

Summary
Background
Despite preventive vaccines for oncogenic human papillomaviruses (HPVs), cervical intraepithelial neoplasia (CIN) is common, and current treatments are ablative and can lead to long-term reproductive morbidity. We assessed whether VGX-3100, synthetic plasmids targeting HPV-16 and HPV-18 E6 and E7 proteins, delivered by electroporation, would cause histopathological regression in women with CIN2/3.
Methods
Efficacy, safety, and immunogenicity of VGX-3100 were assessed in CIN2/3 associated with HPV-16 and HPV-18, in a randomised, double-blind, placebo-controlled phase 2b study. Patients from 36 academic and private gynaecology practices in seven countries were randomised (3:1) to receive 6 mg VGX-3100 or placebo (1 mL), given intramuscularly at 0, 4, and 12 weeks. Randomisation was stratified by age (<25 vs ≥25 years) and CIN2 versus CIN3 by computer-generated allocation sequence (block size 4). Funder and site personnel, participants, and pathologists were masked to treatment. The primary efficacy endpoint was regression to CIN1 or normal pathology 36 weeks after the first dose. Per-protocol and modified intention-to-treat analyses were based on patients receiving three doses without protocol violations, and on patients receiving at least one dose, respectively. The safety population included all patients who received at least one dose. The trial is registered at ClinicalTrials.gov (number NCT01304524) and EudraCT (number 2012-001334-33).
Findings
Between Oct 19, 2011, and July 30, 2013, 167 patients received either VGX-3100 (n=125) or placebo (n=42). In the per-protocol analysis 53 (49.5%) of 107 VGX-3100 recipients and 11 (30.6%) of 36 placebo recipients had histopathological regression (percentage point difference 19.0 [95% CI 1.4–36.6]; p=0.034). In the modified intention-to-treat analysis 55 (48.2%) of 114 VGX-3100 recipients and 12 (30.0%) of 40 placebo recipients had histopathological regression (percentage point difference 18.2 [95% CI 1.3–34.4]; p=0.034). Injection-site reactions occurred in most patients, but only erythema was significantly more common in the VGX-3100 group (98/125, 78.4%) than in the placebo group (24/42, 57.1%; percentage point difference 21.3 [95% CI 5.3–37.8]; p=0.007).
Interpretation
VGX-3100 is the first therapeutic vaccine to show efficacy against CIN2/3 associated with HPV-16 and HPV-18. VGX-3100 could present a non-surgical therapeutic option for CIN2/3, changing the treatment outlook for this common disease.
Funding
Inovio Pharmaceuticals.
Introduction
In 2008, one in six new cancer diagnoses worldwide was attributable to an infectious pathogen.1 Human papillomavirus (HPV) causes one-third of infection-associated cancers. Although prophylactic vaccines provide protection against HPV-16 and HPV-18, the genotypes that cause 70% of cervical cancer, uptake has been disappointing in several countries, including the USA, leaving many women at risk. Exposure to HPV occurs with onset of sexual activity, and, since prophylactic vaccines have no therapeutic effect, HPV infections will likely contribute to the global cancer burden for the foreseeable future. Moreover, the frequency of HPV-associated cancers continues to increase at anatomical sites other than the cervix (vagina, vulva, anus, and oropharynx), where validation of screening strategies is lacking.2 Immunotherapies for early HPV lesions would address a substantive unmet medical need and are likely to yield insights that could inform treatment approaches for other infection-associated malignancies.
HPV is essentially endemic because infection is asymptomatic, and, in immune-competent persons, most cervical infections are controlled without intervention.3 Intraepithelial pre-invasive HPV lesions, cervical intraepithelial neoplasia (CIN) 2/3, evolve from persistent infections, are clinically indolent, and not all CIN2/3 progresses to invasive disease. A subset of CIN2/3 lesions undergoes a presumably immunologically mediated spontaneous complete regression within a timeframe of 15 weeks.4,5 HPV-16 lesions are less likely to undergo regression than lesions caused by other HPV genotypes.4 However, because there is no validated method to predict the likelihood of histopathological regression, the standard of care for CIN2/3 is surgical resection.6
Both cervical squamous cancers and their precursor lesions are associated with integration of the viral genome into the host genome, and subsequent expression of two viral oncoproteins, E6 and E7.7 Although the site in the host genome in which the viral genome integrates varies, the HPV genome is most frequently disrupted in the region coding for E2, which functions normally to regulate expression of E6 and E7.8–10 Additionally, in some HPV-associated tumours in which viral integration is incomplete, methylation of E2 has been reported.11,12 Expression of both E6 and E7 is needed, but not sufficient, for initiation and persistence of disease, providing non-self antigenic targets for immune-based therapies. The indolent clinical behaviour and relatively straightforward accessibility of CIN2/3 lesions present an opportunity for testing immune therapies targeting viral antigens constitutively expressed on pre-neoplastic but not normal cells.
Although many pre-clinical models have supported proof-of-principle for immunotherapeutic targeting of E6 and E7 in HPV-associated malignancies, clinical translation has been incomplete, in part due to the restricted immunogenicity of the vaccines tested to date. VGX-3100 consists of two DNA plasmids encoding optimised synthetic consensus E6 and E7 genes of HPV-16 and HPV-18, using a proprietary design strategy, SynCon (Inovio Pharmaceuticals, Plymouth Meeting, PA, USA), as described previously.13,14 In a phase 1 trial in women in whom CIN2/3 lesions had been resected, intramuscular injection of VGX-3100 followed by electroporation elicited robust adaptive immune responses to E6 and E7 that were a log greater than previously reported immune responses to therapeutic HPV vaccines.15 This phase 2b study was designed to address crucial questions in immunotherapy for malignancies caused by persistent infections: whether therapeutic vaccination can elicit adaptive immune responses in patients with existing disease, and whether T-cell responses elicited by peripheral vaccination can have a therapeutic effect in a mucosal target lesion. Specifically, the trial was designed to assess the efficacy, safety, and immunogenicity of VGX-3100 delivered by electroporation before a planned standard therapeutic resection of CIN2/3 lesions positive for HPV-16 or HPV-18.
Methods
Study design and participants
This study was a multicentre, randomised, double-blind, placebo-controlled phase 2b trial with masked endpoint acquisition and adjudication. Women aged 18–55 years with histologically confirmed HPV-16-positive or HPV-18-positive CIN2/3 were recruited from 36 academic centres and private gynaecology practices in the USA, Estonia, South Africa, India, Canada, Australia, and Georgia. Histopathological diagnoses were confirmed by an independent, masked panel of experts in gynaecological pathology before randomisation. Women who were pregnant, breastfeeding, co-infected with hepatitis B, hepatitis C, or HIV, or having other immune deficient states were excluded. The protocol was approved by the institutional review board or ethics committee at each participating centre, and all patients gave written informed consent. An independent data and safety monitoring board reviewed unmasked safety and histopathological regression data.
Randomisation and masking
Patients were randomly assigned to receive either VGX-3100 (Inovio Pharmaceuticals) or placebo at study weeks 0, 4, and 12. The randomisation sequence was computer-generated by the funder's representative with a blocking factor of 4. Randomisation was stratified by age (<25 years vs ≥25 years) and CIN2 versus CIN3 (3:1, VGX-3100:placebo) in each of the four strata. No upper or lower limits were prespecified for the relative proportions of either CIN severity or age. Study sites were assigned a group of sequential randomisation numbers, and enrolled patients were assigned to a treatment group according to the randomisation sequence. Doses were prepared and labelled in masked syringes by an unmasked study pharmacist who had no other involvement in the study. Investigators, participants, study staff administering treatments, pathologists assessing histopathological regression, and those analysing the data were masked to treatment group assignment.
Procedures
Patients received either 6 mg VGX-3100 (3 mg plasmid targeting HPV-16 E6 and E7, and 3 mg plasmid targeting HPV-18 E6 and E7) or sterile water administered intramuscularly in 1 mL, followed by electroporation using the CELLECTRA (Inovio Pharmaceuticals) constant current device. CELLECTRA generates 52 ms controlled electrical pulses at the injection site, which increase cell membrane permeability and intracellular transfer of VGX-3100 to the nucleus, resulting in enhanced transfection and subsequent immunogenicity.16,17
Injection-site reactions, systemic symptoms, and unexpected adverse events were monitored throughout the study. Patients recorded local and systemic solicited and unsolicited adverse events in a symptom diary for 7 days after each dose. Injection sites were assessed 2 weeks after each dose.
In the first 40 patients enrolled, local pain was assessed with a visual analogue scale immediately after study treatment and 5 min and 10 min later. Creatine phosphokinase testing was done 2 weeks after each dose, and electrocardiograms (ECGs) were done pre-dose and post-dose.
Adverse events were graded according to Common Terminology Criteria for Adverse Events version 4.03, and were assessed by masked trial investigators. Serious adverse events were collected throughout follow-up, and were assessed by both the trial investigators and the funder's masked medical monitor to determine the relation to study treatment. The data and safety monitoring board met quarterly to advise the funder about safety and whether to terminate the study if there was a strong likelihood that histopathological regression in the VGX-3100 group was unacceptably low compared with the placebo group.
A mandatory interim colposcopy was done 12 weeks after the third dose. At the week 36 primary endpoint visit, patients with colposcopic evidence of residual disease underwent standard therapeutic resection. In patients with no evidence of disease, investigators had the option to biopsy the site of the original lesion(s). When all patients had completed their first visit after the primary endpoint visit (week 40), the data were unmasked. Long-term follow-up data were collected on all patients with remaining visits. Patients and study site personnel remained masked to treatment assignments until all study data were collected and finalised.
Whole blood was collected at baseline and at weeks 6, 14, 24, 36, and 88. Peripheral blood mononuclear cells and serum were cryopreserved for immune analysis. T-cell and antibody responses to HPV-16 and HPV-18 E6 and E7 were determined by interferon γ ELISpot, intracellular cytokine staining, and enzyme-linked immunosorbent assay, as described previously.15 ELISpot assays were done using separate E6 and E7 peptide pools from the relevant HPV genotype(s) identified at study entry, and with a single combined pool for the HPV genotype that the patient was not infected with.
Available tissue sections from both the diagnostic biopsy and week 36 specimens were stained by immunohistochemistry for CD8 and p16 after histological diagnoses were finalised. Whole slide image capture was done with a Nanozoomer 1.0-HT digital slide scanner (Hamamatsu Photonics KK, Hamamatsu City, Japan). Quantitative image analyses of the intensity of CD8+ infiltrates in normal and dysplastic epithelium and subjacent stroma were done using Tissue Studio version 3.6 (Definiens, Munich, Germany). HPV genotyping was done on cytological specimens from baseline and at weeks 6, 14, 24, 36, 62, and 88 using the Linear Array HPV assay (Roche, Basel, Switzerland). Cytopathology was assessed at weeks 14, 62, and 88.
Outcomes
The primary efficacy endpoint was histopathological regression to either CIN1 or normal pathology at 36 weeks after the first dose. Non-regressors were defined as patients with a tissue diagnosis of CIN2/3 at week 36. Patients in whom tissue (eg, biopsy) was obtained before week 36 based on suspicion of disease progression were also classified as non-regressors, regardless of histological diagnosis. The secondary endpoint was concomitant histopathological regression and clearance of HPV-16 or HPV-18, or both (viral clearance), at week 36. All histopathology slides were reviewed by a panel of masked, independent gynaecological pathologists.
Statistical analyses
The study was designed to provide 80% power at a two-sided α level of 0.05 to detect about a 27 percentage point difference in histopathological regression, assuming the proportion with histopathological regression in the placebo group ranged from 25% to 40%. Target enrolment was 148 patients (3:1, VGX-3100:placebo), and allowed for supplementation of enrolment based on projected dropouts. The primary and secondary efficacy endpoints were analysed based on stratified differences in the proportion of patients with histopathological regression between treatment groups.18–20 Safety analyses were based on differences between treatment groups in the proportion of patients who experienced adverse events during the 7-day and 28-day periods after any dose, and for the entire study period.21 Efficacy was analysed in the per-protocol population (patients who received all three doses of VGX-3100 or placebo and excluding patients with protocol violations) and also in a modified intention-to-treat population (patients who received at least one dose of VGX-3100 or placebo). Safety was analysed in all patients who received at least one dose of either VGX-3100 or placebo.
Post-hoc efficacy analyses of histopathological regression to normal and viral clearance were done using the same methods used for the primary and secondary efficacy analyses. Analyses of genotype-specific histopathological regression and concomitant histopathological regression and viral clearance across strata were based on differences of proportions.21 Post-hoc immunogenicity analyses compared responses between treatment groups, within the VGX-3100 group between those with histopathological regression or concomitant histopathological regression and viral clearance and those without, and within the VGX-3100 group between the baseline and post-treatment timepoints. Two-sample t tests or paired t tests were used for tissue immunogenicity analyses, and Wilcoxon-Mann-Whitney rank-sum tests or Wilcoxon signed-rank tests for peripheral immunogenicity analyses, due to the non-normality of the peripheral data. Because all post-hoc analyses were intended to be hypothesis-generating for future studies, p values were not corrected for multiplicity and therefore do not account for type I errors. The trial was registered at ClinicalTrials.gov (number NCT01304524) and EudraCT (number 2012-001334-33).
Role of the funding source
Employees and contractors of the funder were involved in the design, medical monitoring, trial oversight, trial monitoring, data management, analysis, and reporting of the study. All authors had access to the data, which were analysed by CLT, MPM, MD, and MLB. All authors had final responsibility for the decision to submit for publication.
Results
Between Aug 4, 2011, and July 30, 2013, 533 patients were screened (169 eligible, 364 ineligible), 169 were randomised, and 167 received either VGX-3100 (n=125) or placebo (n=42; figure 1). Table 1 summarises the demographics of the 167 patients who received at least one dose of VGX-3100 or placebo beginning on Oct 19, 2011. There were no major differences in age (<25 years vs ≥25 years), ethnic origin, race, or disease severity (CIN2 vs CIN3) between treatment groups. Overall, the relative proportion of CIN severity was skewed toward more severe disease (119/167 [71%] had CIN3), and age was skewed toward older patients (126/167 [75%] were aged ≥25 years). The most commonly identified genotype at study entry was HPV-16 (155/167 [92.8%]).
Figure 1. Trial profile.

*One patient had no viral clearance data and could not be included in the secondary endpoint analysis.
Table 1. Baseline demographics and clinical characteristics in patients receiving at least one dose of VGX-3100 or placebo (modified intention-to-treat population).
| VGX-3100 (n=125) | Placebo (n=42) | Total (n=167) | |
|---|---|---|---|
| Age, years | |||
|
| |||
| Mean (SD) | 29.4 (6.4) | 31.6 (9.3) | 30.0 (7.3) |
| Median (IQR) | 28.0 (25–32) | 28.5 (24–41) | 28.0 (25–34) |
|
| |||
| Ethnic origin | |||
|
| |||
| Hispanic or Latino | 18 (14%) | 8 (19%) | 26 (16%) |
| Not Hispanic, not Latino | 105 (84%) | 34 (81%) | 139 (83%) |
| Not reported | 2 (2%) | 0 | 2 (1%) |
|
| |||
| Race | |||
|
| |||
| Asian | 3 (2%) | 4 (10%) | 7 (4%) |
| Black or African American | 9 (7%) | 4 (10%) | 13 (8%) |
| Native Hawaiian or other Pacific Islander | 1 (1%) | 0 | 1 (1%) |
| White | 95 (76%) | 26 (62%) | 121 (72%) |
| Other | 14 (11%) | 7 (17%) | 21 (13%) |
|
| |||
| Strata | |||
|
| |||
| CIN2, age <25 years | 12 (10%) | 3 (7%) | 15 (9%) |
| CIN2, age ≥25 years | 25 (20%) | 8 (19%) | 33 (20%) |
| CIN3, age <25 years | 18 (14%) | 8 (19%) | 26 (16%) |
| CIN3, age ≥25 years | 70 (56%) | 23 (55%) | 93 (56%) |
|
| |||
| Body-mass index (kg/m2) | |||
|
| |||
| Mean (SD) | 25.6 (6.2) | 25.0 (5.0) | 25.4 (5.9) |
| Median (IQR) | 24.9 (21.5–28.6) | 23.8 (21.2–27.6) | 24.5 (21.4–27.9) |
|
| |||
Data are n (%) unless otherwise stated. CIN=cervical intraepithelial neoplasia.
Treatment with VGX-3100 was well tolerated. The most common adverse events were administration site reactions although only injection-site erythema had a statistically higher incidence in the VGX-3100 group (98/125, 78.4%) than in the placebo group (24/42, 57.1%) within the 28-day period after a dose (percentage point difference 21.3, 95% CI 5.3–37.8; p=0.007; table 2). Other common adverse events included fatigue, headache, myalgia, malaise, nausea, and arthralgia. No abnormalities were observed in creatine phosphokinase concentrations or ECGs in the first 40 patients enrolled. There was no difference in tolerability between VGX-3100 and placebo treatment groups. Four patients discontinued dosing due to an adverse event: three in the VGX-3100 group (two had injection-site pain, one had maculopapular rash), and one in the placebo group (allergic reaction). One additional VGX-3100 recipient with injection-site pain discontinued dosing, to seek surgical resection. One additional placebo recipient discontinued before receiving her third dose when she learned she was pregnant. In the total study population two (1.2%) of 167 patients discontinued because of pain. No related serious adverse events were reported. Two microinvasive cancers were identified in the resection specimens of the VGX-3100 group (two of 114, 1.8%).
Table 2. Systemic and local adverse events with onset within 28 days of a dose (safety population).
| VGX-3100 (n=125) | Placebo (n=42) | Percentage point difference | |
|---|---|---|---|
| Overall | |||
|
| |||
| Any adverse event or injection-site reaction | 123 (98.4%) | 41 (97.6%) | 0.8 |
| Grade 1 adverse event or injection-site reaction | 122 (97.6%) | 41 (97.6%) | 0 |
| Grade 2 adverse event or injection-site reaction | 90 (72.0%) | 25 (59.9%) | 12.5 |
| Grade 3 adverse event or injection-site reaction | 6 (4.8%) | 1 (2.4%) | 2.4 |
| Serious adverse event* | 0 | 0 | 0 |
| Related serious adverse event* | 0 | 0 | 0 |
| Dose-limiting toxicity | 3 (2.4%) | 1 (2.4%) | 0 |
| Discontinued due to adverse event | 3 (2.4%) | 1 (2.4%) | 0 |
|
| |||
| Systemic | |||
|
| |||
| Fatigue | 68 (54.4%) | 20 (47.6%) | 6.8 |
| Headache | 51 (40.8%) | 21 (50.0%) | ‒9.2 |
| Myalgia | 48 (38.4%) | 14 (33.3%) | 5.1 |
| Malaise | 39 (31.2%) | 11 (26.2%) | 5.0 |
| Nausea | 30 (24.0%) | 11 (26.2%) | ‒2.2 |
|
| |||
| Local injection site | |||
|
| |||
| Erythema | 98 (78.4%) | 24 (57.1%) | 21.3† |
| Pain | 114 (91.2%) | 38 (90.5%) | 0.7 |
| Tenderness | 104 (83.2%) | 32 (76.2%) | 7.0 |
| Swelling or oedema | 62 (49.6%) | 14 (33.3%) | 16.3 |
|
| |||
Data are n (%) unless otherwise stated.
Over the entire study period, serious adverse events were reported in seven (5.6%) VGX-3100 recipients and three (7.1%) placebo recipients; none was assessed to be related to treatment.
p=0.007.
In the per-protocol analysis, histopathological regression occurred in 53 (49.5%) of 107 VGX-3100 recipients compared with 11 (30.6%) of 36 placebo recipients (percentage point difference 19.0, 95% CI 1.4–36.6; p=0.034). In the modified intention-to-treat analyses, histopathological regression occurred in 55 (48.2%) of 114 VGX-3100 recipients compared with 12 (30.0%) of 40 placebo recipients (percentage point difference 18.2, 1.3–34.4; p=0.034; figure 2). Concomitant histopathological regression and viral clearance occurred in 43 (40.2%) of 107 VGX-3100 recipients compared with five (14.3%) of 35 placebo recipients in the per-protocol analysis (percentage point difference 25.9, 95% CI 8.0–39.2; p=0.003). In the modified intention-to-treat analyses, concomitant histopathological regression and viral clearance occurred in 45 (39.5%) of 114 VGX-3100 recipients and six (15.4%) of 39 placebo recipients (percentage point difference 24.1, 95% CI 7.3–36.6; p=0.003; figure 2). Thus, among those with histopathological regression, viral clearance was more likely among VGX-3100 recipients (about 80%) than among placebo recipients (about 50%).
Figure 2. Clinical efficacy.

Percentage of patients with histopathological regression or concomitant histopathological regression and viral clearance at week 36 in VGX-3100 and placebo groups in (A) the per-protocol analysis and (B) the modified intention-to-treat analysis. (C) Histopathological regression to normal for per-protocol and modified intention-to-treat analyses. (D) Effect of mixed infections including HPV-16 (left) compared with HPV-16 mono-infection (right) on rates of histopathological regression and viral clearance. HPV=human papillomavirus.
In post-hoc efficacy analyses, histopathological regression to normal occurred in 43 (40.2%) of 107 VGX-3100 recipients compared with six (16.7%) of 36 placebo recipients (percentage point difference 23.5, 95% CI 4.4–37.0; p=0.012) in the per-protocol analysis (figure 2). Viral clearance occurred in 56 (52.3%) of 107 VGX-3100 recipients compared with nine (25.7%) of 35 placebo recipients in the per-protocol analysis (percentage point difference 26.6, 95% CI 6.8–42.2; p=0.006). VGX-3100 recipients with histopathological regression were more likely to regress to normal (43/53, 81.1%) compared with placebo recipients (6/11, 54.5%) in the per-protocol analysis.
Most recipients had mixed infections and had CIN3 at baseline, and the difference in histopathological regression was more evident in patients with CIN3 (table 3). Across strata, histopathological regression in VGX-3100 recipients was similar in lesions caused by HPV-16 alone (51.2%) and those caused by mixed infections (48.4%), whereas spontaneous regression (ie, in placebo recipients) was more likely in lesions caused by mixed infections (37.5%; figure 2). In patients with HPV-16 mono-infection, histopathological regression and viral clearance occurred in 16 (39.0%) of 41 VGX-3100 recipients compared with one (7.1%) of 14 placebo recipients (p=0.027). In patients who had mixed infections, concomitant histopathological regression and clearance of HPV-16 occurred in 25 (40.3%) of 62 VGX-3100 recipients compared with two (12.5%) of 16 placebo recipients (p=0.038; figure 2).
Table 3. Infection distribution at study entry and with respect to clinical efficacy by age and disease severity for all recipients receiving at least one dose (modified intention-to-treat population).
| VGX-3100 | Placebo | |
|---|---|---|
| Single infection at study entry | 47/125 (38%) | 21/42 (50%) |
|
| ||
| Mixed infection at study entry | 78/125 (62%) | 21/42 (50%) |
|
| ||
| Efficacy against cervical intraepithelial neoplasia 2 | ||
| Single infection, <25 years | 2/2 (100%) | 1/1 (100%) |
| Single infection, ≥25 years | 5/9 (56%) | 2/5 (40%) |
| Mixed infection, <25 years | 6/10 (60%) | 1/2 (50%) |
| Mixed infection, ≥25 years | 8/15 (53%) | 2/3 (67%) |
|
| ||
| Efficacy against cervical intraepithelial neoplasia 3 | ||
| Single infection, <25 years | 0/1 | 1/2 (50%) |
| Single infection, ≥25 years | 14/31 (45%) | 2/12 (17%) |
| Mixed infection, <25 years | 10/14 (71%) | 1/5 (20%) |
| Mixed infection, ≥25 years | 10/32 (31%) | 2/10 (20%) |
|
| ||
Data are n/N (%).
In post-hoc immunological analyses, T-cell responses to HPV-16 and HPV-18 E6 and E7 peaked at week 14 for VGX-3100 recipients, with a 9.5 times greater median response than in placebo (p<0.0001; figure 3). In patients with concomitant histopathological regression and viral clearance, the size of median combined T-cell responses to E6 and E7 was greater than in patients who did not achieve concomitant histopathological regression and viral clearance (appendix). The magnitude of T-cell responses to E6 was associated with clinical outcome; responses to E6 differed significantly between those who had concomitant histopathological regression and viral clearance and those who did not (p=0.035; figure 3), whereas responses to E7 were not associated with clinical outcome (appendix).
Figure 3. Immune responses to VGX-3100.
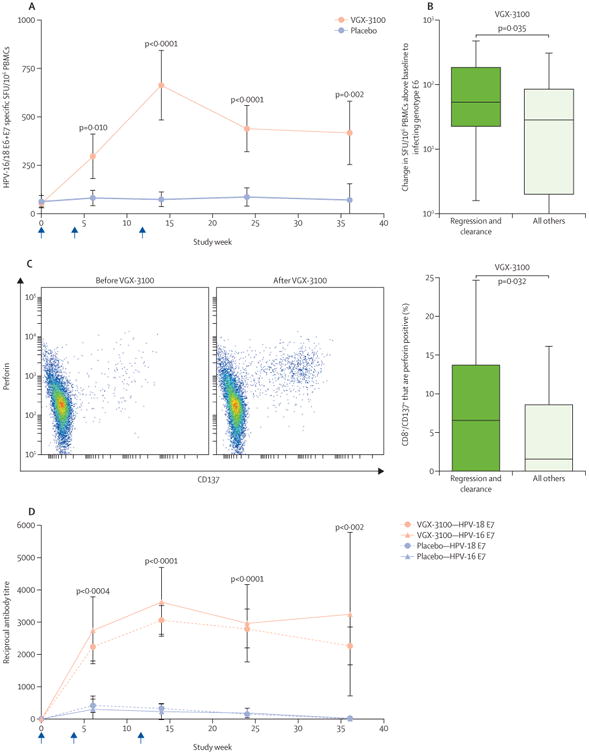
(A) Interferon γ ELISpot responses to HPV-16 and HPV-18 E6 and E7 in VGX-3100 and placebo groups. (B) Change in T-cell responses to infecting genotype E6 in patients treated with VGX-3100 who had both histopathological regression and viral clearance versus those who did not at study week 14. Horizontal lines within boxes show median response. (C) Representative flow cytometric plot of HPV-specific upregulation of CD137 and perforin in CD8 T cells before and after VGX-3100 (left). Frequencies of CD137+ CD8+ T cells expressing perforin in patients treated with VGX-3100 who had both histopathological regression and viral clearance, versus those who did not, at study week 14 (right). Horizontal lines within boxes show median response. (D) Kinetics of antibody responses to HPV-16 and HPV-18 E7 in VGX-3100 and placebo groups; p values for the difference between treatment groups were calculated at each timepoint for both antigens, and the maximum p value is displayed at each timepoint. Arrows show timing of a dose. HPV=human papillomavirus. PBMC=peripheral blood mononuclear cells. SFU=spot-forming units.
VGX-3100 elicited significantly increased frequencies of antigen-specific, activated CD8+ T cells, identified by cell surface expression of CD137,22 that also expressed perforin compared with placebo (p=0.001; appendix). Patients with histopathological regression had higher frequencies of HPV-specific CD8+ CD137+ T cells expressing perforin than those without histopathological regression (p=0.0499; appendix). The difference in frequencies of HPV-specific CD8+ CD137+ T cells that expressed perforin was greatest in patients who had concomitant histopathological regression and viral clearance compared with patients who did not (p=0.032; figure 3).
Humoral responses were also greater in VGX-3100 recipients compared with placebo recipients. Peak responses to E7 occurred at study week 14, and persisted through week 36 (figure 3). Anti-HPV-16 E7 and anti-HPV-18 E7 antibody concentrations in the VGX-3100 and placebo groups were significantly different at study weeks 6, 14, 24, and 36 (p≤0.0023 for both antigens at each visit). VGX-3100 recipients with histopathological regression and viral clearance developed antibody responses to both HPV-16 and HPV-18 E7 that were significantly higher than for those who did not achieve histopathological regression and viral clearance, at the time of peak response (post-dose 3) but also as early as post-dose 2 and as late as week 24 (appendix). Although the median titres of antibodies specific for HPV-16 E6 and HPV-18 E6 in VGX-3100 recipients were higher compared with placebo recipients after day 0, the differences in distributions were only significant for HPV-18 at week 14 (data not shown).
Paired, patient-matched tissue obtained from the diagnostic biopsy and from resection at week 36 was available for digital image analysis in 76 patients, providing the opportunity to do several post-hoc analyses. Representative examples of tissue annotation show p16 and CD8 immunohistochemistry in the diagnostic biopsy specimen (CIN3), compared with the post-treatment resection specimen at week 36, in a patient with no residual disease (appendix). In the pre-treatment specimen, p16 was overexpressed in CIN3 epithelium, and CD8+ infiltrates were scant (appendix). By contrast, in the post-treatment resection specimen, p16 was absent, consistent with normal epithelium, and CD8+ infiltrates were still present 6 months after completing treatment (appendix). In patients treated with VGX-3100 with histopathological regression, CD8+ infiltrates in non-lesional (non-CIN2/3) mucosa increased in both the epithelial and stromal compartments between baseline and week 36 (p=0.002 for epithelial and p=0.003 for stromal compartments; appendix). The increase in stromal CD8+ infiltrates was greater in patients with histopathological regression compared with those without regression (p=0.009; appendix). This result was also recorded for patients with histopathological regression and concomitant viral clearance (p=0.008; appendix).
Discussion
In this phase 2, randomised, double-blind, placebo-controlled clinical trial in women with HPV-16-positive or HPV-18-positive CIN2/3, both histopathological regression and concomitant histopathological regression and viral clearance were significantly greater after therapeutic vaccination with VGX-3100 compared with placebo. Both findings confirmed our hypotheses that therapeutic vaccination with this combination of synthetic plasmids targeting HPV-16 and HPV-18 E6 and E7 delivered by electroporation with the CELLECTRA device would cause histopathological regression and clearance of the infecting HPV genotype(s) in women with HPV-16-positive or HPV-18-positive CIN2/3. Furthermore, immune responses in peripheral blood (both CD8+ T cell and antibody) and cervical tissue correlated with both histopathological regression and concomitant histopathological regression with viral clearance.
Previous attempts to develop a non-surgical treatment for CIN2/3 have had mixed success. Several different immunotherapeutic approaches have targeted the E6 or E7 proteins of oncogenic HPV genotypes, and improvement in HPV-related genitourinary (cervical, vulvar, vaginal) dysplasia has been reported in several uncontrolled open-label studies.23,24 Two different approaches have been assessed recently in randomised placebo-controlled trials. A randomised phase 2 study similar to ours assessed TG4001, a modified vaccinia Ankara viral vector encoding HPV-16 E6 and E7 and the immunoregulatory cytokine interleukin 2.25 Although results have not yet been published, a press release reported significant differences between active vaccine and placebo.26 In TG4001 recipients, the 20% reported efficacy in HPV-16 mono-infected patients did not reach the developer's goal to advance development. In another randomised, phase 2, placebo-controlled trial, vaginal suppositories containing imiquimod, a TLR7/8 agonist, was efficacious in women with CIN2/3.27 Severity of dysplasia was not controlled, and the percentage of patients with CIN3 was higher in the placebo (55%) than the imiquimod (37%) group. The Austrian Gynaecological Oncology Group is doing a non-inferiority study of imiquimod versus conisation for the treatment of recurrent CIN2/3 (NCT01283763). As yet, neither approach is being developed for previously untreated CIN2/3, and no immunological correlation with clinical benefit has been reported. In diseases other than CIN2/3, other antigen-specific immunotherapy approaches have been reported to confer clinical benefit in randomised, placebo-controlled trials. Improvement in overall survival has been reported using two different approaches to antigen-specific immunotherapy for the treatment of castration-resistant prostate cancer, but no T-cell-based immunological correlates have been reported.28,29
This study differs from previous reports of clinical benefit in randomised placebo-controlled immunotherapy trials in several respects: efficacy of VGX-3100 was significantly higher than placebo for both histopathological regression alone and concomitant histopathological regression and viral clearance; efficacy was recorded in a population in which both severity of CIN and age were balanced between the treatment and placebo groups through prespecified stratification; and quantitative immunological measures (blood cellular and humoral; and tissue CD8) were found to correlate with both histopathological regression and concomitant histopathological regression and viral clearance.
One goal of preventive vaccination for other mucosally transmitted viruses is to elicit virus-specific CD8+ T cells that could establish tissue residence in normal tissue.31 In patients treated with VGX-3100 whose lesions regressed, the intensity of CD8+ infiltrates increased in normal mucosa in the stromal compartment, whereas in recipients of the placebo whose lesions regressed, the intensity of CD8+ infiltrates in normal mucosa did not change. This finding suggests a possible protective and therapeutic benefit of this technology. If the increased CD8+ infiltrates include HPV-specific effector T cells that have established tissue residence in normal mucosa, then one would expect that recurrences would be significantly less frequent than in either untreated women, or in women whose CIN2/3 had undergone spontaneous regression.
Patients who received VGX-3100 were substantially more likely to clear their viral infection than those who received placebo. Viral clearance is a critical component of efficacy; women with type-specific persistent HPV infection after excision of CIN3 lesions are much more likely to have recurrent disease needing re-excision than women with undetectable HPV after excision.31 Although a subset of patients mount effective endogenous responses, these naturally occurring responses are not easily detected systemically.32 Peripheral blood T-cell responses to HPV antigens elicited by VGX-3100 were an order of magnitude greater than naturally occurring responses, were detectable without ex-vivo expansion, and expressed significantly more perforin than those elicited by placebo, suggesting that VGX-3100-induced responses were more functional than the endogenous responses.33 Indeed, among placebo recipients with histopathological regression, only half regressed completely to normal, and only half also cleared the infecting HPV genotype. By contrast, about 80% of VGX-3100 recipients with histopathological regression regressed completely to normal, and about 80% also cleared the infecting HPV genotype.
To our knowledge, VGX-3100 is the first therapeutic vaccine to elicit both T-cell and antibody responses to HPV E6 and E7 in patients with pre-invasive cervical disease caused by HPV-16 and HPV-18. Although humoral responses are unlikely to mediate disease regression or elimination of infection directly, these responses do affirm the immunogenicity of VGX-3100. The striking difference in antibody titres between patients with and without clinical responses to VGX-3100 that was evident already at week 6 might provide a systemic correlate of immune activity associated with clinical response.
This study has several strengths. To date, it is the only prospective, randomised, double-blind study of therapeutic vaccination for women with CIN2/3 positive for HPV-16 or HPV-18, and includes centrally adjudicated tissue diagnoses before and after treatment, as well as longitudinal assessment of HPV status. Given the wide variation in reported spontaneous regression of CIN2/3, or CIN2 versus CIN3, we felt that only a randomised, masked, placebo-controlled trial would provide rigorous assessment of the clinical efficacy of VGX-3100.5,34 In unvaccinated persons, 15–30% of CIN2/3 might undergo spontaneous regression (ie, ≤CIN1 in subsequent therapeutic cone resections).
The study has limitations. First, HPV genotyping was based on cervical swabs. Because over half of the lesions were associated with mixed infections, some might have been caused by genotypes other than HPV-16 or HPV-18. Second, this regimen induced histopathological regression and viral clearance in about 40% of women with CIN2/3 positive for HPV-16 or HPV-18, whereas surgical excision would have eliminated the dysplastic tissue in 85–90% of women whose resections had negative surgical margins. However, up to 40–50% of resections that have positive endocervical margins recur. Finally, using a non-surgical treatment approach for CIN2/3 with less effectiveness than what has been reported for surgical resection might raise the concern that microinvasive cancers will develop. However, the frequency of microinvasive cancers identified in resection specimens was consistent with published data; occult, microinvasive carcinoma is a diagnosis typically made at therapeutic conisation, not colposcopy or biopsy.35,36
Our results are bona fide proof-of-principle for this therapeutic approach. Future studies will be directed at strategies to improve efficacy further. In a high-resource setting some women might opt for a trial of immunotherapy before a planned therapeutic resection. With the current VGX-3100 regimen, about half of women with a CIN2/3 positive for HPV-16 or HPV-18 would not only avoid surgery, but also clear the virus. The fact that 80% of the vaccinated recipients who had histopathological regression also had viral clearance is a crucial point, as viral persistence is an absolute requirement for recurrent disease.
Developing immune therapies for HPV disease is important because existing strategies for early detection and prevention of disease are both cumbersome and expensive, needing iterative visits and procedures, and so, disease remains common. The annual cost of the care of HPV-associated disease in the USA is about US$6 billion (appendix).37–41 Because rates of prophylactic vaccination for HPV are suboptimum in the USA (38%),42 the burden of HPV-associated disease is not likely to decrease in the near future.43 In 2013, only 57% of adolescent girls aged 13–17 years had initiated vaccination.44 Among vaccine initiators, only 38% completed the three-dose regimen. Moreover, the incidence of HPV-associated cancers in anatomical sites for which screening algorithms have not yet been validated, particularly in the oropharynx, is increasing steadily, and will likely bypass that of cervical cancer in the near future.45 The existing prophylactic vaccines, Gardasil and Cervarix, have no therapeutic effects.
This trial shows for the first time that peripheral delivery of engineered DNA encoding consensus viral proteins can elicit adaptive immune responses that have therapeutic effects distally in the site of cervical disease. Although a subset of CIN2/3 lesions does undergo spontaneous regression, in 2015, all women with the diagnosis have only a surgical treatment option because prognostic tissue biomarkers that can distinguish the likelihood of regression are unavailable. Our data suggest that VGX-3100 provides a non-surgical option for the treatment of CIN2/3 that could change the approach to care for this very common disease.
Supplementary Material
Research in context.
Evidence before this study
We reviewed the biomedical literature indexed in MEDLINE and searched ClinicalTrials.gov for clinical studies including patients with cervical intraepithelial neoplasia grades 2 or 3 (CIN2/3), using the search terms “HPV”, “therapeutic vaccination”, “intraepithelial neoplasia”, “cervical dysplasia”, “high grade CIN”, “treatment”, “therapy”, “manag”, “efficac”, “phase 2, 3 or II, III”, “controlled”, and “regression”; the search was restricted to articles written in English published between Jan 1, 1994, and April 28, 2015. We assessed all potentially relevant articles for quality and relevance, with the highest quality attributable to randomised trials.
Added value of this study
Recommended treatment for patients with CIN2/3 is therapeutic excision, either a cold knife conisation or a loop electrosurgical excision procedure. Although a subset of CIN2/3 lesions undergo spontaneous regression, because prognostic tissue biomarkers that can distinguish the likelihood of regression are still in development, all women who have CIN2/3 undergo cervical resection (eg, conisation). Previous attempts to develop a non-surgical treatment for CIN2/3 have had mixed results. Here we report, to the best of our knowledge, the first prospective, randomised, double-blind, placebo-controlled trial of a therapeutic vaccine in which women with CIN2/3 associated with HPV-16 and HPV-18 underwent complete histopathological regression and also cleared HPV.
Implications of all the available evidence
VGX-3100 is the first therapeutic vaccine to show efficacy against CIN2/3 associated with HPV-16 and HPV-18, presenting a possible cervical cancer prevention option through HPV-specific immunity without surgery, thereby changing the treatment outlook for this common disease.
Acknowledgments
We thank the patients who participated in this study and the entire HPV-003 clinical team from the participating study sites; Histologix and OracleBio for assistance with immunohistochemistry staining and digital image analysis; FlowMetric for aid in flow cytometry; Thomas C Wright Jr, Mark H Stoler, and Alex Ferenczy for independent review of all histopathology; Robert Park, Warner Huh, Linda Duska, Florian Schodel, and Lisa Taylor for their service on the data and safety monitoring board; Lindsay Sakata, John Stone, Dinah Amante, and Christine Knott for technical execution of humoral assays; Phil Greenberg for critical reading of the manuscript; David He for scientific programming support; and SciStrategy Communications for their editorial assistance in preparing the manuscript for submission, funded by Inovio Pharmaceuticals. This manuscript is dedicated to the memory of Larry Muenz and Christine Knott.
Footnotes
Contributors: CLT, LD, JL, NYS, DBW, and MLB contributed to study design, data analysis, and interpretation. MPM, KAK, XS, JY, AMS, AJS, AK, KEB, JB, and MLB contributed to the execution and analysis of the immunology. MG, ASB, KM-P, DS, AK, KEB, RJJ, TAH, JL, and MLB contributed to the implementation of the study and supervision at the study site. CLT, LE, RLP, and LD were principal investigators. MD led the statistical analysis. All authors contributed to writing and approval of the manuscript.
The HPV-003 Trial Study Team: Guillermo Valenzuela, Kristen Searing, Robert Parker Jr, Julie Jester, Elizabeth Graul, Patti Wall, Allison Jones, Keith Aqua, Polina Kaplun, Katie Smith, Christina Poulin, Janos Tanyi, Naseem Kerr, Francisco Garcia, Susan Vanzzini, Cynthia Goldberg, Nichele Pollock, Gioi Smith-Nguyen, Peggy Smith-Nguyen, Helena Kirkpatrick, Ann Scandariato, James Rice, Denise Richmond, Mark Einstein, Marisol Rivera, Mary Sanvardeker, Lance Edwards, Maureen Hurst, Daniela Naranjo, Ronald Hardy, Kathy Ziehnert, Michael Swor, Gail Wagoner, Jennifer Grube, Elizabeth Altschuler, Micah Harris, Courtney Ingram, Michael Twede, Peggy Genebach, David Young, Jan Horsley, Josefina Romaguera, Janice Diaz, Iris Cruz, Lydie Hazan, Eli Dela Cruz, Milroy Samuel, Sudha Suthanthira, Robert Littleton, Julie Jester, Shari Vincent, Thomas Minnec, Vickie Bannister, Ashley Gerow, Jaclyn Muter, David Wrede, Julie Silvers, Dianne Miller, Susan Keast, Lucy Gilbert, Natacha Albarracin, Lynette Denny, Tracey Adams, Janine Jones, Ayesha Sasman, Marika Japaridze, Maka Katsia, Terje Raud, Jelena Konosina, Katrin Seidelberg, Kätlin Väli, Anneli Mäeots, Kersti Kukk, Varje Ulla, Inga Aavik, Pille Aavik, Katrin Kõster, Annemai Märtson, Kaidi Soonberg, Neerja Bhatla, Shachi Vashist, Partha Basu, Sujoy Dasgupta.
Declaration of interests: MPM, KAK, XS, MD, JY, MG, ASB, KM-P, DS, AMS, AJS, AK, KEB, RJJ, TAH, JB, JL, NYS, and MLB are employees of Inovio Pharmaceuticals, and own shares or have been awarded stock options in the company. DBW has grant funding, participates in industry collaborations, and has received speaking honoraria and fees for consulting; this service includes serving on scientific review committees and advisory boards; remuneration includes direct payments or stock or stock options; DBW reports potential conflicts associated with this work with Pfizer, Bristol-Myers Squibb, Inovio, Merck, VGXI, Aldevron, MedImmune, and Roche. CLT has received an unrestricted research award from Inovio Pharmaceuticals. All other authors declare that they have no competing interests.
Contributor Information
Prof Cornelia L Trimble, Departments of Gynecology/Obstetrics, Oncology, and Pathology, Johns Hopkins University School of Medicine, Baltimore, MD, USA.
Matthew P Morrow, Inovio Pharmaceuticals, Inc, Plymouth Meeting, PA, USA.
Kimberly A Kraynyak, Inovio Pharmaceuticals, Inc, Plymouth Meeting, PA, USA.
Xuefei Shen, Inovio Pharmaceuticals, Inc, San Diego, CA, USA.
Michael Dallas, Inovio Pharmaceuticals, Inc, Plymouth Meeting, PA, USA.
Jian Yan, Inovio Pharmaceuticals, Inc, Plymouth Meeting, PA, USA.
Lance Edwards, Suffolk Obstetrics & Gynecology, LLP, Port Jefferson, NY, USA.
R Lamar Parker, Lyndhurst Gynecologic Associates, Winston-Salem, NC, USA.
Lynette Denny, Department of Obstetrics & Gynaecology, University of Cape Town, Groote Schuur Hospital, Observatory Cape Town, South Africa.
Mary Giffear, Inovio Pharmaceuticals, Inc, Plymouth Meeting, PA, USA.
Ami Shah Brown, Inovio Pharmaceuticals, Inc, Plymouth Meeting, PA, USA.
Kathleen Marcozzi-Pierce, Inovio Pharmaceuticals, Inc, Plymouth Meeting, PA, USA.
Divya Shah, Inovio Pharmaceuticals, Inc, Plymouth Meeting, PA, USA.
Anna M Slager, Inovio Pharmaceuticals, Inc, Plymouth Meeting, PA, USA.
Albert J Sylvester, Inovio Pharmaceuticals, Inc, Plymouth Meeting, PA, USA.
Amir Khan, Inovio Pharmaceuticals, Inc, Plymouth Meeting, PA, USA.
Kate E Broderick, Inovio Pharmaceuticals, Inc, San Diego, CA, USA.
Robert J Juba, Inovio Pharmaceuticals, Inc, Plymouth Meeting, PA, USA.
Timothy A Herring, Inovio Pharmaceuticals, Inc, Plymouth Meeting, PA, USA.
Jean Boyer, Inovio Pharmaceuticals, Inc, San Diego, CA, USA.
Jessica Lee, Inovio Pharmaceuticals, Inc, Plymouth Meeting, PA, USA.
Niranjan Y Sardesai, Inovio Pharmaceuticals, Inc, Plymouth Meeting, PA, USA.
Prof David B Weiner, Department of Pathology and Laboratory Medicine, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA, USA.
Mark L Bagarazzi, Inovio Pharmaceuticals, Inc, Plymouth Meeting, PA, USA.
References
- 1.de Martel C, Ferlay J, Franceschi S, et al. Global burden of cancers attributable to infections in 2008: a review and synthetic analysis. Lancet Oncol. 2012;13:607–15. doi: 10.1016/S1470-2045(12)70137-7. [DOI] [PubMed] [Google Scholar]
- 2.Chaturvedi AK. Beyond cervical cancer: burden of other HPV-related cancers among men and women. J Adolesc Health. 2010;46(suppl):S20–26. doi: 10.1016/j.jadohealth.2010.01.016. [DOI] [PubMed] [Google Scholar]
- 3.Ho GY, Bierman R, Beardsley L, Chang CJ, Burk RD. Natural history of cervicovaginal papillomavirus infection in young women. N Engl J Med. 1998;338:423–28. doi: 10.1056/NEJM199802123380703. [DOI] [PubMed] [Google Scholar]
- 4.Trimble CL, Piantadosi S, Gravitt P, et al. Spontaneous regression of high-grade cervical dysplasia: effects of human papillomavirus type and HLA phenotype. Clin Cancer Res. 2005;11:4717–23. doi: 10.1158/1078-0432.CCR-04-2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Melnikow J, Nuovo J, Willan AR, Chan BK, Howell LP. Natural history of cervical squamous intraepithelial lesions: a meta-analysis. Obstet Gynecol. 1998;92:727–35. doi: 10.1016/s0029-7844(98)00245-2. [DOI] [PubMed] [Google Scholar]
- 6.Massad LS, Einstein MH, Huh WK, et al. the 2012 ASCCP Consensus Guidelines Conference. 2012 updated consensus guidelines for the management of abnormal cervical cancer screening tests and cancer precursors. Obstet Gynecol. 2013;121:829–46. doi: 10.1097/AOG.0b013e3182883a34. [DOI] [PubMed] [Google Scholar]
- 7.Hudson JB, Bedell MA, McCance DJ, Laiminis LA. Immortalization and altered differentiation of human keratinocytes in vitro by the E6 and E7 open reading frames of human papillomavirus type 18. J Virol. 1990;64:519–26. doi: 10.1128/jvi.64.2.519-526.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pett MR, Alazawi WO, Roberts I, et al. Acquisition of high-level chromosomal instability is associated with integration of human papillomavirus type 16 in cervical keratinocytes. Cancer Res. 2004;64:1359–68. doi: 10.1158/0008-5472.can-03-3214. [DOI] [PubMed] [Google Scholar]
- 9.Wentzensen N, Vinokurova S, von Knebel Doeberitz M. Systematic review of genomic integration sites of human papillomavirus genomes in epithelial dysplasia and invasive cancer of the female lower genital tract. Cancer Res. 2004;64:3878–84. doi: 10.1158/0008-5472.CAN-04-0009. [DOI] [PubMed] [Google Scholar]
- 10.Reuschenbach M, Huebbers CU, Prigge ES, et al. Methylation status of HPV16 E2-binding sites classifies subtypes of HPV-associated oropharyngeal cancers. Cancer. 2015;121:1966–76. doi: 10.1002/cncr.29315. [DOI] [PubMed] [Google Scholar]
- 11.Leung TW, Liu SS, Leung RC, Chu MM, Cheung AN, Ngan HY. HPV 16 E2 binding sites 1 and 2 become more methylated than E2 binding site 4 during cervical carcinogenesis. J Med Virol. 2015;87:1022–33. doi: 10.1002/jmv.24129. [DOI] [PubMed] [Google Scholar]
- 12.Chaiwongkot A, Vinokurova S, Pientong C, et al. Differential methylation of E2 binding sites in episomal and integrated HPV 16 genomes in preinvasive and invasive cervical lesions. Int J Cancer. 2013;132:2087–94. doi: 10.1002/ijc.27906. [DOI] [PubMed] [Google Scholar]
- 13.Yan J, Harris K, Khan AS, Draghia-Akli R, Sewell D, Weiner DB. Cellular immunity induced by a novel HPV18 DNA vaccine encoding an E6/E7 fusion consensus protein in mice and rhesus macaques. Vaccine. 2008;26:5210–15. doi: 10.1016/j.vaccine.2008.03.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan J, Reichenbach DK, Corbitt N, et al. Induction of antitumor immunity in vivo following delivery of a novel HPV-16 DNA vaccine encoding an E6/E7 fusion antigen. Vaccine. 2009;27:431–40. doi: 10.1016/j.vaccine.2008.10.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bagarazzi ML, Yan J, Morrow MP, et al. Immunotherapy against HPV16/18 generates potent TH1 and cytotoxic cellular immune responses. Sci Transl Med. 2012;4:155ra38. doi: 10.1126/scitranslmed.3004414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Broderick KE, Humeau LM. Electroporation-enhanced delivery of nucleic acid vaccines. Expert Rev Vaccines. 2015;14:195–204. doi: 10.1586/14760584.2015.990890. [DOI] [PubMed] [Google Scholar]
- 17.Kalams SA, Parker SD, Elizaga M, et al. the NIAID HIV Vaccine Trials Network. Safety and comparative immunogenicity of an HIV-1 DNA vaccine in combination with plasmid interleukin 12 and impact of intramuscular electroporation for delivery. J Infect Dis. 2013;208:818–29. doi: 10.1093/infdis/jit236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mehrotra DV, Railkar R. Minimum risk weights for comparing treatments in stratified binomial trials. Stat Med. 2000;19:811–25. doi: 10.1002/(sici)1097-0258(20000330)19:6<811::aid-sim390>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 19.Mehrotra DV. Stratification Issues with Binary Endpoints. Drug Inf J. 2001;35:1343–50. [Google Scholar]
- 20.Mehrotra DV, Dmitrienko A, Anderson KM. Analysis of clinical trials: theory and applications—continuing education course. American Statistican Association Joint Statistical Meetings; Vancouver, Canada. 2010. [Google Scholar]
- 21.Miettinen O, Nurminen M. Comparative analysis of two rates. Stat Med. 1985;4:213–26. doi: 10.1002/sim.4780040211. [DOI] [PubMed] [Google Scholar]
- 22.Wolfl M, Kuball J, Ho WY, et al. Activation-induced expression of CD137 permits detection, isolation, and expansion of the full repertoire of CD8+ T cells responding to antigen without requiring knowledge of epitope specificities. Blood. 2007;110:201–10. doi: 10.1182/blood-2006-11-056168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daayana S, Elkord E, Winters U, et al. Phase II trial of imiquimod and HPV therapeutic vaccination in patients with vulval intraepithelial neoplasia. Br J Cancer. 2010;102:1129–36. doi: 10.1038/sj.bjc.6605611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kenter GG, Welters MJ, Valentijn AR, et al. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N Engl J Med. 2009;361:1838–47. doi: 10.1056/NEJMoa0810097. [DOI] [PubMed] [Google Scholar]
- 25.Brun JL, Dalstein V, Leveque J, et al. Regression of high-grade cervical intraepithelial neoplasia with TG4001 targeted immunotherapy. Am J Obstet Gynecol. 2011;204:169 e1–8. doi: 10.1016/j.ajog.2010.09.020. [DOI] [PubMed] [Google Scholar]
- 26.Transgene. Strasbourg, France: [accessed April 27, 2015]. Transgene reports randomized phase 2b data with its therapeutic HPV vaccine TG4001 in women with CIN2/3 intraepithelial cervical neoplasia. Press release, May 9, 2012. http://www.transgene.fr/wp-content/uploads/PR/208_en.pdf. [Google Scholar]
- 27.Grimm C, Polterauer S, Natter C, et al. Treatment of cervical intraepithelial neoplasia with topical imiquimod: a randomized controlled trial. Obstet Gynecol. 2012;120:152–59. doi: 10.1097/AOG.0b013e31825bc6e8. [DOI] [PubMed] [Google Scholar]
- 28.Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–22. doi: 10.1056/NEJMoa1001294. the IMPACT Study Investigators. [DOI] [PubMed] [Google Scholar]
- 29.Kantoff PW, Schuetz TJ, Blumenstein BA, et al. Overall survival analysis of a phase II randomized controlled trial of a Poxviral-based PSA-targeted immunotherapy in metastatic castration-resistant prostate cancer. J Clin Oncol. 2010;28:1099–105. doi: 10.1200/JCO.2009.25.0597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Letvin NL. Progress and obstacles in the development of an AIDS vaccine. Nat Rev Immunol. 2006;6:930–39. doi: 10.1038/nri1959. [DOI] [PubMed] [Google Scholar]
- 31.Söderlund-Strand A, Kjellberg L, Dillner J. Human papillomavirus type-specific persistence and recurrence after treatment for cervical dysplasia. J Med Virol. 2014;86:634–41. doi: 10.1002/jmv.23806. [DOI] [PubMed] [Google Scholar]
- 32.Trimble CL, Peng S, Thoburn C, Kos F, Wu TC. Naturally occurring systemic immune responses to HPV antigens do not predict regression of CIN2/3. Cancer Immunol Immunother. 2010;59:799–803. doi: 10.1007/s00262-009-0806-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith C, Elhassen D, Gras S, et al. Endogenous antigen presentation impacts on T-box transcription factor expression and functional maturation of CD8+ T cells. Blood. 2012;120:3237–45. doi: 10.1182/blood-2012-03-420182. [DOI] [PubMed] [Google Scholar]
- 34.Ostör AG. Natural history of cervical intraepithelial neoplasia: a critical review. Int J Gynecol Pathol. 1993;12:186–92. [PubMed] [Google Scholar]
- 35.Ferenczy A, Choukroun D, Arseneau J. Loop electrosurgical excision procedure for squamous intraepithelial lesions of the cervix: advantages and potential pitfalls. Obstet Gynecol. 1996;87:332–37. doi: 10.1016/0029-7844(95)00453-x. [DOI] [PubMed] [Google Scholar]
- 36.Wright TC, Jr, Gagnon S, Richart RM, Ferenczy A. Treatment of cervical intraepithelial neoplasia using the loop electrosurgical excision procedure. Obstet Gynecol. 1992;79:173–78. [PubMed] [Google Scholar]
- 37.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 38.Derkay CS. Task force on recurrent respiratory papillomas. A preliminary report. Arch Otolaryngol Head Neck Surg. 1995;121:1386–91. doi: 10.1001/archotol.1995.01890120044008. [DOI] [PubMed] [Google Scholar]
- 39.Hu D, Goldie S. The economic burden of noncervical human papillomavirus disease in the United States. Am J Obstet Gynecol. 2008;198:500 e1–7. doi: 10.1016/j.ajog.2008.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Insinga RP, Ye X, Singhal PK, Carides GW. Healthcare resource use and costs associated with cervical, vaginal and vulvar cancers in a large U.S. health plan. Gynecol Oncol. 2008;111:188–96. doi: 10.1016/j.ygyno.2008.07.032. [DOI] [PubMed] [Google Scholar]
- 41.Hoy T, Singhal PK, Willey VJ, Insinga RP. Assessing incidence and economic burden of genital warts with data from a US commercially insured population. Curr Med Res Opin. 2009;25:2343–51. doi: 10.1185/03007990903136378. [DOI] [PubMed] [Google Scholar]
- 42.Stokley S, Jeyarajah J, Yankey D, et al. the Immunization Services Division, National Center for Immunization and Respiratory Diseases, CDC, and the Centers for Disease Control and Prevention (CDC) Human papillomavirus vaccination coverage among adolescents, 2007–2013, and postlicensure vaccine safety monitoring, 2006–2014—United States. MMWR Morb Mortal Wkly Rep. 2014;63:620–24. [PMC free article] [PubMed] [Google Scholar]
- 43.Bach PB. Gardasil: from bench, to bedside, to blunder. Lancet. 2010;375:963–64. doi: 10.1016/S0140-6736(09)62029-8. [DOI] [PubMed] [Google Scholar]
- 44.Whiteside MA, Siegel EM, Unger ER. Human papillomavirus and molecular considerations for cancer risk. Cancer. 2008;113(suppl):2981–94. doi: 10.1002/cncr.23750. [DOI] [PubMed] [Google Scholar]
- 45.Jemal A, Simard EP, Dorell C, et al. Annual Report to the Nation on the Status of Cancer, 1975–2009, featuring the burden and trends in human papillomavirus(HPV)-associated cancers and HPV vaccination coverage levels. J Natl Cancer Inst. 2013;105:175–201. doi: 10.1093/jnci/djs491. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.