

Abstract
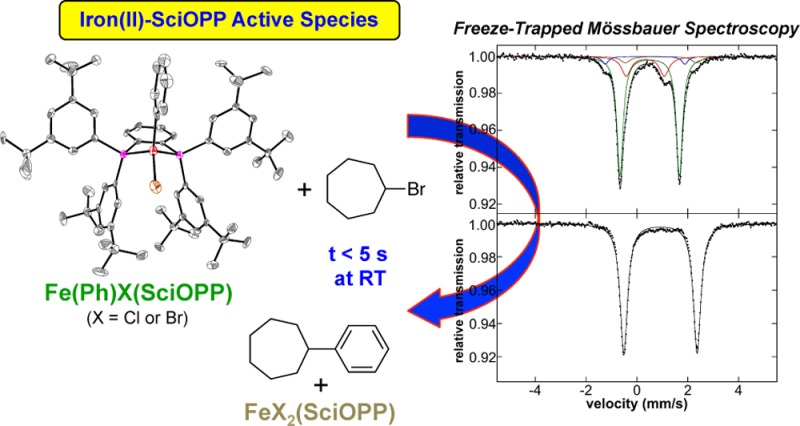
While previous studies have identified FeMes2(SciOPP) as the active catalyst species in iron–SciOPP catalyzed Kumada cross-coupling of mesitylmagnesium bromide and primary alkyl halides, the active catalyst species in cross-couplings with phenyl nucleophiles, where low valent iron species might be prevalent due to accessible reductive elimination pathways, remains undefined. In the present study, in situ Mössbauer and magnetic circular dichroism spectroscopic studies combined with inorganic syntheses and reaction studies are employed to evaluate the in situ formed iron species and identify the active catalytic species in iron–SciOPP catalyzed Suzuki–Miyaura and Kumada cross-couplings of phenyl nucleophiles and secondary alkyl halides. While reductive elimination to form Fe(η6-biphenyl)(SciOPP) occurs upon reaction of FeCl2(SciOPP) with phenyl nucleophiles, this iron(0) species is not found to be kinetically competent for catalysis. Importantly, mono- and bis-phenylated iron(II)–SciOPP species that form prior to reductive elimination are identified, where both species are found to be reactive toward electrophile at catalytically relevant rates. The higher selectivity toward the formation of cross-coupled product observed for the monophenylated species combined with the undertransmetalated nature of the in situ iron species in both Kumada and Suzuki–Miyaura reactions indicates that Fe(Ph)X(SciOPP) (X = Br, Cl) is the predominant reactive species in cross-coupling. Overall, these studies demonstrate that low-valent iron is not required for the generation of highly reactive species for effective aryl-alkyl cross-couplings.
1. Introduction
Iron catalyzed C–C cross-coupling reactions have attracted significant interest as versatile and cost-effective alternatives to traditional precious metal catalysts, including reactions that have proven difficult for precious metal catalysts such as cross-coupling of nonactivated alkyl halides.1−6 Initially developed in the 1970s by Kochi using simple iron salts,7−11 multiple iron-based systems have subsequently been reported which use reaction additives (e.g., TMEDA, N-heterocyclic carbenes, NMP), well-defined mononuclear iron complexes (including those ligated by TMEDA and bisphosphine ligands) or simple iron salts to generate robust catalysts for a wide variety of reactions including iron-catalyzed Kumada, Negishi, and Suzuki–Miyaura cross-coupling.12−34
Well-defined iron–bisphosphines, as well as the combination of simple iron salts and bisphosphine ligands, have been shown to be highly effective cross-coupling catalyst systems in the recent work of Bedford, Chai and Nakamura utilizing dpbz (bis-1,2-(diphenylphosphino)benzene),35−37 Xantphos,38,39 dppe,40,41 and SciOPP ligands42−45 (Scheme 1a). Precatalysts bearing the dpbz ligand have been shown to be highly efficient for Negishi coupling and, importantly, recent studies have isolated a low-spin (S = 1/2) trigonal bipyramidal iron(I) species, (dpbz)2FeX (X = Cl, Br) (Scheme 1b).36 Furthermore, (dpbz)2FeBr was shown to be an effective precatalyst species in Negishi coupling and postulated to be an active on-cycle species. While a specific mechanism was not proposed, these results might suggest a potential Fe(I)/Fe(III) mechanism. Analogous S = 1/2 iron(I) species have also been identified for this Negishi reaction with dppe.40 Nakamura and co-workers have found that the four-coordinate (4C) iron–bisphosphine complex, FeCl2(SciOPP), is an effective precatalyst for Kumada,43,46 Suzuki–Miyaura,42,47,48 Negishi,37 and Sonogashira-type44 couplings. For iron–SciOPP cross-coupling, it has been proposed that catalysis proceeds through an Fe(II)/Fe(III) radical pathway.42,43,49 Recent work from our group investigating iron–SciOPP catalyzed Kumada cross-coupling of mesitylmagnesium bromide (MesMgBr) and primary alkyl halides has identified Fe(Mes)2(SciOPP) as the active catalytic species.50 While an analogous mechanism has been proposed across a range of nucleophiles (e.g., mesityl, phenyl and alkynyl Grignards, as well as aryl-borates) with iron–SciOPP,42−44 the reaction rates, yields, and temperatures vary significantly between reactions, and the possibility that analogous iron(II) active species exist across all of these reactions has not been evaluated in detail. A fundamental understanding of the critical differences between these iron–bisphosphine cross-coupling reactions, including iron speciation, side-product formation, and the mechanisms of catalysis, is critical to broadening our understanding of catalysis in cross-couplings with iron–bisphosphines in order to inspire and facilitate the development of improved catalytic protocols.
Scheme 1. Representative Iron–Bisphosphine Catalyzed Cross-Coupling Reactions.
While it has previously been demonstrated in iron–SciOPP catalyzed cross-coupling that mesityl Grignards result in FeMes2(SciOPP) as the active species and similar bis-phenylated iron(II) species have also been proposed with phenyl nucleophiles, several critical differences exist between mesitylated and phenylated iron-bisphosphines that might lead to important differences in in situ iron speciation and, thus, the underlying mechanisms of catalysis. For example, while several isolated bis-mesitylated iron(II)–bisphosphine compounds have been reported,50,51 there are notably no reports of the analogous phenylated iron(II)–bisphosphine compounds, suggesting a significant difference in stability and/or reactivity with phenyl ligation. The lack of stable phenylated iron(II)–bisphosphines likely reflects the additional reaction pathways accessible for iron species derived from less sterically hindered aryl nucleophiles, including disproportionation and reductive elimination reactions. While the recent study on cross-couplings of MesMgBr and primary alkyl halides with FeCl2(SciOPP) has supported an Fe(II)/Fe(III) redox cycle (Scheme 2),50 the additional reaction pathways available for phenylated iron(II)–bisphosphines would likely yield the formation of additional reduced iron species that are inaccessible in mesityl chemistry. Therefore, it has remained unclear if a similar iron(II) active species would be operative. In fact, it has recently been suggested that lower valent iron species such as iron(I) might be more relevant candidates for the active species in iron cross-couplings involving phenyl nucleophiles.52
Scheme 2. Proposed Mechanism by Nakamura and Co-workers for FeCl2(SciOPP) Catalyzed Cross-Coupling of Aryl Nucleophiles and Alkyl Electrophiles42,43.

Recently, our group has established that an experimental approach combining physical inorganic spectroscopies with synthetic and reaction studies is a powerful method for elucidating mechanistic insight in iron-based cross-coupling reactions.50 Our earlier work demonstrated the utility of 57Fe Mössbauer spectroscopy to monitor in situ formed iron species at freeze-trapped time points during catalytic and stoichiometric reactions to determine the key components of a catalytic mechanism as well as the kinetics of reaction of specific iron species with electrophile. Combined with magnetic circular dichroism (MCD) studies to further define the electronic structure and the local coordination environment of paramagnetic iron species, and electron paramagnetic resonance (EPR), which provides a highly sensitive technique to quantify the presence of Kramers doublets (S = 1/2, 3/2, etc.) formed in situ, this approach is uniquely positioned to provide unprecedented molecular-level insight into the nature of iron–bisphosphine catalyzed cross-coupling with phenyl nucleophiles, including the nature of the active iron species and the potential contributions of disproportionation and reductive elimination reactions in these systems.
In the study presented herein, we utilize this physical inorganic approach to elucidate fundamental structure–activity correlations and mechanistic principles governing iron–SciOPP catalyzed Kumada and Suzuki–Miyaura cross-couplings of phenyl nucleophiles with secondary alkyl halides. While low-valent iron species are formed in this chemistry, they are found to exhibit minimal reactivity toward electrophile. By contrast, transiently formed mono- and bis-phenylated iron(II)–SciOPP species are observed in situ prior to reductive elimination, where both are reactive toward electrophile at catalytically relevant reaction rates. The higher selectivity toward the formation of cross-coupled product observed for the monophenylated species combined with the undertransmetalated nature of the in situ iron species in both Kumada and Suzuki–Miyaura reactions indicates that Fe(Ph)X(SciOPP) (X = Br, Cl) is the predominant reactive species in cross-coupling. Thus, these studies demonstrate that formation of a low-valent iron species is not required for the generation of highly reactive iron–bisphosphine catalysts for aryl-alkyl cross-coupling.
2. Results and Analysis
2.1. Reactivity of FeCl2(SciOPP) with Phenyl Nucleophiles at Room Temperature
Initial studies focused on the evaluation of the iron species formed upon reaction of FeCl2(SciOPP) (1-Cl2) with phenyl nucleophiles at room temperature. Reactions of 1-Cl2 with phenylmagnesium bromide (PhMgBr) and t-butylphenyl pinacolborate (tBuPh-borate)/MgBr2 were performed, which represent the relevant nucleophiles for the Kumada and Suzuki–Miyaura reactions, respectively.42,43 While cross-couplings using both tBuPh-borate/MgBr2 and nBuPh-borate/MgBr2 were reported in the literature depending on the secondary alkyl halide (tBuPh-borate for bromocycloheptane and nBuPh-borate for chlorocycloheptane),42 reaction studies in our group have demonstrated that tBuPh-borate/MgBr2 gives comparable catalytic performance with chlorocycloheptane (vide infra) and is utilized herein. The addition of 20 equiv tBuPh-borate and 6.7 equiv MgBr2 to a 3 mM solution of 57FeCl2(SciOPP) (57Fe–1-Cl2) in THF (or 1:1 THF/2-MeTHF) at 25 °C led to the rapid change of the solution from pale yellow to orange-red within 1 min and further evolution to a dark plum-purple colored solution in ∼10 min. The Mössbauer spectrum of the in situ formed iron species after 1 h of reaction indicates complete conversion to a single iron species with Mössbauer parameters of δ = 0.44 mm/s and ΔEQ = 1.75 mm/s (Figure 1A), and in situ 1H and 31P NMR spectra are consistent with the formation of a diamagnetic iron compound with η6-aryl ligation (see Supporting Information). Analogous Mössbauer studies indicate that the same plum-purple iron species is formed from the reaction of 57Fe–1-Cl2 with 2.2 equiv of PhMgBr (Figure 1B, δ = 0.44 mm/s and ΔEQ = 1.75 mm/s) at 25 °C for 1 h. A similar result is achieved with 20 equiv of PhMgBr (Figure S1). A single crystal of this iron species suitable for study by X-ray crystallography was isolated from the reaction of 1-Cl2 with 2.2 equiv of PhMgBr that permitted the assignment of this iron species. The crystal structure (Figure 1C) reveals the formation of Fe(η6-biphenyl)(SciOPP) (2), an iron(0) complex with bidentate coordination by the SciOPP ligand to the iron as well as coordination to an η6-biphenyl ligand.53 While the disorder observed in this structure precludes a more detailed discussion of the structural parameters, the overall connectivity, geometry and identity of this species are unambiguous.
Figure 1.
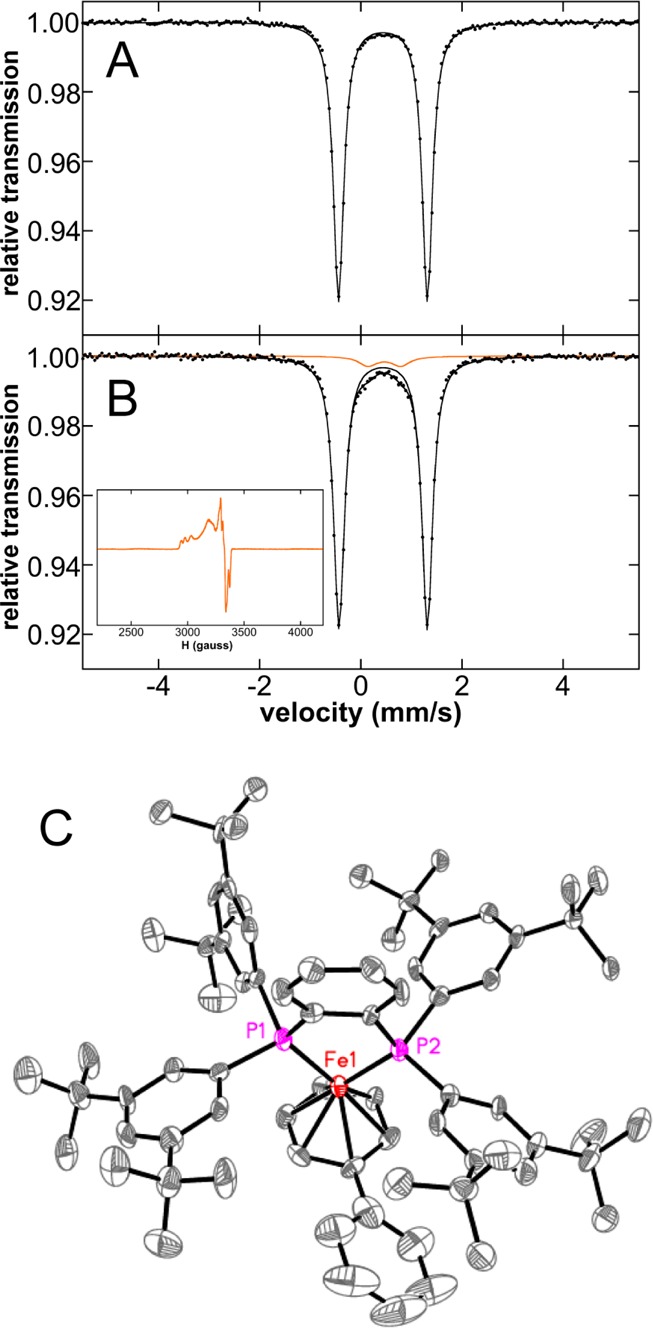
Formation of low-valent iron species upon reaction of FeCl2(SciOPP) (1-Cl2) with phenyl nucleophiles. The 80 K Mössbauer spectra of the in situ iron species from reaction of 57Fe–1-Cl2 with (A) 20 equiv tBuPh-borate/6.7 equiv MgBr2 and (B) 2 equiv PhMgBr at 25 °C. The minor S = 1/2 component (3) observed by 10 K EPR spectroscopy upon reaction with PhMgBr (∼5% of all iron present) is shown in the inset of (B). (C) X-ray crystal structure of Fe(η6-biphenyl)(SciOPP) (2) with thermal ellipsoids shown at 50% probability.
An additional iron side product (3) (Figure 1B and Figure S1, orange component, δ = 0.46 mm/s and ΔEQ = 0.65 mm/s) is also observed by Mössbauer to be present in ∼5% yield for reactions with PhMgBr to form 2. The observed parameters are similar to those previously reported for the S = 1/2 iron(I) complex Fe(dpbz)2Cl (δ = 0.43 mm/s and ΔEQ = 0.61 mm/s),54 suggesting 3 might be a S = 1/2 species. This is confirmed by 10 K EPR spectroscopy which indicates the presence of a phosphine hyperfine split, axial S = 1/2 (g ∼ 2) iron species which spin quantitates to ∼5% of the iron in solution (Figure 1B, inset), consistent with the quantitation by Mössbauer. While this species cannot be observed by Mössbauer spectroscopy for the corresponding reaction with tBuPh-borate/MgBr2, spin quantitated EPR indicates <0.5% of 3 is also formed in this reaction. While a similar S = 1/2 iron species was previously observed using EPR by Bedford and co-workers in reactions of FeBr2L (L = SciOPP analogue with SiMe3 groups replacing the tBu substituents) with 20 equiv PhMgBr at room temperature (RT),41 no spin quantitation was performed, and the results herein show that this species is formed as only a very minor component in solution. While 3 could be consistent with a low-spin iron(I) species as proposed by Bedford and co-workers,41 a mixed-valent iron dimer (or other multinuclear site) is also possible though the very minor amounts generated in solution preclude a more detailed characterization.
2.2. Evaluation of the Reactivity of Fe(η6-biphenyl)(SciOPP)
The formation of 2 upon reaction of 1-Cl2 with phenyl nucleophiles represents a low-valent, in situ formed species not considered in previous mechanistic proposals by Nakamura nor observed as a byproduct in our previous spectroscopic studies with mesityl nucleophiles. Thus, the potential reactivity of this species with electrophile and its role in catalysis were investigated through both catalytic and pseudo single turnover reaction studies.
Catalytic reactions were performed using tBuPh-borate/MgBr2 and chlorocycloheptane following the reaction protocol previously reported in the literature (Scheme 1) using both 1-Cl2 and 2 as precatalysts (Table 1). Using 3 mol % 1-Cl2 as the precatalyst in a 4 h reaction at 25 °C, an 89% yield (by GC–FID) of the cross-coupled product phenyl-cycloheptane was obtained, similar to the 93% yield reported in the literature with nBuPh-borate/MgBr2.42 For comparison, an identical catalytic reaction was performed using 3 mol % 2 as the precatalyst, generated from the reaction of tBuPh-borate/MgBr2 and 1-Cl2 as previously described. With 2 as the precatalyst, 63% yield of the cross-coupled product was observed after 4 h of reaction at 25 °C, which increased to 83% yield after a total reaction time of 6 h. Thus, these results demonstrate that 2 can serve as an effective precatalyst for cross-coupling, though an elongated reaction time is required to achieve high product yield.
Table 1. Suzuki–Miyaura Cross-Couplings with FeCl2(SciOPP) (1-Cl2) and Fe(η6-Biphenyl)(SciOPP) (2) Precatalysts.

| catalyst | R | time (h) | Chp-Ph yield (%) | ChpCl (unreacted) (%) |
|---|---|---|---|---|
| 1-Cl2 | n-butyl | 4 | 93a | 0 |
| t-butyl | 4 | 89 | 2 | |
| 2 | t-butyl | 4 | 63 | 32 |
| t-butyl | 6 | 83 | 17 |
The results with R = n-butyl are from ref (42).
Due to the sluggish results of the catalytic reaction using 2 as precatalyst, pseudo single turnover studies of the reaction of 2 with excess electrophile were performed to evaluate its kinetic competence as a potential active catalyst for cross-coupling. 2 was formed in situ through the reaction of 2 equiv of PhMgBr with 57Fe–1-Cl2 for 1 h at room temperature as confirmed by Mössbauer (Figure 2). After the generation of 2, excess chlorocycloheptane (20 equiv) was added and the subsequent reaction was followed by freeze-trapped Mössbauer spectroscopy as a function of reaction time (Figure 2). While consumption of 2 is observed, only ∼1/3 of the iron has reacted and converted to 1-X2 (X = Cl or Br due to halide exchange) within 45 min (Table S1). Following the amount of 2 present in solution as a function of reaction time, an observed rate constant of 9.0(5) × 10–3 min–1 at room temperature can be estimated via a first-order kinetic fit (Figure S2). By contrast, under catalytic reaction conditions a rate of ∼8 turnovers/h is expected for Suzuki–Miyaura coupling42 at 25 °C, whereas Kumada cross-couplings proceed on the order of multiple turnovers per minute.43 Furthermore, GC analysis indicates that cycloheptene is the predominant product formed in this reaction (see Supporting Information). From our kinetic analysis, it is clear that 2 reacts far too slowly with electrophile to be the active iron species in catalysis. In addition, the rate of formation of the iron(0) species from reaction of 57Fe–1-Cl2 with 2 equiv PhMgBr at RT is estimated from freeze-trapped Mössbauer studies to occur at 0.12(2) min–1 (vide infra). Thus, it forms at a rate much lower than that for turnover in the Kumada cross-coupling. Lastly, 3 remains unperturbed throughout the reaction with chlorocycloheptane (i.e., it remains ∼5% of all iron throughout the 45 min of reaction by Mössbauer and EPR (see Supporting Information)), demonstrating that it is significantly less reactive toward electrophile than 2 and, hence, not kinetically competent to serve as the active species in catalysis.
Figure 2.
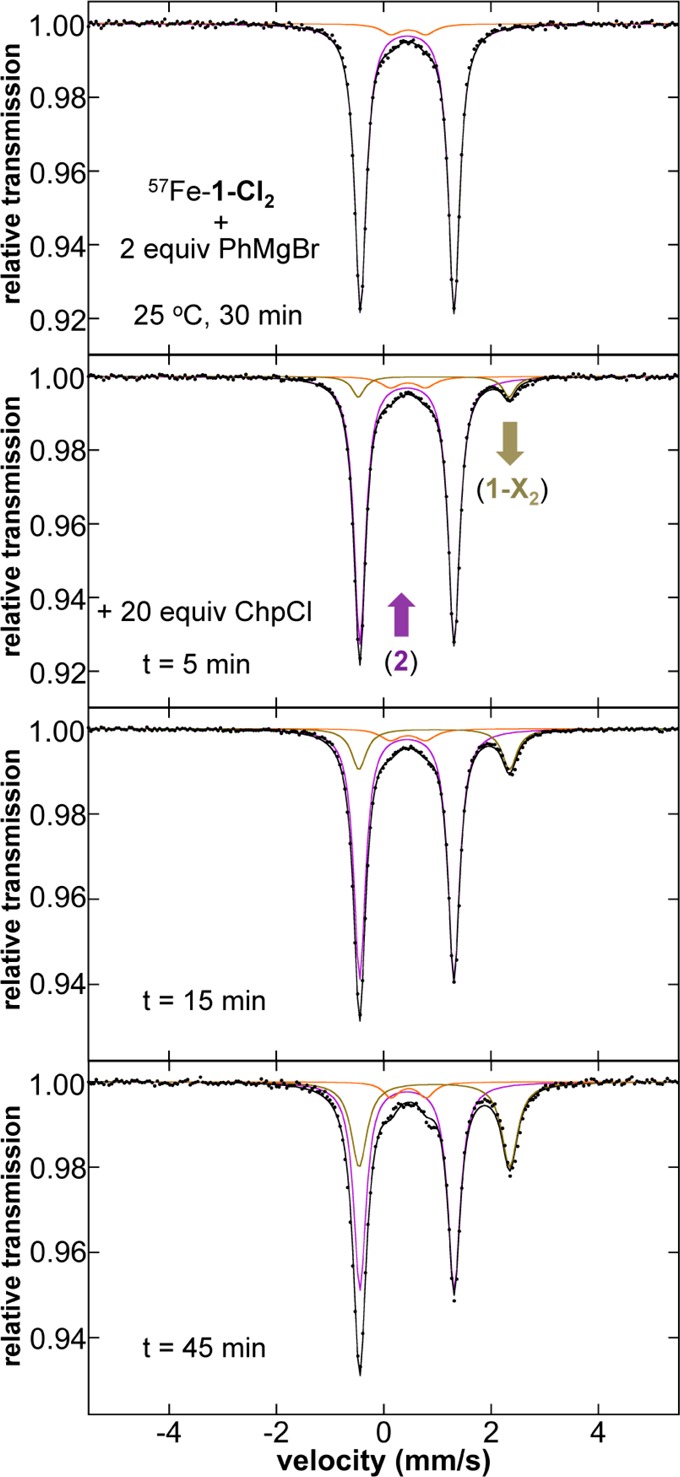
The 80 K Mössbauer spectra of the in situ iron species as a function of reaction time during the reaction of Fe(η6-biphenyl)(SciOPP) (2) with chlorocycloheptane. The individual Mössbauer components are identified as 2 (purple), 1-X2 (beige), and 3 (orange).
2.3. Iron Species Formed in Situ Prior to Reductive Elimination
2.3.1. Bis-phenylated Iron(II)–SciOPP Species
While 2 is not kinetically competent for catalysis, its formation from the reaction of 1-Cl2 with PhMgBr (or tBuPh-borate/MgBr2) suggests that a bis-phenylated iron(II) species is likely formed prior to reductive elimination. Rapid freeze-trapping of the in situ formed iron species from the reaction of a 3 mM solution of 57Fe–1-Cl2 with 2 equiv PhMgBr at 25 °C in 1:1 THF/2-MeTHF (total reaction time ∼30 s) enabled the evaluation of the iron species formed prior to reductive elimination. The 80 K Mössbauer spectrum of the resulting frozen solution (Figure 3A) indicates the presence of two dominant iron species that together comprise ∼94% of the iron in solution. One major component (4a) is characterized by Mössbauer parameters of δ = 0.33 mm/s and ΔEQ = 1.50 mm/s (62%, red component) and the second (4b) by δ = 0.32 mm/s and ΔEQ = 3.13 mm/s (32%, blue component). There is a third minor species present characterized by δ = 0.46 mm/s and ΔEQ = 0.65 mm/s (6%, orange component). A similar iron distribution can also be obtained from the analogous reaction performed at 0 °C for 5 min (Figure 3B). The 5 K Mössbauer spectrum of the 0 °C reaction yields the same iron speciation and distribution (Figure S3). Addition of free SciOPP ligand had no effect on the iron species generated in situ (Figure S4). Spin quantitated EPR spectroscopy indicates that the δ = 0.46 mm/s component is the S = 1/2 species 3 (Figure S16).
Figure 3.
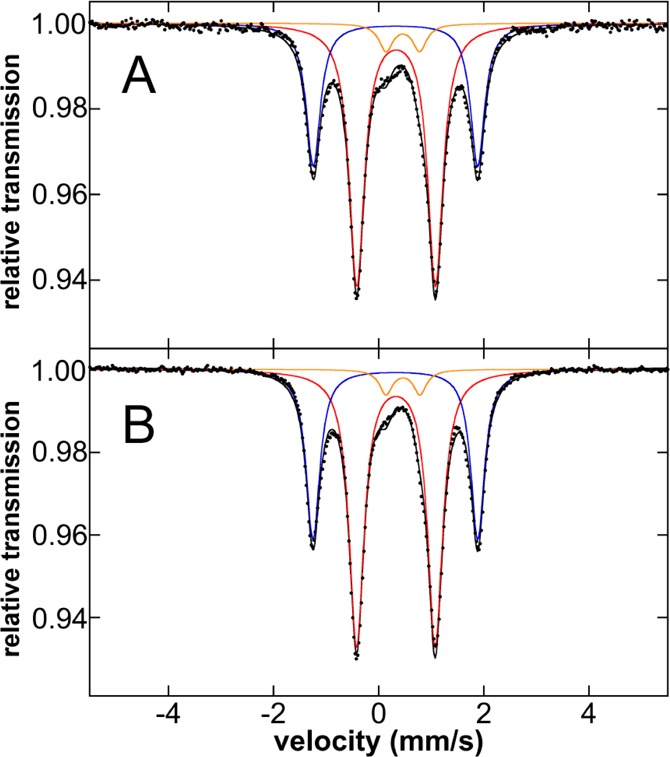
The 80 K Mössbauer spectra of the in situ generated iron species upon reaction of 57FeCl2(SciOPP) (57Fe–1-Cl2) with 2 equiv PhMgBr in 1:1 THF/2-MeTHF at (A) 25 °C freeze-trapped after 30 s of reaction time and (B) 0 °C freeze-trapped after 5 min of reaction time. The individual Mössbauer components are identified as 4a (red), 4b (blue), and 3 (orange).
The generation of 2 as a function of time at 25 °C could be followed by freeze-trapped Mössbauer spectroscopy (Figure 4). Over the course of 10 min, 56% of 2 forms from consumption of the two major iron species initially present upon transmetalation (4a and 4b) (Table S2). No additional intermediates are observed consistent with direct reduction from these species to form 2. Importantly, 3 is formed upon initial reaction with nucleophile and remains constant within error throughout this reaction by both Mössbauer and EPR (Figure S16), arguing against its intermediacy in the reduction to form 2. This is also consistent with the observation that the amount of 3 initially formed upon reaction with nucleophile (∼6%) is identical within error to the amount present in solution upon complete reduction to 2 as previously discussed. A first-order kinetic fit yields an estimated rate of 0.12(2) min–1 for the formation of 2 at 25 °C (Figure S5). An analogous result was obtained for the formation of 2 at 0 °C (Figure S6 and Table S3), though the reaction proceeded much more slowly (∼28% 2 after 2.5 h of reaction).
Figure 4.
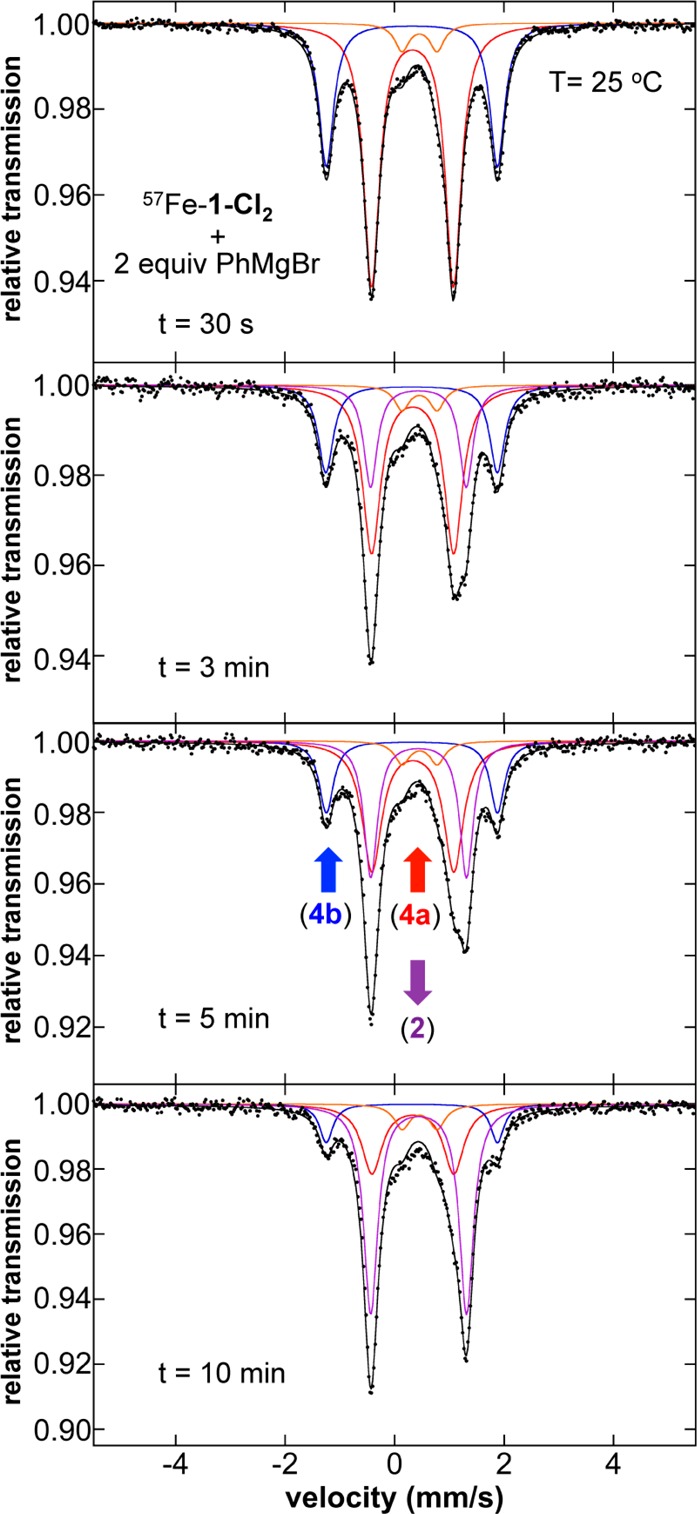
The 80 K Mössbauer spectra of the in situ iron species as a function of reaction time at 25 °C from reaction of 57FeCl2(SciOPP) (57Fe–1-Cl2) with 2 equiv PhMgBr in 1:1 THF/2-MeTHF. The individual Mössbauer components are identified as 4a (red), 4b (blue), 3 (orange), and 2 (purple).
Identification of the remaining two major iron species (4a and 4b) which together comprise ∼94% of all the iron in solution and are consumed to form 2 was possible using the observed Mössbauer parameters combined with MCD spectroscopy. Previous Mössbauer studies of mesitylated iron(II)–bisphosphine complexes provide insight into the two major species formed upon reaction of 1-Cl2 with PhMgBr prior to reduction (Table 2). In our previous work, isolated distorted square planar FeMes2(SciOPP) and distorted tetrahedral FeMesBr(SciOPP) in frozen solution samples exhibited Mössbauer parameters of δ = 0.28 mm/s and ΔEQ = 3.67 mm/s and δ = 0.52 mm/s and ΔEQ = 2.12 mm/s, respectively.50 Chirik and co-workers have also previously reported that the square planar complexes Fe(Mes)2(PEt2Ph)2 and Fe(Mes)2(dppe) are characterized by Mössbauer parameters of δ = 0.31 mm/s and ΔEQ = 4.63 mm/s and δ = 0.33 mm/s and ΔEQ = 4.53 mm/s, respectively.51 Chirik also reported that the bisphosphine complex Fe(Mes)2(depe) exhibited a similar isomer shift (δ = 0.39 mm/s) but a much lower quadrupole splitting (ΔEQ = 1.71 mm/s) which was attributed to the distorted tetrahedral geometry of this complex.51 From these parameters, the two major species freeze-trapped prior to reductive elimination to the iron(0) product both exhibit isomer shifts in the range expected for bisarylated iron(II)–bisphosphine complexes. It should be noted that −30 °C 31P NMR studies indicate that no free SciOPP ligand is present in this mixture (i.e., no phosphine resonances are observed), inconsistent with the formation of homoleptic iron-ate complexes.
Table 2. Mössbauer Parameters of Mesitylated Iron(II)–Bisphosphine Complexes50,51.
| complex | geometry | sample | δ (mm/s) | ΔEQ (mm/s) |
|---|---|---|---|---|
| FeMes2(SciOPP) | sq. planar | solid | 0.29 | 3.58 |
| froz. soln. | 0.28 | 3.67 | ||
| FeMes2(PEt2Ph)2 | sq. planar | solid | 0.31 | 4.63 |
| FeMes2(dppe) | sq. planar | solid | 0.33 | 4.53 |
| FeMes2(depe) | dist. tetra. | solid | 0.39 | 1.71 |
| FeMesBr(SciOPP) | dist. tetra | solid | 0.52 | 1.97 |
| froz. soln. | 0.52 | 2.12 |
The observation of nearly identical isomer shifts but quite distinct quadrupole splittings for 4a and 4b might indicate that they differ in either their geometries and/or coordination numbers. MCD spectroscopy is able to identify and characterize geometric distortions and differences in coordination number in more detail than Mössbauer as the iron d orbitals (and, hence, the LF transitions) are very sensitive to coordination geometry. The near-infrared (NIR) MCD spectrum of the iron species formed in situ from reaction of 1-Cl2 with 2 equiv of PhMgBr, freeze-trapped after 5 min of reaction at 0 °C in 1:1 THF/2-MeTHF (Figure 5C) shows the presence of four LF transitions at ∼5600, 6560, 8480, and 13500 cm–1, consistent with the presence of at least two different major iron(II) species in solution (as previously determined by Mössbauer). The presence of multiple LF transitions at low energy (<10 000 cm–1) is indicative of the presence of a distorted tetrahedral high-spin iron(II) component.55,56 The band at ∼13 500 cm–1 is too high in energy to be consistent with a tetrahedral species and is indicative of a 5-coordinate distorted square-pyramidal, high-spin iron(II) species.55 Saturation magnetization data collected at 5260 and 13333 cm–1 are both well-fit to S = 2 ground state models, but with very different ground state parameters (Figure 5E,F). The band at 5260 cm–1 is well-fit to a negative zero-field split (−ZFS) S = 2 ground state model with δ = 3.0 ± 0.2 cm–1 and g∥ = 9.5 ± 0.2, corresponding to D = −11 ± 1 cm–1 and |E/D| = 0.31 ± 0.02, whereas the band at 13 333 cm–1 is well-fit to a +ZFS S = 2 ground state model with D = 8 ± 1 cm–1 and |E/D| = 0.22 ± 0.03. The different observed ground-states are consistent with each transition being associated with a distinct high-spin iron(II) complex. Combined with the previous analysis of the LF transition energies, the NIR MCD data indicate that the two major bis-phenylated iron(II) species observed by Mössbauer correspond to a distorted tetrahedral iron(II) species and a distorted square pyramidal iron(II) species. Lastly, the corresponding 5 K, 7 T UV–vis MCD spectrum (Figure S15) of the mixture contains charge transfer (CT) bands at ∼19 500 and ∼24 850 cm–1, which represent new CT transitions due to phenyl ligation. It should be noted that because the bis-phenylated iron(II) species can access two distinct geometries in solution, multiple CT bands are expected from both complexes, consistent with the observation of multiple, broad CT transitions.
Figure 5.

Mössbauer and MCD characterization of the in situ formed iron species from the reaction of 57FeCl2(SciOPP) (57Fe–1-Cl2) with 2 equiv PhMgBr at 0 °C. (A and B) The 80 K Mössbauer and (C and D) 5 K, 7T NIR MCD spectra of the in situ iron species from reaction in (A and C) 1:1 THF/2-MeTHF and (B and D) 1:1 Et2O/isopentane. Saturation magnetization data (dots) and best fit (lines) collected at (E) 5260 cm–1 and (F) 13333 cm–1 in 1:1 THF/2-MeTHF and at (G) 5320 cm–1 in 1:1 Et2O/isopentane. The individual Mössbauer components in (A) are identified as 4a (red), 4b (blue), and 3 (orange).
The observation of a five-coordinate iron(II) component raises the question of the origin of the fifth ligand in this complex. As the reaction occurs in a potentially coordinating solvent mixture (THF/2-MeTHF), freeze-trapped Mössbauer studies of the in situ formed iron species upon reaction of 57Fe–1-Cl2 with 2 equiv PhMgBr at 0 °C were performed as a function of solvent. A similar mixture of iron species is formed in pure THF (Figure S7). By contrast, in diethyl either or 1:1 diethyl ether/isopentane (Figure S7 and Figure 5B) only a single iron species is formed with Mössbauer parameters (δ = 0.33 mm/s and ΔEQ = 1.36 mm/s) similar to the parameters for 4a observed in THF/2-MeTHF. The 5 K, 7 T NIR MCD spectrum of this species contains LF transitions at ∼5660, 6730, and 8770 cm–1, consistent with a distorted tetrahedral iron(II) species where all degeneracy in the 5T2 derived excited stated has been removed.55 Saturation magnetization data collected at 5320 cm–1 are well-fit to a −ZFS S = 2 ground-state model with parameters (δ = 2.8 ± 0.2 cm–1 and g∥ = 9.4 ± 0.2, corresponding to D = −11 ± 1 cm–1 and |E/D| = 0.30 ± 0.03) identical within error to those determined for the low energy component in THF/2-MeTHF (Figure 5G). Combined, the ligand-field energies, ground state parameters and Mössbauer parameters of this species indicate that it is the distorted tetrahedral component in the mixture of bis-phenylated iron(II)–SciOPP species formed in THF or THF/2-MeTHF. Furthermore, the lack of formation of the distorted five-coordinate component in diethyl ether or diethyl ether/isopentane indicates that the additional ligand in THF containing solvents is likely THF.
Thus, these extensive spectroscopic studies indicate that the two major iron species formed upon reaction of 1-Cl2 and 2 equiv PhMgBr prior to reductive elimination are Fe(Ph)2(SciOPP) (4a) and Fe(Ph)2(THF)(SciOPP) (4b). These assignments are further supported by the following observations: (1) both species reduce to form 2 (vide supra), (2) both species are consumed at the same rate during reduction consistent with a rapid equilibrium due to solvent coordination, and (3) both species are consumed at the same rate upon reaction with electrophile to generate Fe(Ph)X(SciOPP) (5-X) (vide infra).
2.3.2. Fe(Ph)X(SciOPP)
Upon the basis of the proposed mechanism by Nakamura42,43 and our previous studies,50 the formation of a monophenylated iron(II)–SciOPP species would also be expected to be accessible. The reaction of 57FeBr2(SciOPP) (57Fe–1-Br2) with 1 equiv PhMgBr in 1:1 THF/2-MeTHF at 0 °C for 3 min was performed and found to generate a major iron species in solution with Mössbauer parameters of δ = 0.50 mm/s and ΔEQ = 2.37 mm/s (77% of all iron) (Figure 6A, green component), similar to those previously observed for FeMesBr(SciOPP). The two observed minor species correspond to 16% of unreacted 1-Br2 as well as ∼7% of 4a. No EPR active iron species were observed from 10 K EPR studies. A similar major species is also observed upon the analogous reaction of 57Fe-1-Cl2 with 1 equiv PhMgBr at 25 °C for 1 min or 0 °C for 5 min (Figure S8). The 5 K, 7 T NIR MCD spectrum of this species is also very similar to that previously reported for FeMesBr(SciOPP),50 exhibiting two ligand-field (LF) transitions at 6530 and 7870 cm–1 (Figure 6C), consistent with the formation of a distorted tetrahedral S = 2 iron(II) species. The observation of the transitions for the major species is consistent with its dominant presence in solution combined with the very large Δε value previously observed for monoarylated, distorted tetrahedral iron(II)–SciOPP species (i.e., FeMesBr(SciOPP) relative to other iron(II)–bisphosphine species).50 The saturation magnetization data for this species collected at 6100 cm–1 is well-fit to a −ZFS S = 2 ground-state model with δ = 1.6 ± 0.2 cm–1 and g∥ = 8.5 ± 0.2, corresponding to D = −8 ± 1 cm–1 and |E/D| = 0.27 ± 0.03. Notably, reaction of 57Fe–1-Br2 with 1 equiv of PhMgBr in 1:1 diethyl ether/isopentane yields an analogous major iron species as confirmed by Mössbauer and NIR MCD (Figure 6, panels B and D, respectively) with Mössbauer parameters of δ = 0.50 mm/s and ΔEQ = 2.32 mm/s (77% of all iron) and LF bands at ∼6640 and 8260 cm–1. The small observed differences in these parameters are consistent with minor geometric distortions as a function of solvent. The saturation magnetization data for this species collected at 6120 cm–1 is well-fit to a negative −ZFS S = 2 ground-state model with D = −7 ± 1 cm–1 and |E/D| = 0.28 ± 0.03, within error of the ground state parameters in THF/2-MeTHF. On the basis of the Mössbauer and MCD data, the major iron species formed can be assigned as Fe(Ph)Br(SciOPP) (5-Br). This assignment is confirmed by X-ray crystallography of single crystals obtained from the reaction of 1-FeCl2 with 1 equiv PhMgBr in diethyl ether. The X-ray crystal structure (Figure 5E) reveals the formation of the distorted tetrahedral complex Fe(Ph)Br(SciOPP) (5-Br). The isolation of the brominated species demonstrates that halide exchange is facile, a result which is consistent with our previous characterization of monomesitylated iron(II)–SciOPP species.50
Figure 6.

Spectroscopic and structural characterization of Fe(Ph)Br(SciOPP) (5-Br). (A and B) The 80 K Mössbauer and (C and D) 5 K, 7T NIR MCD spectra of the in situ iron species from reaction of 1-Br2 with 1 equiv PhMgBr for 3 min at 0 °C in (A and C) 1:1 THF/2-MeTHF and (B and D) 1:1 Et2O/isopentane. Saturation magnetization data (dots) and best fit (lines) collected at 6100 cm–1 (C, inset) and 6120 cm–1 (D, inset). (E) X-ray crystal structure of 5-Br with thermal ellipsoids shown at 50% probability. The individual Mössbauer components are identified as 5-Br (green), 4a (red), and 1-Br2 (beige).
Upon identification of monophenylated iron(II)–SciOPP, the stability of the complex was studied as a function of time at 25 °C to evaluate its potential disproportionation. By the generation of 5-X in situ (X = Br or Cl due to halide exchange, vide supra) by addition of 1 equiv of PhMgBr to 57Fe–1-Cl2 in 1:1 THF/2-MeTHF at 25 °C and monitoring its change with time at this temperature over the course of 1 h, the 80 K Mössbauer spectra (Figure S9 and Table S4) indicate the consumption of 5-X (72% at t = 1 min to 46% at t = 60 min) with the simultaneous generation of 2 and 1-X2. The observation of these iron products (where formation of 2 is consistent with generation of the bis-phenylated iron(II) species and their rapid reductive elimination to generate the iron(0) product) is indicative of disproportionation of 5-X. By a second-order kinetic fit of the Mössbauer data, an estimated rate of 4.7(6) × 10–3 mM–1 min–1 for the disproportionation of 5-X at 25 °C was determined (Figure S10). At 0 °C, no disproportionation of this species is observed over the course of 1.5 h (Figure S11).
2.3.3. Iron Species Formed in Situ Utilizing Borate Nucleophile
While the previous evaluation of the phenylated iron(II)–SciOPP species formed in situ prior to reductive elimination focused on reactions with PhMgBr (relevant to Kumada cross-coupling), investigation of the species formed upon reaction with tBuPh-borate/MgBr2 prior to reduction is also important in order to evaluate if similar species are formed upon transmetalation with nucleophiles relevant to Suzuki–Miyaura cross-coupling. The 80 K Mössbauer spectrum of the freeze-trapped reaction of 57Fe–1-Cl2 with 20 equiv tBuPh-borate/6.7 equiv MgBr2 at 25 °C for 6 min is shown in Figure 7A. This spectrum contains contributions from four iron species with identical Mössbauer parameters to species already investigated: 5-X (62%, green component), 4a (18%, red component), 4b (11%, blue component), and 1-X2 (9%, beige component). The analogous reaction at 0 °C proceeds much more slowly, and after 2 h of reaction, 48% of 5-X is present by Mössbauer as well as 32% and 20% of 4a and 4b, respectively (Figure 7B). Lastly, while not observable by Mössbauer, 3 is detectable by EPR at both reaction temperatures and quantitates to <0.5% of all iron in solution, consistent with the amount of this component observed in solution upon reduction to form 2 (vide supra). Importantly, these studies demonstrate that (a) the same phenylated iron–SciOPP species are formed prior to reductive elimination with phenyl nucleophiles for both Kumada and Suzuki–Miyaura reactions and (b) transmetalation with tBuPh-borate/MgBr2 occurs much more slowly than the corresponding transmetalation with PhMgBr as a significant amount of undertransmetalated iron is present even after 6 min of reaction at 25 °C with excess tBuPh-borate/MgBr2.
Figure 7.
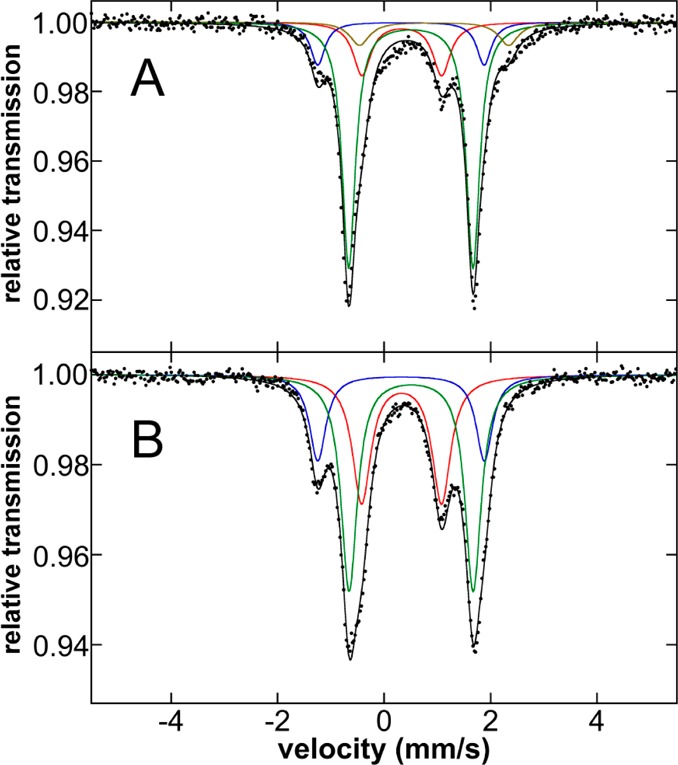
The 80 K Mössbauer spectra of the in situ formed iron species upon reaction of 57FeCl2(SciOPP) (57Fe–1-Cl2) with 20 equiv tBuPh-borate/6.7 equiv MgBr2 freeze-trapped after reaction at (A) 25 °C for 6 min and (B) 0 °C for 2 h. The individual Mössbauer components are identified as 4a (red), 4b (blue), 5-X (green), and 1-X2 (beige).
2.4. Iron Species Present in Situ during Catalysis
The previously described spectroscopic and synthetic studies have defined the iron species formed in situ upon reaction of 1-Cl2 with PhMgBr and tBuPh-borate/MgBr2 (Table 3). Evaluation of the iron species present during catalysis is important in order to ascertain if any additional iron species may form and to determine the extent of transmetalation present during pseudo-steady-state turnover. The 80 K Mössbauer spectrum of the Kumada catalytic reaction freeze-trapped at 5 min into the 30 min reaction is shown in Figure 8A. Only iron species corresponding to 2 (22% of iron, purple component) and 1-X2 (X = Br or Cl due to halide exchange, δ = 0.81 mm/s and ΔEQ = 2.60 mm/s (21%), δ = 0.88 mm/s and ΔEQ = 3.00 mm/s (58%), brown and orange components, respectively) are present and no phenylated iron species are observable. The 10 K EPR spectroscopy does indicate the presence of ∼5% of 3 which is not observable by Mössbauer within the noise of the data. For the Suzuki–Miyaura catalytic reaction freeze-trapped at 45 min into the 4 h reaction, the 80 K Mössbauer spectrum indicates the presence of 6% 2 (purple component), 29% 5-X (green component) and 60% of 1-Cl2 as well as ∼5% of a second 1-X2 species (X = Br, Cl due to halide exchange) (Figure 8B). The 10 K EPR spectroscopy indicates that ∼0.5% of 3 is also present in the Suzuki–Miyaura reaction. Thus, for both the Kumada and Suzuki–Miyaura reactions, it is clear that during catalysis iron is highly undertransmetalated, consistent with the slow rate of nucleophile addition (e.g., Grignard via syringe pump or the slowly transmetalating borate nucleophile) used in the reported reaction protocols.
Table 3. Mössbauer Parameters of in Situ Formed Iron Species with Phenyl Nucleophiles in 1:1 THF/2-MeTHF.
| complex | spin state | δ (mm/s) | ΔEQ (mm/s) |
|---|---|---|---|
| FeX2(SciOPP) (1-X2)a | S = 2 | 0.94 | 2.80 |
| Fe(η6-biphenyl)(SciOPP) (2) | S = 0 | 0.44 | 1.75 |
| S = 1/2 species (3) | S = 1/2 | 0.46 | 0.65 |
| Fe(Ph)2(SciOPP) (4a) | S = 2 | 0.33 | 1.50 |
| Fe(Ph)2(THF)(SciOPP) (4b) | S = 2 | 0.32 | 3.13 |
| Fe(Ph)X(SciOPP)a (5-X) | S = 2 | 0.51 | 2.35 |
X = Cl or Br due to halide exchange.
Figure 8.
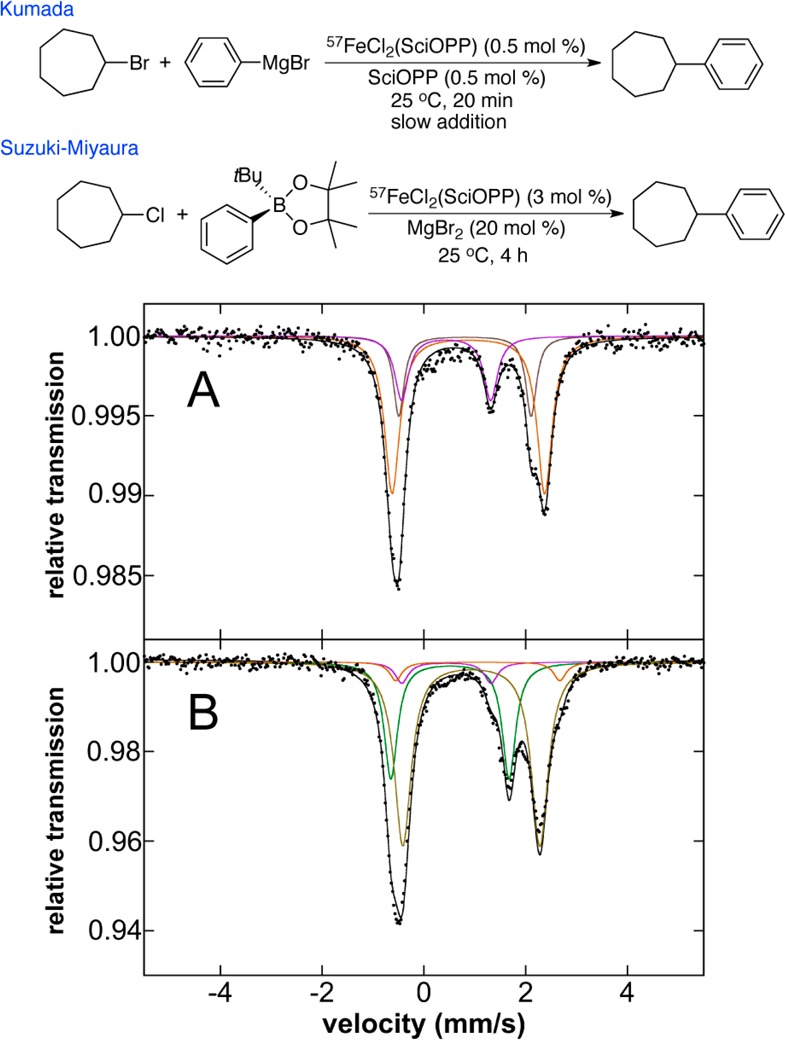
The 80 K Mössbauer spectra of the iron species in solution during catalysis upon freeze-trapping during (A) the Kumada reaction at t = 5 min and (B) the Suzuki–Miyaura reaction at t = 45 min of reaction time. The individual Mössbauer components are identified as 2 (purple), 5-X (green), and 1-X2 species (beige/orange).
2.5. Evaluation of the Reactivity of Phenylated Iron(II)–SciOPP Species with Electrophile
The previous reaction studies of 2 with electrophile indicated that this species is not kinetically competent to be the active catalyst species. In addition, these studies also demonstrated that 3 is even less reactive toward electrophile at RT. Therefore, evaluation of the potential reactivity of the phenylated iron(II)–SciOPP species toward electrophile was performed in order to determine their catalytic relevance. While the low stability of these species precluded direct reaction studies with electrophile using isolated solids of the phenylated species, the previous freeze-trapped Mössbauer studies demonstrated that these species could be formed in situ at both 25 and 0 °C from reactions of 1-Cl2 with 1 or 2 equiv PhMgBr, with stabilities ranging from >30 s and >1 min at 25 °C for the bis- and monophenylated species, respectively (vide supra) to >10 min for both species at 0 °C. Following the reaction of 57Fe–1-Cl2 with 1 equiv PhMgBr at 25 °C for 1 min to generate 5-X, 20 equiv of bromocycloheptane was added and the subsequent reaction was quenched after reaction for 5, 30, and 90 s (Table 4). GC analysis indicated that the reaction was complete within 5 s, generating cross-coupled product phenylcycloheptane (84% with respect to iron) as well as some cycloheptene (Table 4), consistent with the reported catalytic Kumada reaction with this substrate (84–92% phenylcycloheptene, 9–14% cycloheptene).43 The analogous experiment at 0 °C with bromocycloheptane was found to form a similar product distribution after reaction for 5 min (see Supporting Information). Reactions with chlorocycloheptane at both 25 and 0 °C also yielded phenylcycloheptane and slightly less cycloheptene, though extended reaction times were required (see Supporting Information). Freeze-trapped solution Mössbauer spectroscopy for the reaction with 1 equiv PhMgBr and 20 equiv bromocycloheptane at 25 °C demonstrates the generation of 1-X2 as the iron product formed upon reaction of 5-X with electrophile (Figure 9). The same iron transformation is also observed over an elongated reaction time with 20 equiv chlorocycloheptane at 0 °C (Figure S12).
Table 4. GC Analysis of the Products Formed upon Reaction of in Situ Generated Mono- and Bis-Phenylated Iron(II)–SciOPP with Bromocycloheptane at 25 °Ca.
| PhMgBr (equiv) | time (s) | Chp-Ph (%) | cycloheptene (%) |
|---|---|---|---|
| 1 | 5 | 84 | 22 |
| 1 | 30 | 84 | 22 |
| 1 | 90 | 84 | 22 |
| 2 | 5 | 87 | 93 |
| 2 | 30 | 88 | 95 |
| 2 | 90 | 89 | 96 |
Yields are with respect to iron. A minor amount of biphenyl is also observed (∼3% and 10% for the 1 equiv and 2 equiv PhMgBr reactions, respectively).
Figure 9.
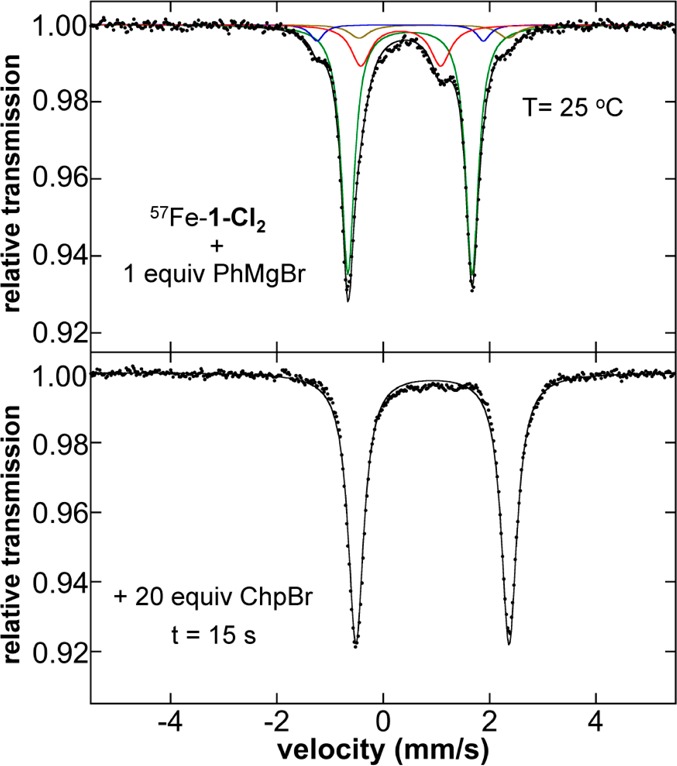
The 80 K Mössbauer spectra of the iron species in solution (A) following reaction of 57FeCl2(SciOPP) (57Fe–1-Cl2) with 1 equiv PhMgBr at 25 °C in 1:1 THF/2-MeTHF for 1 min and (B) after subsequent addition of 20 equiv bromocycloheptane and reaction for 15 s. The individual Mössbauer components are identified as 4a (red), 4b (blue), 5-X (green), and 1-X2 (beige).
By contrast, the in situ generation of bis-phenylated iron(II)–SciOPP (4a and 4b) from reaction of 1-Cl2 with 2 equiv of PhMgBr at 25 °C for 30 s followed by addition of 20 equiv bromocycloheptane was found to be less selective toward cross-coupled product, yielding an ∼1:1 mixture of cross-coupled product to cycloheptene (Table 4). The total product yield indicates ∼2 turnovers per iron within 5 s of reaction time. A similar result was obtained for the analogous reaction at 0 °C (see Supporting Information). As was observed for reactions with 1 equiv PhMgBr, reactions with chlorocycloheptane at both 25 and 0 °C also yielded phenylcycloheptane and slightly less cycloheptene (see Supporting Information). Following the reaction by freeze-trapped Mössbauer at 0 °C upon reaction with 2 equiv chlorocycloheptane (in order to reduce the rate of reaction to allow for resolution of the iron transformations), both bis-phenylated iron(II)–SciOPP species are observed to be consumed at the same approximate rate (i.e., their ratio remains constant) to generate 5-X (X = Cl or Br due to halide exchange) where Mossbauer spectra collected at both 80 K and 5 K gave the same result (Figure S13 and Figure S14, respectively). Spin quantitated EPR spectra also indicate no observable change (within error) in the amount of 3 present in situ during reaction (Figure S17). At long reaction time, further reaction to form 1-X2 is also observed, consistent with the further reaction of the 5-X product species with electrophile. Reactions with chlorocycloheptane are consistent with the rate of reactivity of the bis-phenylated species being higher than that for the monophenylated species.
It is noteworthy that the time dependent product formation in reactions using 1 equiv PhMgBr and chlorocycloheptane (see Supporting Information) indicates that cycloheptene is formed at early time points and does not further increase with reaction time, in contrast to the continued increase in the amount of phenylcycloheptane generated with time. Furthermore, the freeze-trapped Mössbauer studies of the in situ iron speciation (see Supporting Information) indicate that 4a and 4b are consumed rapidly (consistent with their higher reactivity, vide supra), where their consumption correlates with the time period of cycloheptene product. Furthermore, the total amount of cycloheptene produced in the 1 equiv PhMgBr reaction correlates within error to the amount of bis-phenylated iron present prior to electrophile addition. Overall, these observation suggest that the cycloheptene formed in these reactions likely derives predominately from reactions with bis-phenylated iron(II)–SciOPP.
Lastly, biphenyl is also observed as a product in our GC studies. While contributions from reductive elimination are possible, the large amounts of biphenyl observed in reactions where incomplete conversion had occurred (Table S9) led to the hypothesis that the chemical quenching of unreacted phenylated iron(II) species might also lead to biphenyl generation. By preforming 4a and 4b at 0 °C as previously described and directly quenching with either dilute HCl in THF or aqueous NaHSO4 (i.e., no electrophile was added), we observe a significant amount of biphenyl (∼50% with respect to iron) was formed from the bis-phenylated iron(II) species. Since no reduction to 2 had occurred prior to quenching (∼94% of iron was iron(II), vide supra), this result demonstrates that the quenching process itself can lead to biphenyl formation from the decomposition of phenylated iron(II)–bisphosphines, contributing to the biphenyl observed in incomplete reactions. Such artificial contributions to biphenyl generation independent of the presence of reduced iron species in solution are important to be aware of since biphenyl counting methods to estimate the average oxidation state of iron in solution have been previously employed in the literature.36,57
3. Discussion
Obtaining insight into the in situ formed iron species and mechanisms of catalysis in iron–SciOPP catayzed cross-coupling reactions with phenyl nucleophiles represents a significant characterization challenge as potential phenylated iron(II)–SciOPP species may be short-lived and undergo reductive elimination and disproportionation reactions that can contribute to in situ speciation. Furthermore, the cross-coupling reactions with phenyl nucleophiles such as PhMgBr can be very rapid with multiple turnovers per minute observed.43 Despite these substantial challenges, herein we have demonstrated that our experimental methodology combining freeze-trapped Mössbauer, MCD and EPR investigations with synthetic and reaction studies can enable the elucidation of the in situ formed iron species, including the identification of the catalytically active iron species, with phenyl nucleophiles. The major iron species identified to form in situ and their observed transformations are summarized in Scheme 3.
Scheme 3. Major Iron Species Formed in Situ and Their Reaction Pathways for the FeCl2(SciOPP) (1-Cl2) Catalyzed Kumada and Suzuki–Miyaura Cross-Couplings of Phenyl Nucleophiles and Secondary Alkyl Halides.

Estimated rates are given for the transformations at 25 °C.
Low-valent iron species are readily accessible upon reaction of FeCl2(SciOPP) (1-Cl2) with phenyl nucleophiles (PhMgBr or tBuPh-borate/MgBr2) at 25 °C, the temperature employed in catalysis. Fe(η6-biphenyl)(SciOPP) (2) is found to be the dominant low-valent iron species formed with iron–SciOPP, consistent with reductive elimination of a bis-phenylated iron(II)–SciOPP species. Formation of this iron(0) species is in stark contrast to iron-dpbz and iron-dppe chemistry where S = 1/2 iron(I) is the dominant low-valent iron species observed to form upon reduction with aryl nucleophile.36,40 While a S = 1/2 iron species (3) is also observed with iron–SciOPP and phenyl nucleophiles, it represents a very minor species in solution (∼5% with PhMgBr and <0.5% with tBuPh-borate/MgBr2), demonstrating the critical importance of spin quantitation when using EPR due to the high sensitivity of this method to low concentrations of EPR active species (including S = 1/2 iron species). The increased amount of this species with PhMgBr (which results in more rapid transmetalation than tBuPh-borate/MgBr2) and its immediate formation upon transmetalation suggest that it may represent an initial kinetic product in transmetalation reactions. It has been previously suggested that low-valent iron species are likely candidates for the active catalytic species in iron–bisphosphine cross-coupling reactions with phenyl nucleophiles.52 However, reaction studies with electrophile clearly demonstrate that 2 reacts far too slowly to be catalytically relevant, forms predominately cycloheptene during reaction with electrophile and, hence, represents an off cycle species in catalysis. Furthermore, the observed rate of formation of 2 from reaction of 1-Cl2 with PhMgBr at 25 °C (kobs = 0.12(2) min–1) is slow relative to the average turnover time in the Kumada cross-coupling reaction, inconsistent with it serving an active role in the catalytic cycle. 3, which forms immediately upon transmetalation and is not an intermediate in the reduction pathway to form 2, exhibits no observable reactivity toward electrophile at catalytically relevant reaction times, consistent with it also being an off cycle species. While the very low yields of 3 preclude a more detailed characterization of this species, it may represent an iron(I)–SciOPP complexes as suggested by the observed phosphorus hyperfine in its EPR spectrum and its similar Mössbauer parameters to previously characterized, isolable iron(I)–dpbz complexes.54
Phenylated iron(II)–SciOPP species formed in situ upon reaction of 1-Cl2 and phenyl nucleophiles (both Kumada and Suzuki–Miyaura) could be identified using a combination of freeze-trapped Mössbauer and MCD spectroscopies for reactions at both 25 and 0 °C. These species include the distorted tetrahedral S = 2 species Fe(Ph)X(SciOPP) (5-X) (X = Br or Cl due to halide exchange) as well as the bis-phenylated S = 2 species Fe(Ph)2(SciOPP) (4a) and Fe(Ph)2(THF)(SciOPP) (4b). While Fe(Ph)Br(SciOPP) (5-Br) could be further characterized by X-ray crystallography, the reduced stability of the bis-phenylated species (even at low temperature) precluded their structural characterization. However, the combination of detailed spectroscopic studies, solvent effects on in situ iron speciation and reaction studies confirm their assignments. Specifically, the bis-phenylated iron(II)–SciOPP species 4a and 4b are observed to directly reduce to form 2 and both species are consumed at the same rate to form 5-X upon reactions with electrophile at 0 °C, consistent with a rapid equilibrium between the solvent coordinated and uncoordinated species.
Pseudo single turnover studies of the reaction of 5-X and 4a/4b with electrophile at 25 °C indicate that both mono- and bis-phenylated iron(II)–SciOPP species are exceptionally reactive toward electrophile to generate cross-coupled product (complete reaction in <5 s at 25 °C with bromocycloheptane corresponding to an observed rate of >12 min–1). However, reactions using 1 equiv PhMgBr (to form 5-X) yield predominately cross-coupled product, whereas reactions with 2 equiv PhMgBr at 25 °C (to form 4a and 4b) are less selective and yield an ∼1:1 ratio of cross-coupled product and cycloheptene. Thus, while both 5-X and 4a/4b react with electrophile at catalytically relevant rates suggesting both could contribute to catalysis, the higher cross-coupled product selectivities observed with 5-X indicate that this is likely the dominant active species in both the Kumada and Suzuki–Miyaura cross-coupling reactions. This is consistent with the observation of highly undertransmetalated iron–SciOPP species during pseudo-steady-state catalysis. Furthermore, the slow addition of PhMgBr for Kumada cross-coupling (∼1 equiv every 5 s) combined with the relatively slow rate of transmetalation from the borate nucleophile in the Suzuki–Miyaura reaction would serve to maximize the formation of 5-X while minimizing the formation of the less selective bis-phenylated species in order to yield highly selective cross-couplings. The observation of 5-X by Mössbauer during the steady-state Suzuki–Miyaura reaction is consistent with the slower rate of reaction of 5-X with the chlorocycloheptane employed in the Suzuki–Miyaura reaction compared to reactions with bromocycloheptane. Furthermore, these results represent an interesting contrast to the Kumada cross-coupling of MesMgBr and primary alkyl halides with 1-Cl2, where slow Grignard addition was found to be important for minimizing in situ formation of FeMes3– (which is reactive but not highly selective in catalysis) through SciOPP ligand dissociation in the presence of excess MesMgBr.50 Lastly, the observation that reaction of 5-X with electrophile results in the formation of 1-X2 and cross-coupled product is consistent with previous mechanistic proposals involving an Fe(II)/Fe(III) redox cycle in iron–SciOPP catalyzed cross-coupling with aryl nucleophiles.39,43,50
While previous studies demonstrated that FeMes2(SciOPP) was the active catalytic species in Kumada cross-couplings of MesMgBr with primary alkyl halides,50 the bulky mesityl nucleophile and the stability of the in situ formed FeMesBr(SciOPP) and FeMes2(SciOPP) species have led to suggestions that less bulky aryl nucleophiles might more likely result in low-valent iron(I) or iron(0) iron active species in catalysis.52 Consistent with this hypothesis, iron(I)–dpbz complexes including the arylated species (dpbz)2Fe(4-tolyl) as well as (dpbz)2FeBr have been isolated which can serve as effective precatalysts for Negishi cross-coupling with (4-tolyl)2Zn nucleophile.36 However, the observation of highly reactive phenylated iron(II)–SciOPP species in the present study that react with electrophile to form cross-coupled products at rates exceeding that for reductive elimination to form 2 clearly demonstrates that low-valent iron is not required to generate highly reactive arylated iron–bisphosphine species for effective cross-coupling. While it is possible that iron–dpbz cross-coupling does involve iron(I) active species due to the prevalence of this system for reduction to iron(I) compared to reduction to iron(0) with the more sterically bulky SciOPP ligand, it is clear that iron(II)–bisphosphines complexes can serve as active species for cross-couplings with phenyl nucleophiles. Therefore, iron(II) complexes as well as low-valent iron species must be considered as potential active species in mechanistic studies of iron-based cross-coupling reactions.
Lastly, the identification of highly reactive iron(II) species such as 5-Br in Kumada and Suzuki–Miyaura cross-coupling of phenyl nucleophiles with secondary alkyl halides was possible due to the combination of freeze-trapped spectroscopic studies sensitive to iron speciation and reaction/GC studies derived from reactions of electrophile with known iron distributions present in situ. By contrast, chemical quenching alone to track biphenyl formation was found to be inherently unreliable as such quenchings of phenylated iron(II)–SciOPP species were found to yield biphenyl due to quenching. While biaryl quantitation methods have been used to estimate iron oxidation states present in solution during reactions of iron species with aryl nucleophiles,36,57 such contributions to biaryl formation from chemical quenching methods could lead to overestimation of the amount of reduced iron present in solution. Due to this challenge, direct spectroscopic characterization of the actual iron species present in solution represents a direct and more reliable method for the evaluation of the in situ formed species in cross-coupling.
4. Conclusion
In the present study, a combination of spectroscopic methods (Mössbauer, MCD, EPR) and synthetic and reaction studies have enabled elucidation of the in situ formed iron species, identification of the active iron species and insight into the mechanism of catalysis in iron-catalyzed Kumada and Suzuki–Miyaura cross-couplings of phenyl nucleophiles and secondary alkyl halides. While low valent iron species are observed to form in this chemistry, these species do not react with electrophile at catalytically relevant rates and predominately yield cycloheptene. Mono- and bis-phenylated iron(II)–SciOPP species are identified that form prior to reductive elimination and exhibit reactivity with electrophile at catalytically relevant rates. The higher selectivity toward the formation of cross-coupled product observed for the monophenylated species combined with the undertransmetalated nature of the in situ iron species in both Kumada and Suzuki–Miyaura reactions indicates that Fe(Ph)X(SciOPP) (X = Br, Cl) is the predominant reactive species in cross-coupling. Overall, these studies demonstrate that low-valent iron is not required for the generation of highly reactive species for effective aryl-alkyl cross-couplings. The further application of this research methodology to additional iron-based cross-couplings systems should evaluate the broader relevance of iron(II) active species in these reactions.
5. Experimental Section
5.1. General Considerations
All reagents were purchased from commercial sources. All air and moisture sensitive manipulations were carried out in an MBraun inert-atmosphere (N2) drybox equipped with a direct liquid nitrogen inlet line. All anhydrous solvents were further dried using activated alumina/4 Å molecular sieves and stored under inert-atmosphere over molecular sieves. 31P NMR resonances are referenced to the external standard phosphorus signal of 85% H3PO4. 57FeCl2(SciOPP) and 57FeBr2(SciOPP) were prepared following literature methods from 57FeCl2·1.5THF and 57FeBr2, respectively.4257FeCl2·1.5THF and 57FeBr2 were synthesized following literature procedures58 using 57Fe metal (95% enriched) purchased from Isoflex. t-Butylphenyl pinacolborate (tBuPh-borate) was synthesized according to literature methods.42
5.2. Synthesis of Fe(η6-Biphenyl)(SciOPP) (2)
A 0.978 mmol (1.00 g) sample of FeCl2(SciOPP) was added to approximately 25 mL of a 6:1 (v/v) solution of THF and 1,4-dioxane. The resulting pale yellow solution was chilled to −30 °C on a cold plate prior to the slow addition of 2.2 equiv of phenylmagnesium bromide (2.15 mL of 1.0 M THF solution) with stirring. As the addition progressed, the reaction mixture turned orange, with precipitation of magnesium salts. These salts were removed by filtering the reaction mixture through a pad of Celite and, as the filtrate warmed to room temperature, it took on a deep, plum-purple color. The filtrate was concentrated to near-dryness under vacuum, and the resulting residue, which is exceedingly soluble in every common hydrocarbon solvent, was redissolved in 5 mL of a ∼ 1:1 (v/v) solution of hexane and pentane solution and refiltered through Celite. The filtrate was concentrated to dryness under vacuum to yield a purple solid. 1H NMR (500 MHz, THF-d8: δ 7.40–7.30 (m, 14H), 7.09 (br, 2H), 6.88–6.76 (m, 5H), 5.66 (s, 1H), 4.48 (s, 4H), 1.21 (s, 72H). 31P NMR (202 MHz, THF-d8): δ 88.98 (s). 80 K Mössbauer: (powder) δ = 0.44 mm/s, ΔEQ = 1.81 mm/s; (1:1 THF/2-MeTHF) δ = 0.44 mm/s, ΔEQ = 1.75 mm/s. Calcd for C74H98P2Fe with half an equivalent of 1,4-dioxane: 79.42 C, 8.94 H. Found: 79.36 C, 9.03 H. Dissolution of the isolated powder in 5 mL of a ∼ 1:1 (v/v) solution of hexane and benzene yielded, after 3 days at −30 °C, a moderately air-sensitive purple, crystal suitable for X-ray diffraction.
5.3. Synthesis of Fe(Ph)Br(SciOPP) (5-Br)
A 20 mL scintillation vial was charged with FeCl2(SciOPP) (49 mg, 0.048 mmol) and 7 mL diethyl ether. The resulting colorless solution was chilled with stirring to −30 °C and 1.0 equiv of phenylmagnesium bromide (96 μL of a 0.5 M solution in diethyl ether) was added dropwise over 30 s. The resulting orange solution was allowed to stir for an additional 5 min 30 s, and with the temperature maintained at −30 °C, the solvent volume was reduced in vacuo to ∼1 mL. At this time 1 mL more of chilled diethyl ether (−30 °C) was added to the mixture and the suspension filtered through a pad of Celite (prechilled to −30 °C). The concentrated brown-orange filtrate was allowed to slowly evaporate at −30 °C resulting in the formation of highly temperature and air sensitive pale yellow needles suitable for X-ray diffraction analysis within 24 h. 80 K Mössbauer (1:1 THF/2-MeTHF): δ = 0.50 mm/s, ΔEQ = 2.37 mm/s. VTVH-MCD: S = 2, D = −8 ± 1 cm–1, |E/D| = 0.27 ± 0.03. NIR MCD: d–d bands at 6530 and 7870 cm–1.
5.4. Mössbauer Spectroscopy
Solution samples for 57Fe Mössbauer spectroscopy were prepared from 3 mM 57FeCl2(SciOPP) or 57FeBr2(SciOPP) in 1:1 (v/v) THF/2-MeTHF to enable the simultaneous preparation of Mössbauer and MCD samples or in only THF for catalytic reaction measurements. All samples were prepared in an inert atmosphere glovebox equipped with a liquid nitrogen fill port to enable sample freezing to 77 K within the glovebox. Each sample was loaded into a Delrin Mössbauer sample cup for measurements and loaded under liquid nitrogen. Low temperature 57Fe Mössbauer measurements were performed using a See Co. MS4 Mössbauer spectrometer integrated with a Janis SVT-400T He/N2 cryostat for measurements at 80 K with a 0.07 T applied magnetic field. Isomer shifts were determined relative to α-Fe at 298 K. All Mössbauer spectra were fit using the program WMoss (SeeCo). Errors of the fit analyses were the following: δ ± 0.02 mm/s and ΔEQ ± 3%. For multicomponent fits, the quantitation errors were ±3% (e.g., 70 ± 3%).
5.5. Magnetic Circular Dichroism Spectroscopy
All samples for MCD spectroscopy were prepared in an inert atmosphere glovebox equipped with a liquid nitrogen fill port to enable sample freezing to 77 K within the glovebox. MCD samples were prepared in 1:1 (v/v) THF/2-MeTHF (to form low temperature optical glasses) in copper cells fitted with quartz disks and a 3 mm gasket. Low temperature MCD experiments were conducted using two Jasco spectropolarimeters. Both instruments utilize a modified sample compartment incorporating focusing optics and an Oxford Instruments SM4000-7T superconducting magnet/cryostat. This setup permits measurements from 1.6 to 290 K with magnetic fields up to 7 T. A calibrated Cernox sensor directly inserted in the copper sample holder is used to measure the temperature at the sample to 0.001 K. UV–visible MCD spectra were collected using a Jasco J-715 spectropolarimeter and a shielded S-20 photomultiplier tube. Near-infrared (NIR) data were collected with a Jasco J-730 spectropolarimeter and a liquid nitrogen cooled InSb detector. All MCD spectra were baseline-corrected against zero-field scans. VTVH-MCD spectra were analyzed using previously reported fitting procedures.55,59
5.6. Electron Paramagnetic Resonance Spectroscopy
All samples for EPR spectroscopy were prepared in an inert atmosphere glovebox equipped with a liquid nitrogen fill port to enable sample freezing to 77 K within the glovebox. EPR samples were prepared in 4 mm OD Suprasil quartz EPR tubes from Wilmad Labglass. Samples for spin integration utilized high precision Suprasil quartz tubes to allow for direct comparison of intensities between different samples. X-band EPR spectra were recorded on a Bruker EMXplus spectrometer equipped with a 4119HS cavity and an Oxford ESR-900 helium flow cryostat. The instrumental parameters employed for all samples were as follows: 1 mW power; time constant 41 ms; modulation amplitude 8 G; 9.38 GHz (10 K spectra)/9.83 GHz (298 K spectra); modulation frequency 100 kHz. Spin integration was performed on samples exhibiting S = 1/2 EPR spectra and were spin integrated using a 3 mM CuSO4 standard under nonsaturating conditions. Identical instrumentation parameters were used for both the iron and standard samples.
5.7. Reactions of FeCl2(SciOPP) and 1 or 2 equiv of PhMgBr with Electrophile for in Situ Spectroscopic Studies
As an example of the general procedure employed, the reaction of 57FeCl2(SciOPP) with 1 equiv of PhMgBr and 20 equiv of bromocycloheptane is described. To a 1:1 THF/2-MeTHF solution of 57FeCl2(SciOPP) was added dropwise 1.0 equiv PhMgBr at RT to generate 3 mM 57Fe(Ph)Cl(SciOPP) in solution. The solution was stirred for 30 s and then a Mössbauer sample was prepared and frozen in liquid nitrogen within an anaerobic glovebox (samples prepared at 0 °C were stirred for 5 min prior to electrophile addition). To the remaining solution was added 20 equiv of bromocycloheptane and a sample was immediately frozen in liquid nitrogen to freeze trap the reaction at the desired time point (15 s).
5.8. Reaction of FeCl2(SciOPP) and 1 equiv of PhMgBr with Bromocycloheptane
To a solution of FeCl2(SciOPP) (0.012 mmol) in 1:1 THF/2-MeTHF (3.16 mL) was added dodecane (240 μL, 0.1 M in THF) and 1 equiv of PhMgBr (120 μL, 0.1 M in THF) and stirred for 30 s at RT. Then, bromocylcoheptane (480 μL, 0.5 M in THF) was added quickly at room temperature. At 5, 30, and 90 s time points, an aliquot of the reaction mixture was quenched with 10–5 M HCl in THF (or with aqueous 1 M NaHSO4; no difference in product yields was observed between quenching solvents) and diluted with THF and filtered through a pad of Florisil (<200 mesh, Sigma-Aldrich). Product yields and recovery of bromocycloheptane were determined by quantitative GC analysis using dodecane as an internal standard.
Acknowledgments
This work was supported by a grant from the National Institutes of Health (R01GM111480 to M.L.N.). The authors thank Dr. William W. Brennessel for assistance in the collection and analysis of X-ray crystallographic data.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b06648.
The authors declare no competing financial interest.
Supplementary Material
References
- Bolm C.; Legros J.; Paih J. L.; Zani L. Chem. Rev. 2004, 104, 6217. 10.1021/cr040664h. [DOI] [PubMed] [Google Scholar]
- Sherry B. D.; Furstner A. Acc. Chem. Res. 2008, 41, 1500. 10.1021/ar800039x. [DOI] [PubMed] [Google Scholar]
- Czaplik W. M.; Mayer M.; Cvengros J.; Wangelin A. J. v. ChemSusChem 2009, 2, 396–417. 10.1002/cssc.200900055. [DOI] [PubMed] [Google Scholar]
- Jana R.; Pathak T. P.; Sigman M. S. Chem. Rev. 2011, 111, 1417–1492. 10.1021/cr100327p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer I.; Knolker H.-J. Chem. Rev. 2015, 115, 3170–3387. 10.1021/cr500425u. [DOI] [PubMed] [Google Scholar]
- Nakamura E.; Yoshikai N. J. Org. Chem. 2010, 75, 6061–6067. 10.1021/jo100693m. [DOI] [PubMed] [Google Scholar]
- Tamura M.; Kochi J. K. J. Am. Chem. Soc. 1971, 93, 1487–1489. 10.1021/ja00735a030. [DOI] [Google Scholar]
- Tamura M.; Kochi J. K. J. Organomet. Chem. 1971, 31, 289–309. 10.1016/S0022-328X(00)86239-7. [DOI] [Google Scholar]
- Neumann S. M.; Kochi J. K. J. Org. Chem. 1975, 40, 599–606. 10.1021/jo00893a013. [DOI] [Google Scholar]
- Smith R. S.; Kochi J. K. J. Org. Chem. 1976, 41, 502–509. 10.1021/jo00865a019. [DOI] [Google Scholar]
- Tamura M.; Kochi J. K. Bull. Chem. Soc. Jpn. 1971, 44, 3063–3073. 10.1246/bcsj.44.3063. [DOI] [Google Scholar]
- Nakamura M.; Matsuo K.; Ito S.; Nakamura E. J. Am. Chem. Soc. 2004, 126, 3686–3687. 10.1021/ja049744t. [DOI] [PubMed] [Google Scholar]
- Guerinot A.; Reymond S.; Cossy J. Angew. Chem., Int. Ed. 2007, 46, 6521–6524. 10.1002/anie.200702206. [DOI] [PubMed] [Google Scholar]
- Cahiez G.; Duplais C.; Moyeaux A. Org. Lett. 2007, 9, 3253–3254. 10.1021/ol7016092. [DOI] [PubMed] [Google Scholar]
- Noda D.; Sunada Y.; Hatakeyama T.; Nakamura M.; Nagashima H. J. Am. Chem. Soc. 2009, 131, 6078–6079. 10.1021/ja901262g. [DOI] [PubMed] [Google Scholar]
- Bedford R. B.; Brenner P. B.; Carter E.; Cogswell P. M.; Haddow M. F.; Harvey J. N.; Murphy D. M.; Nunn J.; Woodall C. H. Angew. Chem., Int. Ed. 2014, 53, 1804–1808. 10.1002/anie.201308395. [DOI] [PubMed] [Google Scholar]
- Hedstroem A.; Izakian Z.; Vreto I.; Wallentin C.-J.; Norrby P.-O. Chem.–Eur. J. 2015, 21, 5946–5953. 10.1002/chem.201406096. [DOI] [PubMed] [Google Scholar]
- Bedford R. B.; Betham M.; Bruce D. W.; Danopoulos A. A.; Frost R. M.; Hird M. J. Org. Chem. 2006, 71, 1104–1110. 10.1021/jo052250+. [DOI] [PubMed] [Google Scholar]
- Hatakeyama T.; Nakamura M. J. Am. Chem. Soc. 2007, 129, 9844–9845. 10.1021/ja073084l. [DOI] [PubMed] [Google Scholar]
- Hatakeyama T.; Hashimoto S.; Ishizuka K.; Nakamura M. J. Am. Chem. Soc. 2009, 131, 11949–11963. 10.1021/ja9039289. [DOI] [PubMed] [Google Scholar]
- Ghorai S.; Jin M.; Hatakeyama T.; Nakamura M. Org. Lett. 2012, 14, 1066–1069. 10.1021/ol2031729. [DOI] [PubMed] [Google Scholar]
- Guisan-Ceinos M.; Tato F.; Bunuel E.; Calle P.; Cardenas D. J. Chem. Sci. 2013, 4, 1098–1104. 10.1039/c2sc21754f. [DOI] [Google Scholar]
- Li B.-J.; Xu L.; Wu Z.-H.; Guan B.-T.; Sun C.-L.; Wang B.-Q.; Shi Z.-J. J. Am. Chem. Soc. 2009, 131, 14656–14657. 10.1021/ja907281f. [DOI] [PubMed] [Google Scholar]
- Furstner A.; Martin R.; Krause H.; Seidel G.; Goddard R.; Lehmann C. W. J. Am. Chem. Soc. 2008, 130, 8773–8787. 10.1021/ja801466t. [DOI] [PubMed] [Google Scholar]
- Kuzmina O. M.; Steib A. K.; Flubacher D.; Knochel P. Org. Lett. 2012, 14, 4818–4821. 10.1021/ol302136c. [DOI] [PubMed] [Google Scholar]
- Kuzmina O. M.; Steib A. K.; Markiewicz J. T.; Flubacker D.; Knochel P. Angew. Chem., Int. Ed. 2013, 52, 4945–4949. 10.1002/anie.201210235. [DOI] [PubMed] [Google Scholar]
- Agrawal T.; Cook S. P. Org. Lett. 2013, 15, 96–99. 10.1021/ol303130j. [DOI] [PubMed] [Google Scholar]
- Mo Z.; Zhang Q.; Deng L. Organometallics 2012, 31, 6518–6521. 10.1021/om300722g. [DOI] [Google Scholar]
- Steib A.; Thaler T.; Komeyama K.; Mayer P.; Knochel P. Angew. Chem., Int. Ed. 2011, 50, 3303–3307. 10.1002/anie.201007187. [DOI] [PubMed] [Google Scholar]
- Bauer G.; Wodrich M. D.; Scopelliti R.; Hu X. Organometallics 2015, 34, 289–298. 10.1021/om501122p. [DOI] [Google Scholar]
- Furstner A.; Leitner A.; Mendez M.; Krause H. J. Am. Chem. Soc. 2002, 124, 13856–13863. 10.1021/ja027190t. [DOI] [PubMed] [Google Scholar]
- Scheiper B.; Bonnekessel M.; Krause H.; Furstner A. J. Org. Chem. 2004, 69, 3943–3949. 10.1021/jo0498866. [DOI] [PubMed] [Google Scholar]
- Nagano T.; Hayashi T. Org. Lett. 2004, 6, 1297–1299. 10.1021/ol049779y. [DOI] [PubMed] [Google Scholar]
- Martin R.; Furstner A. Angew. Chem., Int. Ed. 2004, 43, 3955–3957. 10.1002/anie.200460504. [DOI] [PubMed] [Google Scholar]
- Bedford R. B.; Huwe M.; Wilkinson M. C. Chem. Commun. 2009, 600–602. 10.1039/B818961G. [DOI] [PubMed] [Google Scholar]
- Adams C. J.; Bedford R. B.; Carter E.; Gower N. J.; Haddow M. F.; Harvey J. N.; Huwe M.; Cartes M. A.; Mansell S. M.; Mendoza C.; Murphy D. M.; Neeve E. C.; Nunn J. J. Am. Chem. Soc. 2012, 134, 10333–10336. 10.1021/ja303250t. [DOI] [PubMed] [Google Scholar]
- Hatakeyama T.; Kondo Y.; Fujiwara Y.; Takaya H.; Ito S.; Nakamura E.; Nakamura M. Chem. Commun. 2009, 1216–1218. 10.1039/b820879d. [DOI] [PubMed] [Google Scholar]
- Dongol K. G.; Koh H.; Sau M.; Chai C. L. L. Adv. Synth. Catal. 2007, 349, 1015–1018. 10.1002/adsc.200600383. [DOI] [Google Scholar]
- Hatakeyama T.; Hashimoto T.; Kathriarachchi K. K. A. D. S.; Zenmyo T.; Seike H.; Nakamura M. Angew. Chem., Int. Ed. 2012, 51, 8834–8837. 10.1002/anie.201202797. [DOI] [PubMed] [Google Scholar]
- Bedford R. B.; Carter E.; Cogswell P. M.; Gower N. J.; Haddow M. F.; Harvey J. N.; Murphy D. M.; Neeve E. C.; Nunn J. Angew. Chem., Int. Ed. 2013, 52, 1285–1288. 10.1002/anie.201207868. [DOI] [PubMed] [Google Scholar]
- Bedford R. B.; Brenner P. B.; Carter E.; Carvell T. W.; Cogswell P. M.; Gallagher T.; Harvey J. N.; Murphy D. M.; Neeve E. C.; Nunn J.; Pye D. R. Chem. - Eur. J. 2014, 20, 7935–7938. 10.1002/chem.201402174. [DOI] [PubMed] [Google Scholar]
- Hatakeyama T.; Hashimoto T.; Kondo Y.; Fujiwara Y.; Seike H.; Takaya H.; Tamad Y.; Ono T.; Nakamura M. J. Am. Chem. Soc. 2010, 132, 10674–10676. 10.1021/ja103973a. [DOI] [PubMed] [Google Scholar]
- Hatakeyama T.; Fujiwara Y.; Okada Y.; Itoh T.; Hashimoto T.; Kawamura S.; Ogata K.; Takaya H.; Nakamura M. Chem. Lett. 2011, 40, 1030–1032. 10.1246/cl.2011.1030. [DOI] [Google Scholar]
- Hatakeyama T.; Okada Y.; Yoshimoto Y.; Nakamura M. Angew. Chem., Int. Ed. 2011, 50, 10973–10976. 10.1002/anie.201104125. [DOI] [PubMed] [Google Scholar]
- Sun C.-L.; Krause H.; Furstner A. Adv. Synth. Catal. 2014, 356, 1281–1291. 10.1002/adsc.201301089. [DOI] [Google Scholar]
- Kawamura S.; Nakamura M. Chem. Lett. 2013, 42, 183–185. 10.1246/cl.2013.183. [DOI] [Google Scholar]
- Nakagawa N.; Hatakeyama T.; Nakamura M. Chem. Lett. 2015, 44, 486–488. 10.1246/cl.141167. [DOI] [Google Scholar]
- Hashimoto T.; Hatakeyama T.; Nakamura M. J. Org. Chem. 2012, 77, 1168–1173. 10.1021/jo202151f. [DOI] [PubMed] [Google Scholar]
- Takaya H.; Nakajima S.; Nakagawa N.; Isozaki K.; Iwamoto T.; Imayoshi R.; Gower N. J.; Adak L.; Hatakeyama T.; Honma T.; Masafumi M.; Sunada T.; Nagashima H.; Hashizume D.; Takahashi O.; Nakamura M. Bull. Chem. Soc. Jpn. 2015, 88, 410–418. 10.1246/bcsj.20140376. [DOI] [Google Scholar]
- Daifuku S. L.; Al-Afyouni M. H.; Snyder B. E. R.; Kneebone J. L.; Neidig M. L. J. Am. Chem. Soc. 2014, 136, 9132–9143. 10.1021/ja503596m. [DOI] [PubMed] [Google Scholar]
- Hawrelak E. J.; Bernskoetter W. H.; Lobkovsky E.; Yee G. T.; Bill E.; Chirik P. J. Inorg. Chem. 2005, 44, 3103–3111. 10.1021/ic048202+. [DOI] [PubMed] [Google Scholar]
- Bedford R. B. Acc. Chem. Res. 2015, 48, 1485–1493. 10.1021/acs.accounts.5b00042. [DOI] [PubMed] [Google Scholar]
- The crystal structure of 2 contains π-ligand disorder resulting from ligand exchange of biphenyl for benzene (i.e., solvent used for crystallization). The occupancy ratio of η6-biphenyl/η6-benzene ligation is 0.67:0.33.
- Bedford R. B.; Brenner P. B.; Carter E.; Clifton J.; Cogswell P. M.; Gower N. J.; Haddow M. F.; Harvey J. N.; Kehl J. A.; Murphy D. M.; Neeve E. C.; Neidig M. L.; Nunn J.; Snyder B. E. R.; Taylor J. Organometallics 2014, 33, 5767–5780. 10.1021/om500518r. [DOI] [Google Scholar]
- Pavel E. G.; Kitajima N.; Solomon E. I. J. Am. Chem. Soc. 1998, 120, 3949–3962. 10.1021/ja973735l. [DOI] [Google Scholar]
- Fillman K. L.; Przyojski J. A.; Al-Afyouni M. H.; Tonzetich Z.; Neidig M. L. Chem. Sci. 2015, 6, 1178–1188. 10.1039/C4SC02791D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedstrom A.; Lindstedt E.; Norrby P.-O. J. Organomet. Chem. 2013, 748, 51–55. 10.1016/j.jorganchem.2013.04.024. [DOI] [Google Scholar]
- Winter G.; Thompson D. W.; Loehe J. R. Inorg. Synth. 1973, 14, 99–104. [Google Scholar]
- Neese F.; Solomon E. I. Inorg. Chem. 1999, 38, 1847–1865. 10.1021/ic981264d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.