin A5 (Genge et al. 2007). Annexins were originally isolated from avian growth plate cartilage‐derived MVs as Ca2+‐dependent membrane‐binding proteins and were later identified as crucial MV nucleation core components due to their capacity to bind calcium in conjunction with PS (Genge et al. 1989; Wu et al. 1993). It has also been suggested that annexin A5 can form a voltage‐gated calcium channel (Rojas et al. 1990). However, this notion remains controversial as annexins lack transmembrane domains and calcium transport activity was not directly detected in native MVs (Gerke et al. 2005). Treatment of chondrocytes with osteogenic conditions in vitro results in elevated cytosolic calcium levels followed by accumulation of annexins A2, A5 and A6 in calcifying MVs (Kirsch et al. 1997; Wang & Kirsch, 2002). These data are in agreement with elevated cytosolic calcium detected in chondrocytes in the growth plate hypertrophic zone colocalizing with increased levels of calcifying MVs just before the onset of mineralization (Buckwalter et al. 1987; Iannotti & Brighton, 1989). Apart from annexins, accumulation of alkaline phosphatase (AP) in calcifying chondrocyte‐derived MVs has been observed in a number of studies and these data directly implicate osteogenic transcription factors in the regulation of MV composition, though the exact links are still unclear (Anderson, 1995; Kirsch et al. 1997; Wang & Kirsch, 2002). Currently the cellular origin of these chondrocyte‐derived MVs is unknown, however, the evidence that they are regulated by SMPD3 and comparative proteomics suggests that at least a subset may be of exosomal origin, but this requires further detailed investigation (Aubin et al. 2005; Kapustin et al. 2015).
Secretion of calcifying exosomes by VSMCs is also driven by pathological changes in cytosolic calcium homeostasis that triggers dramatic changes in exosome composition including enrichment with nucleating PS/AnxA6 complexes, loss of MGP and appearance of amorphous calcium phosphate (Kapustin et al. 2011, 2015). The role of AP in exosome‐mediated VSMC calcification is not definitive as no or modest changes in AP activity in EVs isolated from the media of bovine or human aortic VSMCs after short‐term treatment in calcifying conditions have been reported (Chen et al. 2008; Kapustin et al. 2011). On the contrary, MVs isolated from the media of coronary artery VSMCs after long‐term treatment in osteogenic media or MVs obtained from bovine VSMC matrix by collagenase digestion were enriched with AP (Chen et al. 2008; Hutcheson et al. 2014 a). These data suggest that osteogenic conditions may affect the VSMC EV repertoire and/or composition in a cell and context‐specific manner and the exact role of osteogenic transcription factors in the production of calcifying EVs by VSMCs is yet to be determined. Recent studies showing that BMP2‐activated Runx2 upregulates SMPD3 expression in C2C12 myoblasts and chondrocytes directly links the exosome biogenesis machinery with osteogenic master genes and further studies are required to clarify this link (Chae et al. 2009; Kakoi et al. 2014).
To summarize, VSMC exosomes are novel players in vascular calcification and their composition rapidly changes in response to environmental stresses (e.g. calcifying conditions) and alterations in exosomal lipid and protein composition can either facilitate or ameliorate the calcification process.
Are VSMC exosomes a novel trigger of vascular coagulation?
A role for EVs in the activation of coagulation pathways was originally observed for circulating platelet‐derived EVs (Wolf, 1967; Sims et al. 1988). It has been hypothesized that tumour cell‐derived EVs also contribute to high levels of vascular thrombotic events in cancer patients (Gardiner et al. 2015), and recently the levels of circulating EVs associated with tissue factor (TF), an important activator of extrinsic coagulation pathways, was linked to the increased risk of venous thromboembolism in cancer patients (Tesselaar et al. 2007). Vascular thrombosis due to atherosclerotic plaque rupture is the main cause of vascular occlusion events including myocardial infarction, unstable angina, stroke and sudden cardiac death. Exposure of TF, which is expressed by VSMCs, activated monocytes and endothelial cells, upon plaque rupture initiates the extrinsic coagulation pathway with TF forming a complex with factors VII and VIIa and thus activating factors IX and X (Nemerson, 1988; Wilcox et al. 1989; Toschi et al. 1997; Crawley et al. 2000; Mann, 2011). Vitamin K‐dependent factor X in turn binds to PS exposed on the membrane on the activated platelets or apoptotic VSMCs, macrophages or endothelial cells and forms the membrane‐associated prothrombinase complex, consisting of activated protease factor Xa, co‐factor Va and calcium (Krishnaswamy et al. 1992; Walker & Krishnaswamy, 1994; Bombeli et al. 1997; Flynn et al. 1997; Mallat et al. 1999). Prothrombinase converts prothrombin to thrombin, which catalyses fibrin clot formation and platelet activation and also stimulates protease‐activated receptor‐dependent signalling pathways regulating VSMC contraction, proliferation and migration (McNamara et al. 1993; Seasholtz et al. 1999; Patterson et al. 2001; Mann, 2011; Sevigny et al. 2011).
We reported earlier the presence of externalized PS on VSMC exosomes, which can provide the catalytic surface for binding and activation of coagulation cascade factors (Kapustin et al. 2011). Although VSMCs are one of the main sources of TF in the vasculature, it is unclear if TF is secreted associated with VSMC exosomes. Interestingly, almost all TF activity in the atherosclerotic plaque has been associated with EVs released by apoptotic or activated cells (Mallat et al. 1999). Similar to circulating EVs, vascular EVs are enriched with TF and PS and are highly procoagulant (Mallat et al. 1999; Leroyer et al. 2007). Thrombogenic EVs in the plaque have a heterogeneous origin and EVs from leukocytes, erythrocytes, VSMCs and endothelial cells, but not platelets, have been detected in the plaque (Leroyer et al. 2007). Prominently, plaque‐derived EVs are more thrombogenic than plasma‐derived EVs and this is most likely due to the presence of TF‐enriched EVs secreted by apoptotic VSMCs (Leroyer et al. 2007). Plasma membrane TF is thought to be packed into the EVs budding from the plasma membrane upon cell activation or cell apoptosis (Mallat et al. 1999; Del Conde et al. 2005; Ettelaie et al. 2014; Gardiner et al. 2015). However, immunocytochemical analysis of intracellular TF distribution in fibroblasts, VSMCs and monocytes revealed that TF resides both on the plasma membrane and within intracellular organelles, including endosomes (Schecter et al. 1997; Egorina et al. 2005; Mandal et al. 2006). Moreover, Mulder et al. (1996) utilized immunogold‐labelling to locate TF in intracellular lysosome‐like structures that exhibited high morphological resemblance to MVBs with intraluminal exosomes. Thus these data suggest that TF can be loaded into exosomes originating from VSMC MVBs. In agreement with this hypothesis, Mark Taubman's group and others showed that TF is secreted by VSMCs in small (less than 200 nm) vesicles and this secretion is tightly regulated by platelet‐derived growth factor (PDGF) and tumour necrosis factor‐α (TNFα) (Schecter et al. 1997, 2000; Llorente‐Cortes et al. 2004). These data correlate well with our recent observation showing that VSMCs secrete exosomes with an average size of ∼150 nm and their production is stimulated by PDGF and TNFα (Kapustin et al. 2015). Thus, it is likely that TF can be released by non‐apoptotic VSMCs on exosomes, which may then contribute to vascular thrombosis events.
Exosome secretion by VSMCs is modulated by phenotypic transition
Exosome secretion is a tightly regulated process that begins at the plasma membrane upon oligomerization or clustering of signalling transmembrane proteins such as the heparin sulphate proteoglycan syndecan or sortilin (Fang et al. 2007; Baietti et al. 2012; Wilson et al. 2014). Upon endocytosis these proteins are delivered to early and late endosomes where they are sorted for either degradation or exosomal recycling. Protein sorting and intraluminal vesicle biogenesis at MVBs is orchestrated by ESCRT components including Alix, Tsg101, HRS and STAM1, and protein post‐translational modifications such as ubiquitination or glycosylation can be involved in protein targeting (Buschow et al. 2005; Baietti et al. 2012; Colombo et al. 2013). Following MVB trafficking to the cellular periphery, subsequent docking to the plasma membrane is regulated by the Rab family of small GTPases and Rab11, Rab27a and Rab27b in particular (Savina et al. 2005; Ostrowski et al. 2010; Baietti et al. 2012). Lipids have also been implicated in exosome biogenesis and it has been shown that SMPD3 generates ceramide required for inward budding of the MVB membrane (Trajkovic et al. 2008). In addition, alterations in cytosolic calcium also stimulate exosome secretion, probably by regulating calcium‐dependent fusion events or by activation of calcium‐dependent calpains, proteases involved in the remodelling of the cortical cytoskeleton required for plasma membrane dynamics (Savina et al. 2005; Mellgren et al. 2007).
Environmental factors inducing VSMC phenotypic modulation include oxidized phospholipids (oxLDLs), growth factors PDGF‐BB and PDGF‐DD and proinflammatory cytokines such as TNFα or IL1β (Pidkovka et al. 2007; Deaton et al. 2009; Thomas et al. 2009; Gomez & Owens, 2012). These factors modulate expression and activity of the myocardin family of transcriptional coactivators for serum response factor (SRF) driving expression of contractile VSMC proteins (Gomez & Owens, 2012). Similar environmental factors, including oxLDLs, TNFα and oxidative stress stimulate SMPD3 activity and proliferation of VSMCs and fibroblasts (Auge et al. 1999; Tellier et al. 2007; Cinq‐Frais et al. 2015). Given the role of the SMPD3 pathway in exosome biogenesis, we tested if SMPD3 is involved in exosome secretion by VSMCs and found that inhibition of SMPD3 reduced exosome production (Trajkovic et al. 2008; Kapustin et al. 2015). Importantly, PDGF‐BB treatment reduced expression of VSMC contractile markers and increased exosome secretion whilst TGFβ1 induced VSMC marker expression and nearly abolished exosome secretion (Kapustin et al. 2015). Thus, there is an inverse correlation between VSMC phenotypic marker expression and exosome secretion, most likely driven by the SMPD3‐dependent pathway. Changes in cell phenotype, namely the epithelial‐to‐mesenchymal transition, were previously linked to elevated exosome‐like vesicle secretion though it is not clear what the links between phenotypic changes and exosome production are (Garnier et al. 2012). Interestingly, VSMC phenotype transition is driven by cytoskeleton remodelling and single mutations in the contractile proteins ACTA2 or MYH11 are enough to cause the dilatation of the thoracic aorta and aortic dissections (Guo et al. 2007; Kuang et al. 2012). Actin cytoskeletal proteins are also downstream targets of Rab27A and Rab27B, which play crucial roles in exosome secretion by regulating MVB trafficking (Seabra & Coudrier, 2004; Ostrowski et al. 2010). These data suggest the cytoskeletal changes upon VSMC phenotypic modulation might enable MVB trafficking and exosome secretion, but further studies are required to fully establish the spatiotemporal links between exosome production, VSMC phenotypic switching and cytoskeleton remodelling.
Perspectives and conclusions
Exosomes are novel intercellular communication messengers acting both in a paracrine and an autocrine manner by delivering biologically active proteins, lipids and RNA species (Raposo & Stoorvogel, 2013). In the cardiovascular system exosomes are secreted by endothelial cells, VSMCs, monocytes and cardiac progenitor cells and they act as stimulators of endothelial cell migration, proliferation and cell survival (Vrijsen et al. 2010; Li et al. 2013; van Balkom et al. 2013; Deng et al. 2015; Kapustin et al. 2015). VSMC phenotypic modulation is a hallmark of vascular repair and remodelling processes and elevated exosome secretion by ‘synthetic’ VSMCs, upon activation of SMPD3 and cytoskeletal rearrangements, highlights a potentially important physiological role for VSMC exosomes in vascular repair (Fig. 1 A and B). On one side VSMC exosomes are enriched with miR‐143 and proteins regulating cell adhesion and migration and can be involved in the regulation of cell proliferation and migration in an autocrine and paracrine manner with overall exosome effects an integer of activation of multiple signalling pathways by different exosome cargos (Deng et al. 2015; Kapustin et al. 2015). On the other hand, VSMC exosomes can contribute to pathological processes such as vascular calcification when the balance between calcification activators and inhibitors is lost (Kapustin et al. 2011, 2015). The exposure of PS on VSMC exosomes along with VSMC TF expression also implicates exosomes in the activation of coagulation cascades (Wilcox et al. 1989; Schecter et al. 2000; Leroyer et al. 2007; Kapustin et al. 2011) with further characterization required to determine the exact role of VSMC exosomes in vascular thrombosis. Importantly, environmental cues may define the composition and secretion levels of exosomes by VSMCs thus modulating their functional role and proposed contribution to vascular repair processes. Current developments in this area are limited by a number of unknowns including (1) the exact mechanisms of exosome cargo loading and (2) a lack of gold‐standard markers for different EV populations. Indeed, multiple organelle proteins are detected in exosome preparations isolated from different cell types and it is not clear whether these are loaded in the exosome or secreted as independent entities that are then co‐purified with exosomes during analytical purification. In addition, different catabolic membrane trafficking processes, such has exocytosis and authophagy, can cross‐talk with each other increasing the level of complexity and the difficulty in determining EV origins and function.
Figure 1. Functional roles for VSMC exosomes .
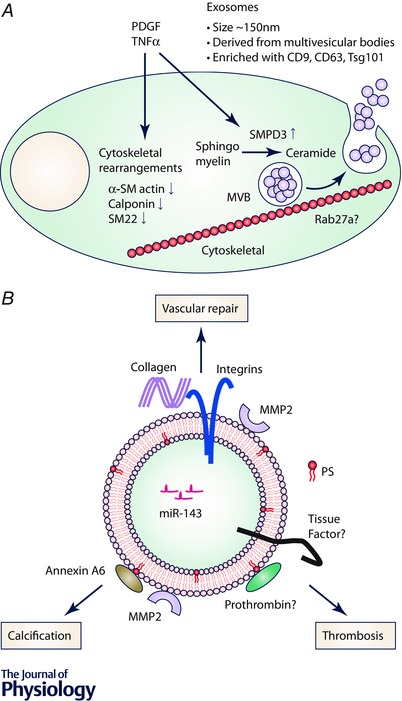
A, cytokines and growth factors induce phenotypic conversion of VSMCs accompanied by cytoskeletal remodelling and SMPD3 activation. These changes enable budding of exosomes in MVBs and MVB trafficking and docking to the plasma membrane. Exosomes are released in the extracellular space upon the fusion of MVBs and the plasma membrane. B, exosome composition defines the role of exosomes in vascular repair. Extracellular matrix proteins, integrins and miRNA stimulate vascular repair by stimulating cell adhesion and migration. Externalized PS forms the nidus for calcification by forming a complex with annexin A6. Vascular calcification is also facilitated by matrix degradation catalysed by matrix metalloproteinase‐2 (MMP2). Exosomal PS can also bind coagulation factors and may stimulate vascular thrombosis activated by tissue factor expressed by VSMCs.
Although the spatiotemporal and mechanistic relationships between pathological factors, phenotypic VSMC conversion and activation of exosome biogenesis are still unknown, the modulation of exosome biogenesis may be a novel therapeutic approach to facilitate improvement in vascular repair. Inhibition of exosome production or introduction of specifically designed cargos targeted to exosomes might prevent exosomal nucleation core formation and/or binding of coagulating factors thus blocking excessive thrombosis and unwanted calcification. However, these approaches will have to be carefully considered to ensure that the physiological roles of exosomes in vascular repair are not compromised. Finally, EVs are also promising novel diagnostic makers and their use to assess cardiovascular risk is an emerging field (Loyer et al. 2014).
Additional information
Competing interests
None of the authors has any conflicts of interests.
Authors contribution
A.N.K.: conception, manuscript writing (sections 1–4). C.M.S.: manuscript writing and editing (sections 1–4). Both authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by a Programme grant (RG/11/14/29056) from the British Heart Foundation (BHF) awarded to C.M.S.
Acknowledgements
We thank Dr Andrew Cobb for his constructive comments on this manuscript.
Biography
Catherine M. Shanahan obtained a PhD in Genetics from the University of Adelaide, Australia. Her research in cardiovascular biology began in the Biochemistry Department, University of Cambridge, UK. From there she became British Heart Foundation Lecturer and later Senior Fellow in the Department of Medicine at Cambridge. In 2007 she became Chair of Cellular Signalling in the Cardiovascular Division at King's College London. Her work focuses on mechanisms of vascular smooth muscle cell dysfunction in ageing and disease. Alexander Kapustin received his postgraduate degree from the Moscow Lomonosov State University and The Russian Cardiology Research and Production Complex (Russia). In 2007 he joined C. M. Shanahan's group at Cambridge University to study the role of matrix vesicles in vascular calcification and then moved together with the group to King's College London as a research associate. His main research interests are mechanisms of exosome biogenesis and their role in vascular repair processes.

This review was presented at ‘Extracellular vesicles, exosomes and microparticles in cardiovascular disease’ which took place at Physiology 2015, Cardiff, UK, between 6–8 July 2015.
References
- Anderson HC (1995). Molecular biology of matrix vesicles. Clin Orthop Relat Res, 266–280. [PubMed] [Google Scholar]
- Aubin I, Adams CP, Opsahl S, Septier D, Bishop CE, Auge N, Salvayre R, Negre‐Salvayre A, Goldberg M, Guenet JL & Poirier C (2005). A deletion in the gene encoding sphingomyelin phosphodiesterase 3 (Smpd3) results in osteogenesis and dentinogenesis imperfecta in the mouse. Nat Genet 37, 803–805. [DOI] [PubMed] [Google Scholar]
- Auge N, Nikolova‐Karakashian M, Carpentier S, Parthasarathy S, Negre‐Salvayre A, Salvayre R, Merrill AH Jr & Levade T (1999). Role of sphingosine 1‐phosphate in the mitogenesis induced by oxidized low density lipoprotein in smooth muscle cells via activation of sphingomyelinase, ceramidase, and sphingosine kinase. J Biol Chem 274, 21533–21538. [DOI] [PubMed] [Google Scholar]
- Baietti MF, Zhang Z, Mortier E, Melchior A, Degeest G, Geeraerts A, Ivarsson Y, Depoortere F, Coomans C, Vermeiren E, Zimmermann P & David G (2012). Syndecan‐syntenin‐ALIX regulates the biogenesis of exosomes. Nat Cell Biol 14, 677–685. [DOI] [PubMed] [Google Scholar]
- Bobryshev YV, Killingsworth MC, Huynh TG, Lord RS, Grabs AJ & Valenzuela SM (2007). Are calcifying matrix vesicles in atherosclerotic lesions of cellular origin? Basic Res Cardiol 102, 133–143. [DOI] [PubMed] [Google Scholar]
- Bombeli T, Karsan A, Tait JF & Harlan JM (1997). Apoptotic vascular endothelial cells become procoagulant. Blood 89, 2429–2442. [PubMed] [Google Scholar]
- Buckwalter JA, Mower D & Schaeffer J (1987). Differences in matrix vesicle concentration among growth plate zones. J Orthop Res 5, 157–163. [DOI] [PubMed] [Google Scholar]
- Buschow SI, Liefhebber JM, Wubbolts R & Stoorvogel W (2005). Exosomes contain ubiquitinated proteins. Blood Cells Mol Dis 35, 398–403. [DOI] [PubMed] [Google Scholar]
- Chae YM, Heo SH, Kim JY, Lee JM, Ryoo HM & Cho JY (2009). Upregulation of smpd3 via BMP2 stimulation and Runx2. BMB Rep 42, 86–90. [DOI] [PubMed] [Google Scholar]
- Chamley‐Campbell J, Campbell GR & Ross R (1979). The smooth muscle cell in culture. Physiol Rev 59, 1–61. [DOI] [PubMed] [Google Scholar]
- Chen NX, O'Neill KD, Chen X & Moe SM (2008). Annexin‐mediated matrix vesicle calcification in vascular smooth muscle cells. J Bone Miner Res 23, 1798–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinq‐Frais C, Coatrieux C, Savary A, D'Angelo R, Bernis C, Salvayre R, Negre‐Salvayre A & Auge N (2015). Annexin II‐dependent actin remodelling evoked by hydrogen peroxide requires the metalloproteinase/sphingolipid pathway. Redox Biol 4, 169–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo M, Moita C, van Niel G, Kowal J, Vigneron J, Benaroch P, Manel N, Moita LF, Thery C & Raposo G (2013). Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J Cell Sci 126, 5553–5565. [DOI] [PubMed] [Google Scholar]
- Crawley J, Lupu F, Westmuckett AD, Severs NJ, Kakkar VV & Lupu C (2000). Expression, localization, and activity of tissue factor pathway inhibitor in normal and atherosclerotic human vessels. Arterioscler Thromb Vasc Biol 20, 1362–1373. [DOI] [PubMed] [Google Scholar]
- Deaton RA, Gan Q & Owens GK (2009). Sp1‐dependent activation of KLF4 is required for PDGF‐BB‐induced phenotypic modulation of smooth muscle. Am J Physiol Heart Circ Physiol 296, H1027–H1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Conde I, Shrimpton CN, Thiagarajan P & Lopez JA (2005). Tissue‐factor‐bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood 106, 1604–1611. [DOI] [PubMed] [Google Scholar]
- Deng L, Blanco FJ, Stevens H, Lu R, Caudrillier A, McBride MW, McClure JD, Grant JS, Thomas M, Frid MG, Stenmark KR, White K, Seto AG, Morrell NW, Bradshaw AC, MacLean MR & Baker AH (2015). miR‐143 activation regulates smooth muscle and endothelial cell crosstalk in pulmonary arterial hypertension. Circ Res 117, 870–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egorina EM, Sovershaev MA, Bjorkoy G, Gruber FX, Olsen JO, Parhami‐Seren B, Mann KG & Osterud B (2005). Intracellular and surface distribution of monocyte tissue factor: application to intersubject variability. Arterioscler Thromb Vasc Biol 25, 1493–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettelaie C, Collier ME, Maraveyas A & Ettelaie R (2014). Characterization of physical properties of tissue factor‐containing microvesicles and a comparison of ultracentrifuge‐based recovery procedures. J Extracell Vesicles 3, doi: 10.3402/jev.v3.23592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewence AE, Bootman M, Roderick HL, Skepper JN, McCarthy G, Epple M, Neumann M, Shanahan CM & Proudfoot D (2008). Calcium phosphate crystals induce cell death in human vascular smooth muscle cells: a potential mechanism in atherosclerotic plaque destabilization. Circ Res 103, e28–e34. [DOI] [PubMed] [Google Scholar]
- Fang Y, Wu N, Gan X, Yan W, Morrell JC & Gould SJ (2007). Higher‐order oligomerization targets plasma membrane proteins and HIV gag to exosomes. PLoS Biol 5, e158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn PD, Byrne CD, Baglin TP, Weissberg PL & Bennett MR (1997). Thrombin generation by apoptotic vascular smooth muscle cells. Blood 89, 4378–4384. [PubMed] [Google Scholar]
- Gardiner C, Harrison P, Belting M, Boing A, Campello E, Carter BS, Collier ME, Coumans F, Ettelaie C, van Es N, Hochberg FH, Mackman N, Rennert RC, Thaler J, Rak J & Nieuwland R (2015). Extracellular vesicles, tissue factor, cancer and thrombosis – discussion themes of the ISEV 2014 Educational Day. J Extracell Vesicles 4, 26901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnier D, Magnus N, Lee TH, Bentley V, Meehan B, Milsom C, Montermini L, Kislinger T & Rak J (2012). Cancer cells induced to express mesenchymal phenotype release exosome‐like extracellular vesicles carrying tissue factor. J Biol Chem 287, 43565–43572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genge BR, Wu LN & Wuthier RE (1989). Identification of phospholipid‐dependent calcium‐binding proteins as constituents of matrix vesicles. J Biol Chem 264, 10917–10921. [PubMed] [Google Scholar]
- Genge BR, Wu LN & Wuthier RE (2007). In vitro modeling of matrix vesicle nucleation: synergistic stimulation of mineral formation by annexin A5 and phosphatidylserine. J Biol Chem 282, 26035–26045. [DOI] [PubMed] [Google Scholar]
- Gerke V, Creutz CE & Moss SE (2005). Annexins: linking Ca2+ signalling to membrane dynamics. Nat Rev Mol Cell Biol 6, 449–461. [DOI] [PubMed] [Google Scholar]
- Gomez D & Owens GK (2012). Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc Res 95, 156–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo DC, Pannu H, Tran‐Fadulu V, Papke CL, Yu RK, Avidan N, Bourgeois S, Estrera AL, Safi HJ, Sparks E, Amor D, Ades L, McConnell V, Willoughby CE, Abuelo D, Willing M, Lewis RA, Kim DH, Scherer S, Tung PP, Ahn C, Buja LM, Raman CS, Shete SS & Milewicz DM (2007). Mutations in smooth muscle α‐actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet 39, 1488–1493. [DOI] [PubMed] [Google Scholar]
- Harding C, Heuser J & Stahl P (1983). Receptor‐mediated endocytosis of transferrin and recycling of the transferrin receptor in rat reticulocytes. J Cell Biol 97, 329–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Wang T, Wright AC, Yang J, Zhou S, Li L, Yang J, Small A & Parmacek MS (2015). Myocardin is required for maintenance of vascular and visceral smooth muscle homeostasis during postnatal development. Proc Natl Acad Sci USA 112, 4447–4452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutcheson JD, Goettsch C, Pham T, Iwashita M, Aikawa M, Singh SA & Aikawa E (2014. a). Enrichment of calcifying extracellular vesicles using density‐based ultracentrifugation protocol. J Extracell Vesicles 3, 25129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutcheson JD, Maldonado N & Aikawa E (2014. b). Small entities with large impact: microcalcifications and atherosclerotic plaque vulnerability. Curr Opin Lipidol 25, 327–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannotti JP & Brighton CT (1989). Cytosolic ionized calcium concentration in isolated chondrocytes from each zone of the growth plate. J Orthop Res 7, 511–518. [DOI] [PubMed] [Google Scholar]
- Kakoi H, Maeda S, Shinohara N, Matsuyama K, Imamura K, Kawamura I, Nagano S, Setoguchi T, Yokouchi M, Ishidou Y & Komiya S (2014). Bone morphogenic protein (BMP) signaling up‐regulates neutral sphingomyelinase 2 to suppress chondrocyte maturation via the Akt protein signaling pathway as a negative feedback mechanism. J Biol Chem 289, 8135–8150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapustin AN, Chatrou ML, Drozdov I, Zheng Y, Davidson SM, Soong D, Furmanik M, Sanchis P, De Rosales RT, Alvarez‐Hernandez D, Shroff R, Yin X, Muller K, Skepper JN, Mayr M, Reutelingsperger CP, Chester A, Bertazzo S, Schurgers LJ & Shanahan CM (2015). Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ Res 116, 1312–1323. [DOI] [PubMed] [Google Scholar]
- Kapustin AN, Davies JD, Reynolds JL, McNair R, Jones GT, Sidibe A, Schurgers LJ, Skepper JN, Proudfoot D, Mayr M & Shanahan CM (2011). Calcium regulates key components of vascular smooth muscle cell‐derived matrix vesicles to enhance mineralization. Circ Res 109, e1–e12. [DOI] [PubMed] [Google Scholar]
- Khavandgar Z, Poirier C, Clarke CJ, Li J, Wang N, McKee MD, Hannun YA & Murshed M (2011). A cell‐autonomous requirement for neutral sphingomyelinase 2 in bone mineralization. J Cell Biol 194, 277–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch T, Nah HD, Shapiro IM & Pacifici M (1997). Regulated production of mineralization‐competent matrix vesicles in hypertrophic chondrocytes. J Cell Biol 137, 1149–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnaswamy S, Field KA, Edgington TS, Morrissey JH & Mann KG (1992). Role of the membrane surface in the activation of human coagulation factor X. J Biol Chem 267, 26110–26120. [PubMed] [Google Scholar]
- Kuang SQ, Kwartler CS, Byanova KL, Pham J, Gong L, Prakash SK, Huang J, Kamm KE, Stull JT, Sweeney HL & Milewicz DM (2012). Rare, nonsynonymous variant in the smooth muscle‐specific isoform of myosin heavy chain, MYH11, R247C, alters force generation in the aorta and phenotype of smooth muscle cells. Circ Res 110, 1411–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroyer AS, Isobe H, Leseche G, Castier Y, Wassef M, Mallat Z, Binder BR, Tedgui A & Boulanger CM (2007). Cellular origins and thrombogenic activity of microparticles isolated from human atherosclerotic plaques. J Am Coll Cardiol 49, 772–777. [DOI] [PubMed] [Google Scholar]
- Li J, Zhang Y, Liu Y, Dai X, Li W, Cai X, Yin Y, Wang Q, Xue Y, Wang C, Li D, Hou D, Jiang X, Zhang J, Zen K, Chen X & Zhang CY (2013). Microvesicle‐mediated transfer of microRNA‐150 from monocytes to endothelial cells promotes angiogenesis. J Biol Chem 288, 23586–23596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorente‐Cortes V, Otero‐Vinas M, Camino‐Lopez S, Llampayas O & Badimon L (2004). Aggregated low‐density lipoprotein uptake induces membrane tissue factor procoagulant activity and microparticle release in human vascular smooth muscle cells. Circulation 110, 452–459. [DOI] [PubMed] [Google Scholar]
- Loyer X, Vion AC, Tedgui A & Boulanger CM (2014). Microvesicles as cell‐cell messengers in cardiovascular diseases. Circ Res 114, 345–353. [DOI] [PubMed] [Google Scholar]
- Mallat Z, Hugel B, Ohan J, Leseche G, Freyssinet JM & Tedgui A (1999). Shed membrane microparticles with procoagulant potential in human atherosclerotic plaques: a role for apoptosis in plaque thrombogenicity. Circulation 99, 348–353. [DOI] [PubMed] [Google Scholar]
- Mandal SK, Pendurthi UR & Rao LV (2006). Cellular localization and trafficking of tissue factor. Blood 107, 4746–4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann KG (2011). Thrombin generation in hemorrhage control and vascular occlusion. Circulation 124, 225–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara CA, Sarembock IJ, Gimple LW, Fenton JW 2nd, Coughlin SR & Owens GK (1993). Thrombin stimulates proliferation of cultured rat aortic smooth muscle cells by a proteolytically activated receptor. J Clin Invest 91, 94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellgren RL, Zhang W, Miyake K & McNeil PL (2007). Calpain is required for the rapid, calcium‐dependent repair of wounded plasma membrane. J Biol Chem 282, 2567–2575. [DOI] [PubMed] [Google Scholar]
- Mulder AB, Smit JW, Bom VJ, Blom NR, Ruiters MH, Halie MR & van der Meer J (1996). Association of smooth muscle cell tissue factor with caveolae. Blood 88, 1306–1313. [PubMed] [Google Scholar]
- Nadra I, Mason JC, Philippidis P, Florey O, Smythe CD, McCarthy GM, Landis RC & Haskard DO (2005). Proinflammatory activation of macrophages by basic calcium phosphate crystals via protein kinase C and MAP kinase pathways: a vicious cycle of inflammation and arterial calcification? Circ Res 96, 1248–1256. [DOI] [PubMed] [Google Scholar]
- Nemenoff RA, Horita H, Ostriker AC, Furgeson SB, Simpson PA, VanPutten V, Crossno J, Offermanns S & Weiser‐Evans MC (2011). SDF‐1alpha induction in mature smooth muscle cells by inactivation of PTEN is a critical mediator of exacerbated injury‐induced neointima formation. Arterioscler Thromb Vasc Biol 31, 1300–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemerson Y (1988). Tissue factor and hemostasis. Blood 71, 1–8. [PubMed] [Google Scholar]
- New SE, Goettsch C, Aikawa M, Marchini JF, Shibasaki M, Yabusaki K, Libby P, Shanahan CM, Croce K & Aikawa E (2013). Macrophage‐derived matrix vesicles: an alternative novel mechanism for microcalcification in atherosclerotic plaques. Circ Res 113, 72–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrowski M, Carmo NB, Krumeich S, Fanget I, Raposo G, Savina A, Moita CF, Schauer K, Hume AN, Freitas RP, Goud B, Benaroch P, Hacohen N, Fukuda M, Desnos C, Seabra MC, Darchen F, Amigorena S, Moita LF & Thery C (2010). Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat Cell Biol 12, 19–30. [DOI] [PubMed] [Google Scholar]
- Patterson C, Stouffer GA, Madamanchi N & Runge MS (2001). New tricks for old dogs: nonthrombotic effects of thrombin in vessel wall biology. Circ Res 88, 987–997. [DOI] [PubMed] [Google Scholar]
- Pidkovka NA, Cherepanova OA, Yoshida T, Alexander MR, Deaton RA, Thomas JA, Leitinger N & Owens GK (2007). Oxidized phospholipids induce phenotypic switching of vascular smooth muscle cells in vivo and in vitro. Circ Res 101, 792–801. [DOI] [PubMed] [Google Scholar]
- Raposo G & Stoorvogel W (2013). Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol 200, 373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds JL, Joannides AJ, Skepper JN, McNair R, Schurgers LJ, Proudfoot D, Jahnen‐Dechent W, Weissberg PL & Shanahan CM (2004). Human vascular smooth muscle cells undergo vesicle‐mediated calcification in response to changes in extracellular calcium and phosphate concentrations: a potential mechanism for accelerated vascular calcification in ESRD. J Am Soc Nephrol 15, 2857–2867. [DOI] [PubMed] [Google Scholar]
- Reynolds JL, Skepper JN, McNair R, Kasama T, Gupta K, Weissberg PL, Jahnen‐Dechent W & Shanahan CM (2005). Multifunctional roles for serum protein fetuin‐a in inhibition of human vascular smooth muscle cell calcification. J Am Soc Nephrol 16, 2920–2930. [DOI] [PubMed] [Google Scholar]
- Robbins CS, Hilgendorf I, Weber GF, Theurl I, Iwamoto Y, Figueiredo JL, Gorbatov R, Sukhova GK, Gerhardt LM, Smyth D, Zavitz CC, Shikatani EA, Parsons M, van Rooijen N, Lin HY, Husain M, Libby P, Nahrendorf M, Weissleder R & Swirski FK (2013). Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat Med 19, 1166–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas E, Pollard HB, Haigler HT, Parra C & Burns AL (1990). Calcium‐activated endonexin II forms calcium channels across acidic phospholipid bilayer membranes. J Biol Chem 265, 21207–21215. [PubMed] [Google Scholar]
- Rong JX, Shapiro M, Trogan E & Fisher EA (2003). Transdifferentiation of mouse aortic smooth muscle cells to a macrophage‐like state after cholesterol loading. Proc Natl Acad Sci USA 100, 13531–13536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sage AP, Lu J, Tintut Y & Demer LL (2011). Hyperphosphatemia‐induced nanocrystals upregulate the expression of bone morphogenetic protein‐2 and osteopontin genes in mouse smooth muscle cells in vitro. Kidney Int 79, 414–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savina A, Fader CM, Damiani MT & Colombo MI (2005). Rab11 promotes docking and fusion of multivesicular bodies in a calcium‐dependent manner. Traffic 6, 131–143. [DOI] [PubMed] [Google Scholar]
- Schecter AD, Giesen PL, Taby O, Rosenfield CL, Rossikhina M, Fyfe BS, Kohtz DS, Fallon JT, Nemerson Y & Taubman MB (1997). Tissue factor expression in human arterial smooth muscle cells. TF is present in three cellular pools after growth factor stimulation. J Clin Invest 100, 2276–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schecter AD, Spirn B, Rossikhina M, Giesen PL, Bogdanov V, Fallon JT, Fisher EA, Schnapp LM, Nemerson Y & Taubman MB (2000). Release of active tissue factor by human arterial smooth muscle cells. Circ Res 87, 126–132. [DOI] [PubMed] [Google Scholar]
- Seabra MC & Coudrier E (2004). Rab GTPases and myosin motors in organelle motility. Traffic 5, 393–399. [DOI] [PubMed] [Google Scholar]
- Seasholtz TM, Majumdar M, Kaplan DD & Brown JH (1999). Rho and Rho kinase mediate thrombin‐stimulated vascular smooth muscle cell DNA synthesis and migration. Circ Res 84, 1186–1193. [DOI] [PubMed] [Google Scholar]
- Sevigny LM, Austin KM, Zhang P, Kasuda S, Koukos G, Sharifi S, Covic L & Kuliopulos A (2011). Protease‐activated receptor‐2 modulates protease‐activated receptor‐1‐driven neointimal hyperplasia. Arterioscler Thromb Vasc Biol 31, e100–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanahan CM, Cary NR, Metcalfe JC & Weissberg PL (1994). High expression of genes for calcification‐regulating proteins in human atherosclerotic plaques. J Clin Invest 93, 2393–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanahan CM, Crouthamel MH, Kapustin A & Giachelli CM (2011). Arterial calcification in chronic kidney disease: key roles for calcium and phosphate. Circ Res 109, 697–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, Swiatlowska P, Newman AA, Greene ES, Straub AC, Isakson B, Randolph GJ & Owens GK (2015). KLF4‐dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med 21, 628–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shroff RC, Shah V, Hiorns MP, Schoppet M, Hofbauer LC, Hawa G, Schurgers LJ, Singhal A, Merryweather I, Brogan P, Shanahan C, Deanfield J & Rees L (2008). The circulating calcification inhibitors, fetuin‐A and osteoprotegerin, but not matrix Gla protein, are associated with vascular stiffness and calcification in children on dialysis. Nephrol Dial Transplant 23, 3263–3271. [DOI] [PubMed] [Google Scholar]
- Sims PJ, Faioni EM, Wiedmer T & Shattil SJ (1988). Complement proteins C5b‐9 cause release of membrane vesicles from the platelet surface that are enriched in the membrane receptor for coagulation factor Va and express prothrombinase activity. J Biol Chem 263, 18205–18212. [PubMed] [Google Scholar]
- Tanimura A, McGregor DH & Anderson HC (1983). Matrix vesicles in atherosclerotic calcification. Proc Soc Exp Biol Med 172, 173–177. [DOI] [PubMed] [Google Scholar]
- Tellier E, Negre‐Salvayre A, Bocquet B, Itohara S, Hannun YA, Salvayre R & Auge N (2007). Role for furin in tumor necrosis factor alpha‐induced activation of the matrix metalloproteinase/sphingolipid mitogenic pathway. Mol Cell Biol 27, 2997–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesselaar ME, Romijn FP, Van Der Linden IK, Prins FA, Bertina RM & Osanto S (2007). Microparticle‐associated tissue factor activity: a link between cancer and thrombosis? J Thromb Haemost 5, 520–527. [DOI] [PubMed] [Google Scholar]
- Thomas JA, Deaton RA, Hastings NE, Shang Y, Moehle CW, Eriksson U, Topouzis S, Wamhoff BR, Blackman BR & Owens GK (2009). PDGF‐DD, a novel mediator of smooth muscle cell phenotypic modulation, is upregulated in endothelial cells exposed to atherosclerosis‐prone flow patterns. Am J Physiol Heart Circ Physiol 296, H442–H452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toschi V, Gallo R, Lettino M, Fallon JT, Gertz SD, Fernandez‐Ortiz A, Chesebro JH, Badimon L, Nemerson Y, Fuster V & Badimon JJ (1997). Tissue factor modulates the thrombogenicity of human atherosclerotic plaques. Circulation 95, 594–599. [DOI] [PubMed] [Google Scholar]
- Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F, Schwille P, Brugger B & Simons M (2008). Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 319, 1244–1247. [DOI] [PubMed] [Google Scholar]
- van Balkom BW, de Jong OG, Smits M, Brummelman J, den Ouden K, de Bree PM, van Eijndhoven MA, Pegtel DM, Stoorvogel W, Wurdinger T & Verhaar MC (2013). Endothelial cells require miR‐214 to secrete exosomes that suppress senescence and induce angiogenesis in human and mouse endothelial cells. Blood 121, 3997–4006, S3991–S3915. [DOI] [PubMed] [Google Scholar]
- Vengrenyuk Y, Nishi H, Long X, Ouimet M, Savji N, Martinez FO, Cassella CP, Moore KJ, Ramsey SA, Miano JM & Fisher EA (2015). Cholesterol loading reprograms the microRNA‐143/145‐myocardin axis to convert aortic smooth muscle cells to a dysfunctional macrophage‐like phenotype. Arterioscler Thromb Vasc Biol 35, 535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrijsen KR, Sluijter JP, Schuchardt MW, van Balkom BW, Noort WA, Chamuleau SA & Doevendans PA (2010). Cardiomyocyte progenitor cell‐derived exosomes stimulate migration of endothelial cells. J Cell Mol Med 14, 1064–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker RK & Krishnaswamy S (1994). The activation of prothrombin by the prothrombinase complex. The contribution of the substrate‐membrane interaction to catalysis. J Biol Chem 269, 27441–27450. [PubMed] [Google Scholar]
- Wang W & Kirsch T (2002). Retinoic acid stimulates annexin‐mediated growth plate chondrocyte mineralization. J Cell Biol 157, 1061–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Wang DZ, Pipes GC & Olson EN (2003). Myocardin is a master regulator of smooth muscle gene expression. Proc Natl Acad Sci USA 100, 7129–7134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox JN, Smith KM, Schwartz SM & Gordon D (1989). Localization of tissue factor in the normal vessel wall and in the atherosclerotic plaque. Proc Natl Acad Sci USA 86, 2839–2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CM, Naves T, Vincent F, Melloni B, Bonnaud F, Lalloue F & Jauberteau MO (2014). Sortilin mediates the release and transfer of exosomes in concert with two tyrosine kinase receptors. J Cell Sci 127, 3983–3997. [DOI] [PubMed] [Google Scholar]
- Wolf P (1967). The nature and significance of platelet products in human plasma. Br J Haematol 13, 269–288. [DOI] [PubMed] [Google Scholar]
- Wu LN, Yoshimori T, Genge BR, Sauer GR, Kirsch T, Ishikawa Y & Wuthier RE (1993). Characterization of the nucleational core complex responsible for mineral induction by growth plate cartilage matrix vesicles. J Biol Chem 268, 25084–25094. [PubMed] [Google Scholar]