

Abstract
Objective(s):
In the present study, C57BL/6 female mice (n=56) were used to explore the neuroprotective effects of riboflavin in motor disability of experimental autoimmune encephalomyelitis (EAE) as a model of multiple sclerosis.
Materials and Methods:
The animals were assigned into 7 groups: sham-operated 1 (SO1), healthy mice receiving PBS (phosphate buffer saline); sham-operated 2 (SO2), healthy mice receiving PBS and riboflavin; sham treatment 1 (ST1), EAE mice receiving water; sham treatment 2 (ST2), EAE mice receiving sodium acetate buffer; treatment 1 (T1), EAE mice receiving interferon beta-1a (INFβ-1a); treatment 2 (T2), EAE mice receiving riboflavin; treatment 3 (T3), EAE mice receiving INFβ-1a and riboflavin. After EAE induction, scoring was performed based on clinical signs. Upon detecting score 0.5, riboflavin at 10 mg/kg of body weight and/or INFβ-1a at 150 IU/g of body weight administration was started for two weeks. The brain and spinal cord levels of brain-derived neurotrophic factor (BDNF), interleukin-6 (IL-6), and interleukin-17A (IL-17A) were studied using real-time PCR and ELISA methods.
Results:
BDNF expression and protein levels were increased in the brain and spinal cord of the T3 group compared with the other groups (P<0.01). IL-6 and IL-17A expressions were increased in the brains of the T3 and T1 groups, respectively, compared to the other groups (P<0.01). The daily clinical score was reduced significantly by riboflavin in both effector and chronic phases of the disease compared with that of the controls (P<0.05).
Conclusion:
Our findings showed that riboflavin is capable of suppressing the neurological disability mediated by BDNF and IL-6.
Keywords: Brain-derived neurotrophic -factor, Experimental autoimmune -encephalomyelitis, Interleukin-17A, Interleukin-6, Motor disability, Riboflavin
Introduction
Multiple sclerosis (MS) is a neuroinflammatory disease mediated by immune-cell in the central nervous system (CNS) (1). Experimental autoimmune encephalomyelitis (EAE) is a commonly employed animal model of MS. Several studies report the critical pathogenic role of inflammatory cytokines in the pathogenesis of MS/EAE (2, 3). In this context, interleukin-6 (IL-6) protects against reactive oxygen species (ROS) excitotoxicity decreasing the neuronal death in MS (4). IL-6 induces differentiation of T-cells into Th17 cells, which secrete interleukin-17A (IL-17A).
IL-6 production in astrocytes is stimulated by IL-17 in a positive feedback loop (4).
Brain-derived neurotrophic factor (BDNF) is an important regulator of neuronal maturation and protection which is expressed in and around MS lesions (5).
Interferon beta (INFβ) is one of the most widely used drugs for MS, which is involved in the regulation of cytokines (6) and inhibition of IL-17 secretion (7). However, a major limitation of this drug is that 30 to 50 percent of MS patients do not respond to INFβ therapy (8).
Riboflavin is an essential nutrient in the human diet (9); it is important in the myelin formation (10-15). Studies have shown that riboflavin deficiency in young chickens leads to general demyelinating polyneuropathy (12, 13, 15-20). Lipids of myelin, cerebrosides, sphingomyelin, and phosphatidylethanolamines, as critical components of the myelin sheath, are reduced in riboflavin deficiency. Riboflavin is also involved in the regulating processes of the nervous system (21). Reduced tissue levels of the riboflavin-based coenzymes flavin adenine dinucleotide (FAD) and flavin mononucleotide (FMN), leading to reduced cellular energy levels, may be associated with neurologic abnormalities characterized by segmental demyelination in riboflavin deficit chickens (15).
Increased production of ROS and oxidative damage have been reported in the pathogenesis of MS and EAE (22, 23). Riboflavin is one of the antioxidant nutrients that may protect the body against oxidative stress via the glutathione redox cycle (24). We assess whether dietary antioxidants along with conventional treatments might be beneficial in MS treatment, and aimed to find whether riboflavin may have neuroprotective effects in motor disability of EAE. Furthermore, it was proposed that these effects can be mediated through gene expression and protein levels of BDNF and immunologic factors including IL-6 and IL-17 in the CNS of EAE. The answers to these questions could eventually be applied to predict the possible role of a new nutrition therapy aspect in MS patients.
Materials and Methods
Animals
Ten-week-old C57BL/6 female adult mice (weighing 20–25 g) were purchased from Pasteur Institute (Tehran, Iran) and housed in standard cages under the following controlled conditions: 12 hr light/dark cycle; temperature 20±2°C and 50–60 % relative humidity with ad libitum access to food and water. Animals were preserved for 1 week for acclimation. Measures to improve welfare assistance and clinical status, as well as endpoint criteria, were established to minimize suffering and ensure animal welfare. Additionally, wet food pellets were placed on the bed-cage when the animals began to develop clinical signs to facilitate access to food and water.
The mice (n=56) were assigned into 7 groups randomly (8 in each) (2) as follows:
1) Sham+ phosphate buffer saline (PBS) (sham operated 1 or SO1), mice receiving PBS as vehicle (Veh) of pertussis toxin (PTX); 2) Sham +PBS+ Riboflavin (sham operated 2 or SO2), mice receiving PBS and riboflavin (Puritan’s Pride Co., 1233 Montauk Highway Oakdale, NY 11769-9001, United States); 3) EAE+Veh1 (EAE Sham treatment 1 or ST1), mice received the same volume of water (as vehicle of riboflavin); 4) EAE+Veh2 (Sham treatment 2 or ST2), EAE mice received sodium acetate buffer (as vehicle of interferon beta-1a (INFβ-1a)); 5) EAE+ INFβ-1a (treatment 1 or T1), EAE mice received INFβ-1a (150 IU/g of body weight, subcutaneously; 6) EAE+ Riboflavin (treatment 2 or T2), EAE mice received riboflavin (at 10 mg/kg of body weight, as gavage); 7) EAE+ INFβ-1a + Riboflavin (treatment 3 or T3), EAE mice received INFβ-1a + riboflavin.
EAE induction and experimental groups
EAE was induced in the female mice at +10 weeks of age by Hooke Kit™ (Hooke labs, EK2110, Lawrence, MA, USA) according to the manufacturer’s guidelines as follows: After inhaled anesthesia with ether to minimize stress, immunization was done by injecting 0.1 ml Myelin Oligodendrocyte Glycoprotein-35-55 (MOG35-55) emulsion in complete Freund’s adjuvant (CFA) to the flanks of each mouse (0.2 ml/mouse) subcutaneously, followed by administration of pertussis toxin (PTX) in PBS within 2 hr and 22–26 hr after injection of the emulsion intraperitoneally (0.1 ml/mouse) (25, 26). Healthy control animals in sham groups were incubated with PBS without PTX and MOG35-55. Treatment started on the first day of clinical signs observation (days 9–14 post-immunization) and consisted of daily oral gavage of riboflavin/water for two weeks (27, 28) or of the medication alone with INFβ-1a (RECIGEN®, CinnaGen Co., Tehran, Iran) /sodium acetate buffer, subcutaneously for the following two weeks (8).
Clinical evaluation
Mice were checked for the clinical symptoms of the EAE daily starting on one day before immunization until 35 days after immunization (dpi). The animals were daily evaluated for the signs of EAE according to Soleimani’s procedure (29) using the 10 score system as follows: 0, no clinical disease; 0.5, partial tail paralysis; 1.0, complete tail paralysis; 1.5, complete tail paralysis and discrete hind limb weakness; 2.0, complete tail paralysis and strong hind limb weakness; 2.5, unilateral hind limb paralysis; 3, complete hind limb paralysis; 3.5, hind limb paralysis and forelimb weakness; 4.0, complete paralysis (tetraplegia); 5.0: moribund or dead. Three clinical parameters were analyzed in order to compare the course of EAE: to detect the severity of disease, cumulative disease index (CDI) score was calculated as the average of the sum of daily clinical scores for each mouse. The disease onset was calculated as the average of the first day of clinical symptom for each mouse in the group. The peak disease score was calculated as the average of the highest individual score for each mouse in the group.
Tissue harvesting and sectioning
At the end of the each stage, animals were anesthetized deeply with ketamine (90 mg/kg) and xylazine (10 mg/kg) (30). Then, the animals were sacrificed; the whole brain and the spinal cord were removed and rinsed in ice-cold PBS (0.02 mol/l, pH 7.0–7.2) to remove excess blood thoroughly. Specimens were placed on an ice-cold surface, cut in half, and weighed. Both brain hemispheres and spinal cord were snap-frozen in liquid nitrogen and stored at −80 °C until further processing (31).
Molecular and biochemical assays
In order to measure all biochemical and molecular parameters in the same region, the whole brain and spinal cord were removed for further assays (32).
RNA preparation and cDNA synthesis
Total RNA was extracted from whole brain and spinal cord with RNeasy® Lipid Tissue Mini Kit (Qiagen®, Hilden, Germany) according to the manufacturer’s instructions. All gene expression results were shown as arbitrary units relative to expression of the gene encoding β-actin. cDNA was synthesized from ≤ 5 µg of total RNA using the oligo(dt)12-18 primers (Invitrogen™ by Life Technologies, Frankfurt, Germany) according to the manufacturer’s protocol.
Quantitative real-time PCR
mRNAs were quantified for BDNF, IL-6, and IL-17A using a step one fluorescence-based real-time PCR (Applied Biosystems, Foster City, CA, USA) using mouse TaqMan® probe (Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s instructions. For each gene, the samples were run in triplicate and the experiments were repeated three times (33). The number of targets, normalized to an endogenous reference was defined by the Ct (threshold cycle) method for comparing the relative expression ratio in real-time PCR between the treatment and the control groups (34).
Enzyme-linked immunosorbent assay (ELISA)
BDNF protein levels were assayed using BDNF EMAX® ImmunoAssay System (Promega, Madison, USA) (35). IL-6 and IL-17A levels were assayed using ELISA kits according to their manufacturer’s instructions (Diacolon, France) (1).
Statistical methods
For analyzing the ELISA, real-time PCR, and clinical observation at different phases of EAE data, ANOVA test followed by LSD post hoc test were applied. (1). GEE (Generalized Estimating Equations) model was used to compare the clinical scores between the experimental groups and evaluate the interaction between riboflavin and INFβ-1a (36). All results were presented as mean and the standard error of the mean (mean± SEM). A P-value less than 0.05 was considered significant. Analyses were conducted using SPSS for Windows® v. 21 (SPSS Inc., Chicago, USA).
All procedures were carried out in accordance with the conventional guidelines for experimentation with animals (NIH Publication, revised 1996) and approved by the Ethics Committee for Animal Experimentation at the Ahvaz Jundishapur University of Medical Sciences (AJUMS, NRC-9208).
Results
Riboflavin supplementation increases BDNF expression in the brain and spinal cord and also IL-6 expression in the brain
Real-time PCR revealed that BDNF expression was dramatically increased (P<0.01) in the brain and spinal cord of the T3 EAE group compared with the intact and EAE mice groups (Figures 1a and 1b). Also, IL-6 expression was increased in the brain of the T3 EAE group compared with intact and EAE mice groups (Figure 1c, P<0.01).
Figure 1.
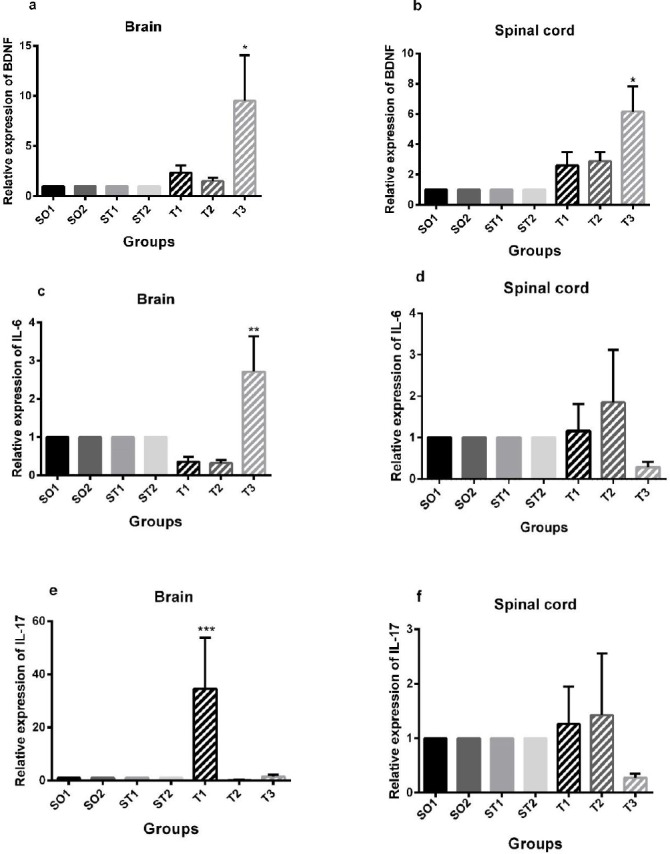
Real-time PCR results of relative expression of brain-derived neurotrophic factor (BDNF), interleukin-6 (IL-6), and interleukin-17A (IL-17A) mRNA in the brain and spinal cord. (a) (c) Animals in the T3 group showed significantly increased relative expression of BDNF and IL-6 in the brain compared to sham operated, sham-treated, and treatment groups (*P<0.01, **P<0.01). (e) Experimental autoimmune encephalomyelitis (EAE) mice in the T1 group showed significantly increased relative expression of IL-17A in the brain compared to sham operated, sham-treated, and treatment groups (***P<0.01). (b) Animals in the T3 group showed significantly increased relative expression of BDNF in the spinal cord compared to sham operated, sham-treated, and treatment groups (*P<0.01). Data are given as mean ± SEM analyzed by one-way ANOVA and LSD’s post hoc test. SO1 (sham operated 1, sham +PBS); SO2 (sham operated 2, sham+ PBS+ riboflavin); ST1 (sham treatment 1, EAE+ Veh1); ST2 (sham treatment 2, EAE +Veh2); T1 (treatment 1, EAE + INFβ-1a); T2 (treatment 2, EAE +riboflavin); T3 (treatment 3, EAE + INFβ-1a + riboflavin); BDNF (brain-derived neurotrophic factor). The experiments were repeated three times in duplicates. (n = 8 for all groups)
INFβ-1a but not riboflavin supplementation increases IL-17A expression in the brain
IL-17A expression was increased (P<0.01) in the brains of the T1 EAE group as compared to that of intact and EAE mice groups (Figure 1e).
Riboflavin supplementation increases protein levels of BDNF in the brain and spinal cord
A markedly increased BDNF protein level in the whole brain of T3 EAE animals was seen compared to ST2, T1, and T2 EAE mice (P= 0.048; P= 0.041, P=0.023, respectively, Figure 2a). Animals in T1 and T2 groups showed significantly increased levels of BDNF in the spinal cord compared to the healthy controls and EAE mice (P <0.01, Figure 2b). In the sham+PBS group, a significant increase in BDNF levels was observed in the spinal cord but not in the brain of the mice compared to the other groups (Sham+PBS+Riboflavin and EAE animals, Figure 2b). No significant differences were found between the experimental groups in terms of IL-6 and IL-17A levels in the brain (Figures 2c and 2e) and spinal cord (Figures 2d and 2f).
Figure 2.
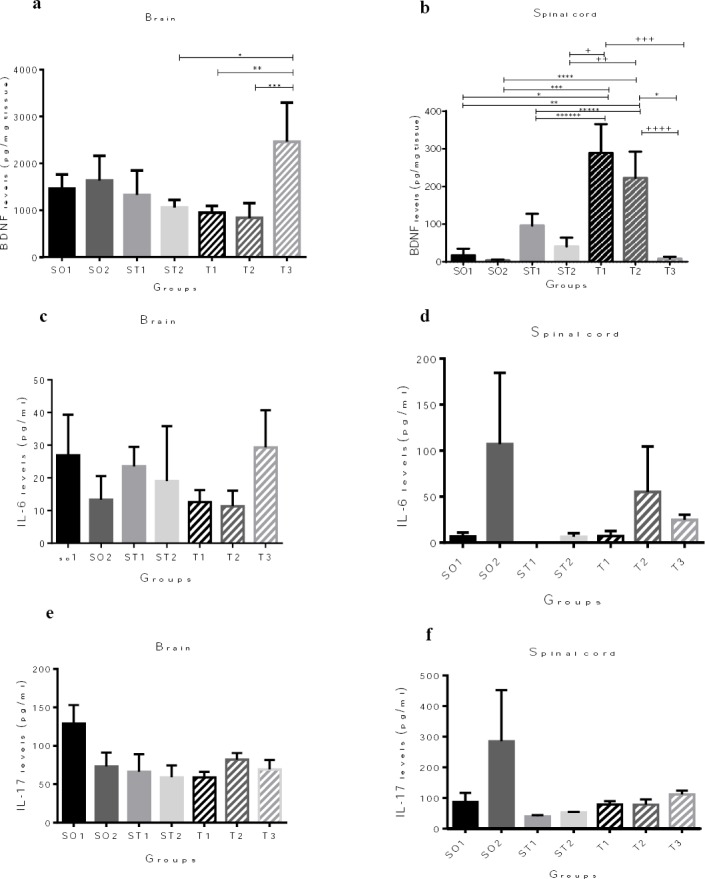
Protein quantification of the brain-derived neurotrophic factor (BDNF), interleukin-6 (IL-6), and interleukin-17A (IL-17A) expression in the whole brain and spinal cord by enzyme-linked immunosorbent assay (ELISA). (a) Animals in T3 group showed significantly increased BDNF protein levels in the brain compared to ST2, T1, and T2 experimental autoimmune encephalomyelitis (EAE) mice (*P = 0.048; ** P = 0.041, *** P =0.023, respectively). (c), (d), (e), and (f) There are no differences between the experimental groups in terms of IL-6 and IL-17 protein levels in the brain and spinal cord. (b) Animals in SO1 and SO2 groups, also EAE animals in ST1, ST2, and T3 groups showed significantly decreased BDNF expression in the spinal cord compared to EAE mice in T1 and T2 groups (*P= 0.001, **P= 0.008, *** P = 0.000; **** P = 0.001, ***** P = 0.003; ****** P= 0.041, + P = 0.001; ++ P = 0.008, +++ P = 0.000, ++++ P =0.001). Results are shown as pg/mg tissue BDNF and pg/ml IL-6 and Il-17. Data are given as mean ± SEM analyzed by one way ANOVA and LSD’s post hoc test. SO1 (sham operated 1, sham+ PBS); SO2 (sham operated 2, sham+ PBS+ riboflavin); ST1 (sham treatment 1, EAE+ Veh1); ST2 (sham treatment 2, EAE +Veh2); T1 (treatment 1, EAE + INFβ-1a); T2 (treatment 2, EAE +Riboflavin); T3 (Treatment 3, EAE + INFβ-1a + Riboflavin); BDNF (brain-derived neurotrophic factor). The experiments were repeated three times in triplicates. (n = 8 for all groups)
Riboflavin supplementation ameliorates motor disability in EAE Mice
There was a significant difference between all study groups in terms of clinical score means in both effector and chronic phases of the disease. Clinical score of the disease was significantly lower in the riboflavin treated group compared with that of the controls at both effector and chronic phases (P<0.05, Table 1).
Table 1.
Comparison of the daily clinical score between the study groups in different phases of experimental autoimmune encephalomyelitis (EAE)
| Disease phases | Groups | Mean± SEM |
|---|---|---|
| Effector | ST1 | 1.8 ±0.0* |
| ST2 | 1.3±0.0* | |
| T1 | 2.7±0.03* | |
| T2 | 1.1±0.02* | |
| T3 | 1.1±0.02* | |
| Chronic | ST1 | 1.8±0.01* |
| ST2 | 1.3±0.0* | |
| T1 | 2.6±0.01* | |
| T2 | 1.20±0.03* | |
| T3 | 1 ±0.01* | |
Daily clinical score was significantly different between all experimental study groups and reduced significantly in the riboflavin treated groups rather than others at both two phase of the EAE (P<0.01). ANOVA followed by the LSD’s post hoc test was applied to analyze data. Effector phase (days 26–35 after immunization); chronic phase (days 30 to 35 post-immunization); ST1 (sham treatment 1, EAE+ Veh1); ST2 (sham treatment 2, EAE +Veh2); T1 (treatment 1, EAE + INFβ-1a); T2 (treatment 2, EAE +riboflavin); T3 (Treatment 3, EAE + INFβ-1a + riboflavin)
Animals in T1 group showed higher EAE disease severity as measured by CDI (41.7± 5.2) compared with ST2, T2, and T3 groups (22.8±6.3, 23.5±2.8, and 19 ± 4.9, respectively; P<0.05, Figure 3a). In the T1 EAE mice, peak disease score means were significantly higher than ST2 and T3 EAE mice (P<0.016, Figure 3b). T2 and T3 EAE mice showed significantly delayed onset of disease compared to ST1 EAE mice (P<0.04, Figure 3c).
Figure 3.
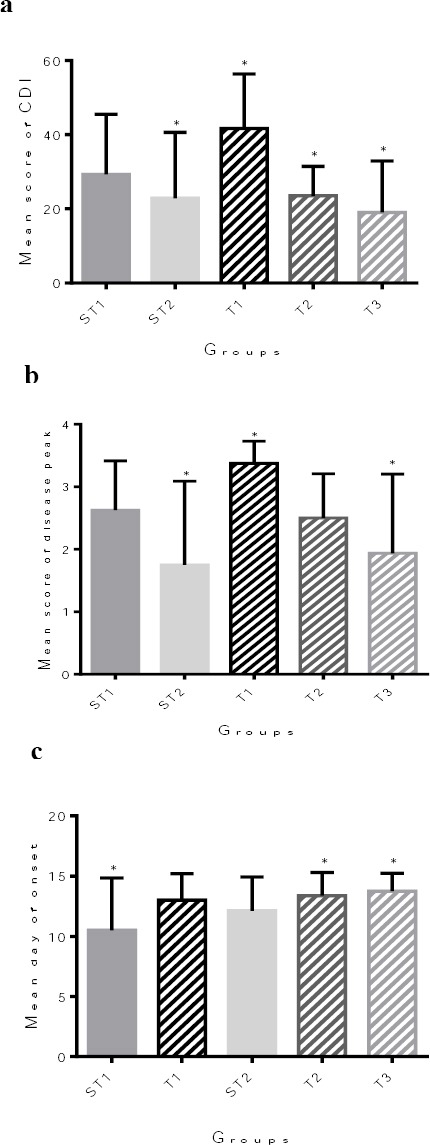
Effects of the riboflavin supplementation on motor disability in experimental autoimmune encephalomyelitis (EAE) mice. (a) Starting from day 7 post immunization (p.i.), T1 EAE mice (n = 8) showed significantly higher mean of the CDI score than ST2 (n = 8), T1 (n = 8), T2 (n = 8), and T3 (n = 8) EAE mice (*P<0.032, one-way ANOVA test). (b) In the T1 and T3 EAE mice, peak disease score means were significantly less than ST2 EAE mice (*P<0.016, one-way ANOVA test). (c) T2 and T3 EAE mice showed significantly delayed onset of disease compared with ST1 EAE mice (n = 10) (*P<0.04, one-way ANOVA test). CDI (cumulative disease index, daily clinical scores for each mouse); Peak disease score (the mean of the highest individual score for each mouse in the group); Disease Onset (averaging the first day of clinical signs for each mouse); ST1 (sham treatment 1, EAE+ Veh1); ST2 (sham treatment 2, EAE +Veh2); T1 (treatment 1, EAE + INFβ-1a); T2 (treatment 2, EAE +Riboflavin); T3 (treatment 3, EAE + INFβ-1a + riboflavin). (n = 8 for all groups)
GEE model showed that INFβ-1a worsens the clinical scores (P=0.249). Riboflavin alone and in combination with INFβ-1a improved the clinical scores (P<0.01, Figure 4).
Figure 4.
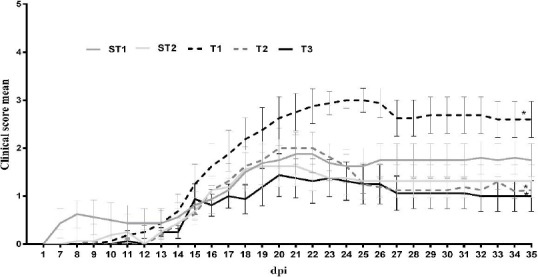
Clinical assessment of experimental autoimmune encephalomyelitis (EAE) mice. Clinical signs (n =8 mice/group) were monitored daily and the EAE mice were compared for 35 dpi. The intervention protocol was started after observation of the first signs of EAE induction and was continued for 2 weeks. Differences were observed between the EAE mice in different groups from 11 to 15 dpi (*P< 0.05). In order to study the interactions between riboflavin and INFβ-1a on motor disability score, GEE (generalized estimating equations) model was used. Test of Model Effect indicated that concomitant use of riboflavin and INFβ-1a has a better effect than the use of INFβ-1a alone. ST1 (sham treatment 1, EAE+ Veh1); ST2 (sham treatment 2, EAE +Veh2); T1 (treatment 1, EAE + INFβ-1a); T2 (treatment 2, EAE +riboflavin); T3 (treatment 3, EAE + INFβ-1a + riboflavin). (n= 8 for all groups)
GEE model showed that INFβ-1a worsens the clinical scores (P=0.249). Riboflavin alone and in combination with INFβ-1a improved the clinical scores (P<0.01, Figure 4).
Discussion
To our knowledge, this is the first report that has surveyed the effect of riboflavin supplementation on the motor disability and the levels of neurotrophins and cytokines in the brain and spinal cord of the EAE model of MS, addressing a new aspect of MS treatment. The results of this study demonstrated that riboflavin has positive effects on the EAE disease process. Riboflavin alone and in concordant use with INFβ-1a diminished the clinical severity of EAE and had an inhibitory effect on the EAE course, started at the onset of the disease and continued to the peak (in both effector and chronic phases). We have previously shown that riboflavin supplementation (10 mg/day) for six months significantly reduced the expanded disability status scale (EDSS) for MS patients by 26.4 % compared with MS patients taking a placebo (15.4 %) (37).
We also evaluated the effect of riboflavin and INFβ-1a combination on neurologic disability. The purpose of this combination was to evaluate the possible interactions with riboflavin supplement. GEE model illustrated that INFβ-1a worsens the clinical scores, whereas combined administration of the INFβ-1a and riboflavin is more effective than the INFβ-1a administration alone (Figure 4). Since, lower CDI scores and late onset were observed in both groups receiving the combination of riboflavin/INFβ-1a and also riboflavin alone, and on the other hand, there were no differences between the groups receiving the combination of riboflavin and INFβ-1a and riboflavin alone in terms of clinical score, it can be concluded that the improvement might be attributed to riboflavin administration independent of INFβ-1a.
These effects were associated with modulation of the neurotrophic and immunologic activities. To investigate the role of immunologic mediators on riboflavin-mediated amelioration of the disease, the cytokines concentrations were evaluated in CNS. We hypothesized that by administrating riboflavin, BDNF protein levels would be increased while the levels of IL-6 (inflammation-regulatory) and IL-17A (pro-inflammatory) in the whole brain and spinal cord of EAE mice would be reduced. To test our hypothesis, we assessed the levels of BDNF, IL-6, and IL-17A in the whole brain and spinal cord of the healthy controls and EAE animals. This analysis seems relevant because cytokines play important roles in the polarization of immune responses. In this context, IL-6 expression was increased in the whole brain of EAE mice receiving both riboflavin and INFβ-1a, and IL-17A expression was increased in the whole brain of EAE mice receiving INFβ-1a compared to those of other experimental groups (Figure 1).
To investigate the role of neurotrophic mediators in riboflavin-related amelioration of the disease, the expression and protein levels of BDNF were evaluated in the CNS. We showed a markedly increased gene expression (P<0.01) and protein levels (P<0.05) of BDNF in the whole brain of EAE mice receiving both riboflavin and INFβ-1a compared to sham and other treated EAE groups.
Studies have shown that riboflavin is involved in the metabolism of essential fatty acids in the brain lipids and the pathological effects of riboflavin deficiency are similar to fatty acid deficiency leading to rapid onset of abnormal development and maturation of the brain (10). Riboflavin selectively interferes in the peripheral nerve myelin synthesis (11). Moreover, mitochondrial dysfunction is known to cause neuronal injury (23). In addition, riboflavin has a role as a cofactor for succinate dehydrogenase in the complex II of mitochondrial respiratory chain generating intracellular adenosine triphosphate (ATP) (38). Since, CNS response to circulating neurotrophins depends on the intracellular ATP availability, enhancement of mitochondrion survival and bio-energy with riboflavin increases its responsiveness to BDNF neurotrophin followed by increased production of myelin in MS (39).
However, in this study, BDNF protein levels were higher in the spinal cord of mice receiving either riboflavin or INFβ-1a compared to the healthy controls and other EAE mice. This unexpected result is inconsistent with our hypothesis about the beneficial effects of IFNβ-1a with riboflavin according to clinical scores of animals. Higher levels of BDNF in the spinal cord of mice receiving IFNβ-1a can be interpreted as follows. As previous studies have mentioned, IFN-β effects are specific for each disease state & immune subsets. IFN-β is the most effective in relapsing-remitting MS (RRMS) regulated by T cells and the least effective in progressive MS, which has chronic plaque with the least number of T-cells and a large number of monocytes (40). This different treatment effect may explain why IFNβ-1a therapy is effective in increasing the levels of BDNF in the spinal cord but not the brain. Moreover, Th17 plays an essential role in the MS process (41). However, no study has evaluated the role of BDNF in the regulation of Th17 cells. It is possible that the differentiation of these cells in the early stages of maturation has been regulated by BDNF. Also, BDNF may act in Th17 cells protection against activation-induced apoptosis (42). At the moment, more studies are needed to confirm the role of BDNF in the regulation of Th17 cells.
Moreover, it seems that mRNA expression and protein levels of BDNF are dramatically up-regulated in inflammatory pain models (43). Peripheral tissue inflammation induces an increased BDNF synthesis in the dorsal root ganglion and an elevated anterograde transport of BDNF to the spinal dorsal horn (44). Here, it may be thought that the gene expression and protein levels of BDNF observed in the treatment groups of the present study have been due to increased BDNF levels in inflammatory pain models. However, given that the EAE sham groups receiving water as Veh 1 (in ST1 group) and sodium acetate buffer as Veh 2 (in ST2 group) showed no significant increase in the gene expression and protein levels of BDNF in the whole brain compared with treatment groups, it could be accepted that the increased BDNF levels in the brain may be caused by riboflavin and/or IFNβ-1a administration.
In this study, we have shown an increased expression of IL-6 coincided with increased expression of BDNF in the whole brain and the spinal cord of EAE mice receiving both riboflavin and INFβ-1a. Another study sought the immune factors that regulate the BDNF levels and affect the survival of neuronal cells in vitro (5). IL-6 probably has a proliferative effect on astrocytes in vitro (45, 46), synergistically with other factors (47). In vitro assays showed that both virus-infected microglia and astrocytes secrete IL6, and that IL-6 induces the secretion of the neurotrophin nerve growth factor (NGF) by the latter (48). Microglia proliferates when stimulated with IL-6 in the culture, and IL-6 probably promotes survival through inducing BDNF (49). It was observed that IL-6 induces the neuronal differentiation of the rat pheochromocytoma PC12 cell line, similar to the prototypical NGF neurotrophin (50, 51). In vivo, IL-6 knockout mice do not up-regulate this neurotrophin in dorsal root ganglia (DRG) following nerve injury (49). As a result, IL-6 behaves in a neurotrophin-like fashion and seemingly makes understandable why the cytokine family is known as neuropoietins (4). Therefore, it was not surprising that both BDNF and IL-6 expression were increased following the administration of riboflavin and INFβ-1a throughout the brain.
In our study, we identified that disability is reduced in EAE mice treated with both riboflavin and riboflavin+INFβ-1a, while EAE mice treated with INFβ-1a did not respond to this medicine and the disease worsened. Studies showed that INFβ is ineffective in treating Th17-induced EAE and symptoms increase in these mice (8). One proposed hypothesis is that these non-responders have the aggressive Th17 disease. To counteract inflammation, their immune system up-regulates INFβ. Since endogenous INFβ levels are already high, INFβ treatment is ineffective. A second hypothesis is that INFβ is pro-inflammatory during Th17 disease (52). Not only INFβ treatment would be ineffective, but it could also worsen the symptoms (8). This result is in concordance with the findings on RRMS, in which the patients with high IL-17F and INFβ show exacerbated disease. Thus, INFβ has polar effects in different contexts, leading to benefit in Th1 conditions, but harm in Th17 conditions (8). Moreover, it has been shown that IFNβ alone could not inhibit IL-17 production in purified naïve CD4 cells (8). Th17 inhibition likely requires the synergistic effects of IFNβ and IFNγ. Indeed, it was found that IFNβ or IFNγ alone could not attenuate IL-17 production, but, IFNβ and IFNγ together reduced IL-17 (8). It is likely that the treatment prescribed in this study has prevented the effect of INFβ-1a on protein levels of IL-17 by reducing the levels of IFNγ in the brain and the spinal cord.
The limitation of our study was lack of a group for controlling the EAE mice receiving the combination of riboflavin and INFβ. However, since there was no statistically significant difference between ST1 (sham treatment 1, EAE+ Veh1) and ST2 (sham treatment 2, EAE +Veh2) groups, this limitation can be bridged.
Results of this study may demonstrate a new approach for the application of riboflavin supplementation in EAE and MS and therefore, suggest further studies to investigate the possible role of using riboflavin in amelioration of severity of MS.
Conclusion
Our findings showed that supplementation of riboflavin increases the gene expression and protein levels of BDNF in the whole brain and the spinal cord as well as an increase in IL-6 gene expression in the whole brain in EAE mice suggesting that IL-6 and BDNF could have a role in the observed beneficial effects of riboflavin on neurological motor disability and therefore, they could be a target of strategic therapies. These findings strongly suggest that riboflavin dietary supplementation can be regarded as a possible therapeutic agent working with INFβ-1a to reduce the deleterious effects of neurological disability in an EAE model of MS.
Acknowledgment
The results reported in this paper were part of Mahshid Naghashpour’s PhD thesis. This study was performed in Physiology and Nutrition/Metabolic Disease Research Centers, Ahvaz, Iran. It was supported by the Vice-Chancellor for Research, Ahvaz Jundishapur University of Medical Sciences (grant No: NRC-9208), Ahvaz, Iran. Also, CinnaGen® Co., Tehran, Iran, provided the RECIGEN®. Funding sources were not involved in the conduct of the research and/or preparation of the article. We wish to thank Ahmad Rouhizadeh for his kind help in biochemical analysis, and Mohammad Reza Rashidi Nooshabadi for his kind help in providing the riboflavin supplement.
Reference
- 1.Bernardes D, Oliveira-Lima OC, Silva TV, Faraco CC, Leite HR, Juliano MA, et al. Differential brain and spinal cord cytokine and BDNF levels in experimental autoimmune encephalomyelitis are modulated by prior and regular exercise. J Neuroimmunol. 2013;264:24–34. doi: 10.1016/j.jneuroim.2013.08.014. [DOI] [PubMed] [Google Scholar]
- 2.Kim do Y, Hao J, Liu R, Turner G, Shi FD, Rho JM. Inflammation-mediated memory dysfunction and effects of a ketogenic diet in a murine model of multiple sclerosis. PLoS One. 2012;7:e35476. doi: 10.1371/journal.pone.0035476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Srivastava P, Mujtaba M, Singhal M. Gene and Cytokines expression of Multiple Sclerosis and its Therapeutic Regimen:A Systemic Review. Int J Drug Dev & Res. 2012;4:55–66. [Google Scholar]
- 4.Erta M, Quintana A, Hidalgo J. Interleukin-6, a major cytokine in the central nervous system. Int J Biol Sci. 2012;8:1254–66. doi: 10.7150/ijbs.4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Azoulay D, Urshansky N, Karni A. Low and dysregulated BDNF secretion from immune cells of MS patients is related to reduced neuroprotection. J Neuroimmunol. 2008;195(1-2) doi: 10.1016/j.jneuroim.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 6.Dhib-Jalbut S, Marks S. Interferon-beta mechanisms of action in multiple sclerosis. Neurology. 2010;74(Suppl 1):S17–24. doi: 10.1212/WNL.0b013e3181c97d99. [DOI] [PubMed] [Google Scholar]
- 7.Ramgolam VS, Sha Y, Jin J, Zhang X, Markovic-Plese S. IFN-beta inhibits human Th17 cell differentiation. J Immunol. 2009;183:5418–27. doi: 10.4049/jimmunol.0803227. [DOI] [PubMed] [Google Scholar]
- 8.Axtell RC, de Jong BA, Boniface K, van der Voort LF, Bhat R, De Sarno P, et al. T helper type 1 and 17 cells determine efficacy of interferon-beta in multiple sclerosis and experimental encephalomyelitis. Nat Med. 2010;16:406–12. doi: 10.1038/nm.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gallager M. The Nutrients and Their Metabolism. In: Mahan L ES, editor. Krausés Food & Nutrition Therapy. Philadelphia: Saunders; 2008. pp. 74–8. 84-6. [Google Scholar]
- 10.Ogunleye AJ OA. The effect of riboflavin deficiency on cerebrum and cerebellum of developing rat brain. J Nutr Sci Vitaminol (Tokyo) 1989;35:193–7. doi: 10.3177/jnsv.35.193. [DOI] [PubMed] [Google Scholar]
- 11.Cai Z, Blumbergs PC, Finnie JW, Manavis J, Thompson PD. Selective vulnerability of peripheral nerves in avian riboflavin deficiency demyelinating polyneuropathy. Vet Pathol. 2009;46:88–96. doi: 10.1354/vp.46-1-88. [DOI] [PubMed] [Google Scholar]
- 12.Johnson WD, Storts RW. Peripheral neuropathy associated with dietary riboflavin deficiency in the chicken. I. Light microscopic study. Vet Pathol. 1988;25:9–16. doi: 10.1177/030098588802500102. [DOI] [PubMed] [Google Scholar]
- 13.Cai Z, Finnie JW, Blumbergs PC. Avian riboflavin deficiency:an acquired tomaculous neuropathy. Vet Pathol. 2006;43:780–1. doi: 10.1354/vp.43-5-780. [DOI] [PubMed] [Google Scholar]
- 14.Wada Y, Kondo H, Itakura C. Peripheral neuropathy of dietary riboflavin deficiency in racing pigeons. J Vet Med Sci. 1996;58(2):161–3. doi: 10.1292/jvms.58.161. [DOI] [PubMed] [Google Scholar]
- 15.Jortner BS, Cherry J, Lidsky TI, Manetto C, Shell L. Peripheral neuropathy of dietary riboflavin deficiency in chickens. J Neuropathol Exp Neurol. 1987;46:544–55. doi: 10.1097/00005072-198709000-00004. [DOI] [PubMed] [Google Scholar]
- 16.Cai Z, Blumbergs PC, Finnie JW, Manavis J, Thompson PD. Novel fibroblastic onion bulbs in a demyelinating avian peripheral neuropathy produced by riboflavin deficiency. Acta Neuropathol. 2007;114:187–94. doi: 10.1007/s00401-007-0215-8. [DOI] [PubMed] [Google Scholar]
- 17.Cai Z, Finnie JW, Blumbergs PC, Manavis J, Ghabriel MN, Thompson PD. Early paranodal myelin swellings (tomacula) in an avian riboflavin deficiency model of demyelinating neuropathy. Exp Neurol. 2006;198:65–71. doi: 10.1016/j.expneurol.2005.10.028. [DOI] [PubMed] [Google Scholar]
- 18.Phillips PH ER. Neuromalacia Associated with Low Riboflavin Diets, a Preliminary Report. Poul Sci. 1983;17:463–5. [Google Scholar]
- 19.Phillips PH ER. The histopathology of neuromalacia and “curled toe” paralysis in the chick fed low riboflavin diets. J Nutr. 1983;16:451–63. [Google Scholar]
- 20.Wyatt RD TH, Donaldson WE, Hamilton PB. A new description of riboflavin deficiency syndrome in chickens. Poul Sci. 1973;52:237–44. doi: 10.3382/ps.0520237. [DOI] [PubMed] [Google Scholar]
- 21.Ghadirian P, Jain M, Ducic S, Shatenstein B, Morisset R. Nutritional factors in the aetiology of multiple sclerosis:a case-control study in Montreal, Canada. Int J Epidemiol. 1998;27:845–52. doi: 10.1093/ije/27.5.845. [DOI] [PubMed] [Google Scholar]
- 22.Mao P, Reddy PH. Is multiple sclerosis a mitochondrial disease? Biochim Biophys Acta. 2010;1802:66–79. doi: 10.1016/j.bbadis.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kalinowska-Lyszczarz A, Losy J. The role of neurotrophins in multiple sclerosis-pathological and clinical implications. Int J Mol Sci. 2012;13:13713–25. doi: 10.3390/ijms131013713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ashoori M, Saedisomeolia A. Riboflavin (vitamin B2) and oxidative stress:a review. Br J Nutr. 2014:1–7. doi: 10.1017/S0007114514000178. [DOI] [PubMed] [Google Scholar]
- 25.Bittner S, Afzali AM, Wiendl H, Meuth SG. Myelin oligodendrocyte glycoprotein (MOG35-55) induced experimental autoimmune encephalomyelitis (EAE) in C57BL/6 mice. J Vis Exp. 2014:1–5. doi: 10.3791/51275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kafami L, Raza M, Razavi A, Mirshafiey A, Movahedian M, Khorramizadeh MR. Intermittent feeding attenuates clinical course of experimental autoimmune encephalomyelitis in C57BL/6 mice. Avicenna J Med Biotechnol. 2010;2:47–52. [PMC free article] [PubMed] [Google Scholar]
- 27.Feitoza FC VL. The response of young and adult rats to the riboflavin supplementation. Braz. arch. biol. technol. (4):855–60. [Google Scholar]
- 28.Liu J, Wu JQ, Yang JJ, Wei JY, Gao WN, Guo CJ. Metabolomic study on vitamins B(1), B(2), and PP supplementation to improve serum metabolic profiles in mice under acute hypoxia based on (1)H NMR analysis. Biomed Environ Sci. 2010;23:312–8. doi: 10.1016/S0895-3988(10)60069-4. [DOI] [PubMed] [Google Scholar]
- 29.Soleimani M, Jameie SB, Barati M, Mehdizadeh M, Kerdari M. Effects of coenzyme Q10 on the ratio of TH1/TH2 in experimental autoimmune encephalomyelitis model of multiple sclerosis in C57BL/6. Iran Biomed J. 2014;18:203–11. doi: 10.6091/ibj.13362.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu Q, Ming Z, Dart AM, Du XJ. Optimizing dosage of ketamine and xylazine in murine echocardiography. Clin Exp Pharmacol Physiol. 2007;34:499–507. doi: 10.1111/j.1440-1681.2007.04601.x. [DOI] [PubMed] [Google Scholar]
- 31.Datta SC, Opp MR. Lipopolysaccharide-induced increases in cytokines in discrete mouse brain regions are detectable using Luminex xMAP technology. J Neurosci Methods. 2008;175:119–24. doi: 10.1016/j.jneumeth.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ogonovszky H, Berkes I, Kumagai S, Kaneko T, Tahara S, Goto S, et al. The effects of moderate-, strenuous- and over-training on oxidative stress markers, DNA repair, and memory, in rat brain. Neurochem Int. 2005;46:635–40. doi: 10.1016/j.neuint.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 33.Nosrat IV, Margolskee RF, Nosrat CA. Targeted taste cell-specific overexpression of brain-derived neurotrophic factor in adult taste buds elevates phosphorylated TrkB protein levels in taste cells, increases taste bud size, and promotes gustatory innervation. J Biol Chem. 2012;287:16791–800. doi: 10.1074/jbc.M111.328476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu W FE, Begum F, Vora P, Au K, Gong Y, MacNeil B, Pillai P, Namaka M. The role of dorsal root ganglia activation and brain-derive neurotrophic factor in multiple sclerosis. J Cell Mol Med. 2012;16:1856–65. doi: 10.1111/j.1582-4934.2011.01481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mieno MN, Yamaguchi T, Ohashi Y. Alternative statistical methods for estimating efficacy of interferon beta-1b for multiple sclerosis clinical trials. BMC Med Res Methodol. 2011;11:80. doi: 10.1186/1471-2288-11-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Naghashpour M, Majdinasab N, Shakerinejad G, Kouchak M, Haghighizadeh MH, Jarvandi F, et al. Riboflavin supplementation to patients with multiple sclerosis does not improve disability status nor is riboflavin supplementation correlated to homocysteine. Int J Vitam Nutr Res. 2013;83:281–90. doi: 10.1024/0300-9831/a000170. [DOI] [PubMed] [Google Scholar]
- 38.Cicek G, Schiltz E, Hess D, Staiger J, Brandsch R. Analysis of mitochondrial antigens reveals inner membrane succinate dehydrogenase flavoprotein subunit as autoantigen to antibodies in anti-M7 sera. Clin Exp Immunol. 2002;128:83–7. doi: 10.1046/j.1365-2249.2002.01816.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reese D, Shivapour ET, Wahls TL, Dudley-Javoroski SD, Shields R. Neuromuscular electrical stimulation and dietary interventions to reduce oxidative stress in a secondary progressive multiple sclerosis patient leads to marked gains in function:a case report. Cases J. 2009;2:7601. doi: 10.4076/1757-1626-2-7601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hamamcioglu K, Reder AT. Interferon-beta regulates cytokines and BDNF:greater effect in relapsing than in progressive multiple sclerosis. Mult Scler. 2007;13:459–70. doi: 10.1177/1352458506069672. [DOI] [PubMed] [Google Scholar]
- 41.El-behi M, Rostami A, Ciric B. Current views on the roles of Th1 and Th17 cells in experimental autoimmune encephalomyelitis. J Neuroimmune Pharmacol. 2010;5:189–97. doi: 10.1007/s11481-009-9188-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.De Santi L, Annunziata P, Sessa E, Bramanti P. Brain-derived neurotrophic factor and TrkB receptor in experimental autoimmune encephalomyelitis and multiple sclerosis. J Neurol Sci. 2009;287:17–26. doi: 10.1016/j.jns.2009.08.057. [DOI] [PubMed] [Google Scholar]
- 43.Pezet S, Malcangio M, McMahon SB. BDNF:a neuromodulator in nociceptive pathways? Brain Res Brain Res Rev. 2002;40:240–9. doi: 10.1016/s0165-0173(02)00206-0. [DOI] [PubMed] [Google Scholar]
- 44.Cho HJ, Kim JK, Zhou XF, Rush RA. Increased brain-derived neurotrophic factor immunoreactivity in rat dorsal root ganglia and spinal cord following peripheral inflammation. Brain Res. 1997;764:269–72. doi: 10.1016/s0006-8993(97)00597-0. [DOI] [PubMed] [Google Scholar]
- 45.Benveniste EN, Whitaker JN, Gibbs DA, Sparacio SM, Butler JL. Human B cell growth factor enhances proliferation and glial fibrillary acidic protein gene expression in rat astrocytes. Int Immunol. 1989;1:219–28. doi: 10.1093/intimm/1.3.219. [DOI] [PubMed] [Google Scholar]
- 46.Selmaj KW, Farooq M, Norton WT, Raine CS, Brosnan CF. Proliferation of astrocytes in vitro in response to cytokines. A primary role for tumor necrosis factor. J Immunol. 1990;144:129–35. [PubMed] [Google Scholar]
- 47.Levison SW, Jiang FJ, Stoltzfus OK, Ducceschi MH. IL-6-type cytokines enhance epidermal growth factor-stimulated astrocyte proliferation. Glia. 2000;32:328–37. doi: 10.1002/1098-1136(200012)32:3<328::aid-glia110>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 48.Frei K, Malipiero UV, Leist TP, Zinkernagel RM, Schwab ME, Fontana A. On the cellular source and function of interleukin 6 produced in the central nervous system in viral diseases. Eur J Immunol. 1989;19:689–94. doi: 10.1002/eji.1830190418. [DOI] [PubMed] [Google Scholar]
- 49.Murphy PG, Borthwick LA, Altares M, Gauldie J, Kaplan D, Richardson PM. Reciprocal actions of interleukin-6 and brain-derived neurotrophic factor on rat and mouse primary sensory neurons. Eur J Neurosci. 2000;12:1891–9. doi: 10.1046/j.1460-9568.2000.00074.x. [DOI] [PubMed] [Google Scholar]
- 50.Nakafuku M, Satoh T, Kaziro Y. Differentiation factors, including nerve growth factor, fibroblast growth factor, and interleukin-6, induce an accumulation of an active Ras.GTP complex in rat pheochromocytoma PC12 cells. J Biol Chem. 1992;267:19448–54. [PubMed] [Google Scholar]
- 51.Satoh T, Nakamura S, Taga T, Matsuda T, Hirano T, Kishimoto T, et al. Induction of neuronal differentiation in PC12 cells by B-cell stimulatory factor 2/interleukin 6. Mol Cell Biol. 1988;8:3546–9. doi: 10.1128/mcb.8.8.3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Galligan CL, Pennell LM, Murooka TT, Baig E, Majchrzak-Kita B, Rahbar R, et al. Interferon-beta is a key regulator of proinflammatory events in experimental autoimmune encephalomyelitis. Mult Scler. 2010;16:1458–73. doi: 10.1177/1352458510381259. [DOI] [PubMed] [Google Scholar]