

We show that JAK2 activation by the pseudokinase V617F mutation is mediated by a community of residues across JAK2. Charge changes on pseudokinase helix αC specifically inhibits V617F-induced activation. Thus, allosteric modulation can lead to specific JAK2 V617F inhibition.
Keywords: JAK2 V617F, JAK-STAT signalling, inhibitor, kinase, myeloproliferative neoplasm (MPN)
Abstract
The mechanisms by which JAK2 is activated by the prevalent pseudokinase (JH2) V617F mutation in blood cancers remain elusive. Via structure-guided mutagenesis and transcriptional and functional assays, we identify a community of residues from the JH2 helix αC, SH2-JH2 linker and JH1 kinase domain that mediate V617F-induced activation. This circuit is broken by altering the charge of residues along the solvent-exposed face of the JH2 αC, which is predicted to interact with the SH2-JH2 linker and JH1. Mutations that remove negative charges or add positive charges, such as E596A/R, do not alter the JH2 V617F fold, as shown by the crystal structure of JH2 V617F E596A. Instead, they prevent kinase domain activation via modulation of the C-terminal residues of the SH2-JH2 linker. These results suggest strategies for selective V617F JAK2 inhibition, with preservation of wild-type function.
INTRODUCTION
JAK-STAT signalling is essential for development, haematopoiesis and immune responses [1]. Inappropriate signalling, especially the gain-of-function mutations of Janus kinases (JAKs) and cytokine receptors, are linked to multiple human malignancies. JAK2 has a pivotal role in numerous signalling pathways [2], and the unique acquired somatic mutation JAK2 V617F is associated with >65% of human myeloproliferative neoplasms (MPN) [3]. Considerable interest has emerged toward V617F-specific JAK2 inhibitors since the discovery of the mutation in 2005 [3–6], nonetheless the mechanism of activation of the mutant at the atomic level remains poorly understood. As a consequence, there are no JAK2 inhibitors that can discriminate between the mutant and the wild-type protein. One JAK2 inhibitor has been approved for clinical use in myelofibrosis [7–9], but the effect is not curative and the allele burden of the mutated clone does not appear to decrease. Hence, there is a critical need for novel V617F-targeted therapeutic approaches.
JAK proteins (JAK1–3 and TYK2) include tandem kinase domains: a pseudokinase (JH2) and a canonical tyrosine kinase (JH1) domain. The latter is responsible for the catalytic activity of the protein whereas the function of JH2, where the V617F mutation is located, is still being defined. JH2 exerts an auto-inhibitory role in JAK regulation [10], possibly by restraining JH1 in an inactive conformation in the absence of cytokine receptor activation [11,12]. Whether this inhibition occurs in cis or trans [13,14] is still unclear, but evidence based on activating/resistance mutations [15] and the recent TYK2 JH1–JH2 crystal structure [12] would suggest a cis mechanism. The JAK2 JH2 domain has been crystallized and was found to adopt a protein kinase fold with features typical of tyrosine kinases, including an activation loop (albeit shorter than usual kinases), a G-loop and the critical helix αC [16]. The crystal structures of the individual JH1 and JH2 domains have been solved for every member of the JAK family [12,16–19] with the exception of JAK3, and in addition, a FERM-SH2 structure of TYK2 (tyrosine kinase 2) was recently determined [20]. However, in the absence of a full-length JAK protein structure, the atomic details of how all of these domains work together are still elusive. This clouds our understanding of pseudokinase-mediated auto-inhibition, and hence the basis of V617F constitutive activation. Indeed, despite significant recent insights into this mechanism [11,19], whether the V617F mutation (which is located in JH2) utilizes the same molecular pathway to activate JAK2 as cytokine-induced stimulation is still unknown.
Two JH2 regions in the vicinity of V617F are essential for its activity. First, we previously demonstrated that a stacking interaction between the aromatic rings of F617 and F595 of the JH2 αC is required for constitutive signalling of JAK2 V617F [21]. This interaction mediates a conformational change that has been visualized on the crystal structure of JH2 V617F [16]. Additionally, the JH2 αC of JAK2 was proposed in a molecular dynamics (MD) simulation [11] to be in the JH1/JH2 interface and was later visualized in this interface in the TYK2 JH1/JH2 crystal structure [12]. Attention has lately turned towards a second region, the SH2-JH2 linker, for understanding the mechanism of auto-inhibition of JAK2 [11,12,19]. This linker has been studied for its role in erythropoietin receptor (EpoR) trafficking [22], its importance in controlling JAK2 activity [23] and most recently as a proposed JH1 interaction site by MD simulations [11]. It also contains the highly conserved F537, which completes the so-called ‘F-F-V triad’ formed by residues F537, F595 and V617. F537 has been shown to be necessary for JAK2 V617F activity [19].
In the present study, we used a comprehensive structure-guided mutagenesis-based approach to identify additional key residues of the JH2 αC and the SH2-JH2 linker that are required for JAK2 V617F activity. Via these methods, we discovered specific residues that, upon charge negation or reversal, result in inhibition of V617F constitutive activity without impeding cytokine-induced activity. Our results enable us to precisely define a V617F activation circuit involving residues in the JH2 αC and SH2-JH2 linker. Confirming this hypothesis, prominent pathological activating mutations in residues that are not involved in this circuit are not sensitive to charge reversals on JH2 αC. Taken together, our results strongly suggest that wild-type JAK2 and JAK2 V617F activation networks are different. Selective targeting of these novel pathways of inhibition could become a valuable approach leading to mutant-specific inhibition.
EXPERIMENTAL
Vector construction and mutagenesis
Generation of point mutations in JAK2 was achieved by overlap-extension PCR-based mutagenesis using synthetic oligonucleotides containing the specific mutations. The reaction was performed using pfuTurbo (Agilent) following the manufacturer's protocol. All the constructs were subcloned in the bicistronic retroviral vector pMX-IRES-GFP [24], enabling a correlation of the level of expression of the gene cloned upstream of the internal ribosomal entry site (IRES) to the level of expression of the GFP. EpoR is subcloned in the pMX-IRES-CD4 [24]. All the constructs were verified by Sanger sequencing.
Cell lines
γ2A cells are human fibrosarcoma cells that are deficient for JAK2 [25]. 11.1 [26,27] and U4C [28] are TYK2 and JAK1 deficient human fibrosarcoma cell lines respectively. The three cell lines were grown in Dulbecco's modified Eagle's medium (DMEM) with FBS (10%). BaF3 are murine bone marrow-derived pro-B lymphocytes that are dependent on IL3 for survival [29]. BaF3 are maintained in Gibco® RPMI 1640 medium supplemented by 10% of FBS and IL-3.
BaF3 transduction
To obtain stable BaF3 expressing EpoR and the JAK2 WT or the JAK2 variants, high-titre helper free retroviruses were generated by transient transfection of BOSC packaging cell line [29]. Supernatant was collected 48 h after transfection and was directly used to infect BaF3 by spin infection for 3 h at 37°C. GFP for JAK2 and CD4 for EpoR were used as a marker of infections. Populations expressing similar levels of markers were sorted by fluorescence activated cell sorting (FACS).
Dual luciferase assay
STAT5 transcriptional activity of the JAK2 WT and the different mutants was measured in γ2A cells by dual luciferase assays with the reporter gene, Spi_Luc [30]. Cells were seeded overnight in a 48-well plate in DMEM+10% FBS. Cells were transiently transfected with EpoR, JAK2, STAT5, Spi_Luc and pRL-TK using Lipofectamine™ 2000 (Invitrogen) with the following ratio: 3:1:1:3:1 in Optimem. Medium was changed 4 h after transfection for DMEM containing or not Epo at 15 unit/ml (Eprex®). The luciferase activity was monitored with the dual Luciferase Reporter Assay System kit (Promega) 24 h after transfection. Emitted light was recorded on a luminescence microplate reader (PerkinElmer).
Proliferation assay
Cells were washed three times and set at a density of 250000 cells/ml in cytokine-free medium. Cells numbers were recorded over a period of six days using either a coulter cell counter or a CellTiter-Glo Luminescent Cell Viability Assay (Promega), where indicated. The later allows quantification of ATP amount within cells, which is directly correlated with the number of living cells per well. Addition of CellTilerGlo® reagents is followed by measurement of the resulting emitted luminescence at 570 nm.
Western blotting
γ2A cells were transfected with cDNAs coding the JAK2 WT or JAK2 mutants and EpoR using FuGene HD (Promega). Cells were plated in 6-well plates the day preceding the transfection in DMEM+10% FBS. Forty-eight hours after transfection, medium was changed to DMEM supplemented or not with Epo. Stimulation was performed for 20 min at 37°C followed by washing cells with PBS. Cells were trypsinized and lysed in 1× Nodinet P-40 (NP40) lysis buffer supplemented with orthovanadate, PMSF and protease inhibitor cocktail (ThermoFischer Scientific). Lysate samples were analysed by SDS/PAGE using a 4–12% gradient NuPage® NOVEX BisTris Gel (Invitrogen) and transferred to a nitrocellulose membrane. Total JAK2 was detected using Jak2 (D2E12) XP® Rabbit mAb (Cell Signaling Technology #3230) and Phopho-JAK2 with phospho-Jak2 Tyr1007/1008 (C80C3) Rabbit mAb (Cell Signaling Technology #3776). Detection was performed using a peroxidase-conjugated anti-rabbit secondary antibody (GE Healthcare) and images were acquired by Fusion FX7 (Vilber Lourmat).
Flow cytometry and phospho flow analysis
After stimulation, BaF3 cells were fixed with formaldehyde solution (1.6%, methanol-free, ThermoFisher Scientific) and permeabilized with PermBuffer III (BD Phospflow, BD Biosciences). After washing with PBS FBS 2%, cells were further stained with pSTAT5-Alexa647 antibody (PY694) (dilution 1:20, BD Biosciences) and phospho-Jak2 (Tyr1007/1008) rabbit antibody (Cell Signaling Technology, dilution 1:400), followed by an anti-rabbit donkey PE antibody (1:400). Flow Cytometry analysis was performed on a BD LSRFortessa cell analyser (BD Biosciences).
Structure determination
The E596A mutation was introduced into a baculovirus expression plasmid encoding the human JAK2 JH2 domain (amino acids 535–812 with a C-terminal thrombin-cleavable His6-tag) harbouring the V617F and three other mutations (W659A, W777A and F794H) [16], and the resulting mutant protein was expressed in Spodoptera frugiperda-9 cells as described previously. Cell pellets were resuspended in 20 mM Tris (pH 8.5), 500 mM NaCl, 0.5 mM Tris (2-carboxyethyl) phosphine (TCEP), 10% glycerol, 1 mM sodium orthovanadate and 20 mM imidazole with protease inhibitors (Roche Diagnostics), and lysed using a microfluidizer. After clarification via centrifugation, the lysate was incubated with Ni-NTA Superflow resin (Qiagen), and the bound protein was eluted in 20 mM Tris/HCl, pH 8.5, 500 mM NaCl, 0.5 mM TCEP, 10% glycerol, 1 mM sodium orthovanadate and 200 mM imidazole. Peak fractions were pooled, diluted 1:11 with 20 mM Tris/HCl, pH 8.5, 0.5 mM TCEP, 10% glycerol, and flowed through a HiTrap Q HP column. After adding ATP and MgCl2 to 1 and 3 mM respectively, the protein was concentrated to 6.8 mg/ml using Amicon Ultra 3K MWCO concentrators (Millipore).
The protein was crystallized by hanging drop vapour diffusion using a reservoir buffer consisting of 100 mM Tris (pH 8.5), 200 mM sodium acetate, 22% PEG 4000 at 4°C. 1.5 μl protein solution was mixed with 1.5 μl reservoir buffer and sealed in a chamber containing 400 μl of reservoir solution. After 1 week, a rod-shaped crystal (∼400 μm × 30 μm × 5 μm) was transferred to a cryoprotectant containing 100 mM Tris (pH 8.5), 200 mM sodium acetate, 22% PEG 4000, 10% glycerol, 1 mM ATP and 3 mM MgCl2, and flash-frozen in liquid nitrogen.
X-ray diffraction data were measured using NE-CAT beamline 24-ID-C at the Advanced Photon Source at Argonne National Laboratory and processed with XDS [31] and AIMLESS from the CCP4 suite [32]. The structure was determined by molecular replacement using PHASER [33] and a partial model of the wild-type JAK2 JH2 domain (PDB: 4FVP with residues 586–600 and 617 deleted). Refinement was performed using PHENIX [34] interspersed with iterative cycles of rebuilding using Moloc [35].
RESULTS
JAK2 V617F activity requires residue E596 of the helix αC of JH2
Previously, our group showed that the V617F mutation, which lies in the JH2 β4–β5 loop, affects the conformation of the neighbouring helix, αC. Specifically, the substituted F617 side chain interacts with those of two αC aromatic residues, F594 and F595 [21,36], causing a conformational change of this helix [16] that likely nucleates mutant protein activity. In order to determine which residues beyond F594 and F595 might be necessary for V617F activity, we conducted an alanine scan of the entire JH2 αC from R588 to S605 in JAK2 V617F. We then analysed the effect of these substitutions in the context of the mutant JAK2 V617F reconstituted in JAK2-deficient cell line (γ2A) together with the EpoR (Figure 1A). STAT5 transcriptional activity was quantified by a dual luciferase assay using Spi_Luc as the reporter. Expression of JAK2 V617F but not wild-type JAK2 resulted in STAT5 transcriptional activity in the absence of exogenous ligand (Epo), and alanine substitutions of F594 and F595 caused a decrease in this basal activity as previously described [21,36]. In addition to these two already described substitutions, E596A significantly reduced the mutant activity (Figure 1A). The levels of expression of the JAK2 variants were assessed by Western blot, allowing us to conclude that the decreased activity is not due to a reduction of the levels of expression (Figure 1A). Strikingly, E596A in the context on the mutant V617F still permitted response to cytokine, reverting the mutant to a wild-type JAK2 phenotype (Figure 1B). These results were confirmed in a functional assay using the IL-3-dependent haematopoietic cell line BaF3 (Figure 1C). Cells were retrovirally transduced with EpoR and the JAK2 variants for stable expression and sorted for comparable levels of CD4 and GFP respectively. BaF3 expressing JAK2 WT, V617F or V617F/E596A and the parental cell line were grown in the absence of cytokine. Although expression of JAK2 V617F in BaF3 led to factor independent growth of BaF3 cells, the E596A mutation abrogated autonomous proliferation (Figure 1C). We then assessed phosphorylation of STAT5 (Y694) and JAK2 (Y1007/Y1008) before and after stimulation with 50 unit/ml of Epo by phospho-flow cytometry. BaF3 cells expressing JAK2 V617F/E596A only exhibit phosphorylation of the JAK-STAT signalling pathway in response to Epo (Figure 1D).
Figure 1. Alanine scanning of the JH2 helix αC of JAK2 V617F.

(A) γ2A cells were transiently transfected with EpoR, STAT5, pRL-TK, STAT5 luciferase reporter Spi_Luc and the JAK2 variants. Transcriptional activity measurement shows that mutation of F594, F595 and E596 affects the basal activity of JAK2 V617F. Shown are means±S.E.M. of the mean for three independent experiments done in triplicate (*P<0.05, **P<0.01, ***P<0.001; Dunnett's multiple comparison test). rlu, relative light units. Levels of total JAK2 were analysed in γ2A cells by Western blot. (B) Transcriptional activity measurement in the presence of Epo revealed that substitution of E596 still allows normal Epo-induced response similar to JAK2 WT. (C) BaF3-EpoR stably expressing JAK2 WT, JAK2 V617F or JAK2 V617F/E596A and the parental cell line were washed and allowed to grow in the absence of cytokine for proliferation assay. Cell numbers were recorded over a period of 6 days with a coulter cell counter (cell numbers=average of triplicates±S.D.). Although JAK2 V617F gives a proliferative advantage (red line) to the BaF3-EpoR cells, the E596A mutation prevents autonomous growth of the oncogenic JAK2 V617F (orange line). (D) The same cell lines were subjected to phospho-flow cytometry to assess the activation of STAT5 and JAK2 in the cells. In the absence of Epo, only BaF3 transfected with JAK2 V617F exhibit phosphorylation of STAT5 and JAK2. The double mutant JAK2 V617F/E596A prevents constitutive activation of STAT5 and JAK2 while triggering good signalling in response to Epo. (E) In JAK2 V617F JH2 domain, F595, F594 and F617 (orange) form an aromatic π–π stacking leading to a change of conformation of JH2 αC responsible for the activation of the kinase domain. E596 (pink) is located on the opposite helical face with two other charged residues R588 and E592 (grey). Position of E596 has been modelled based of the structure of JH2-WT as atoms from its side chain are missing (PDB: 4FVR). (F) Crystal structure of JAK2 JH2 V617F/E596A (green) (PDB: 5I4N) overlapped with that of JAK2 V617F (blue) (PDB: 4FVR). ATP is shown in yellow spheres. The conformations of F537, F595 and F617 (stick diagrams) and the αC (orange) are essentially identical in the two structures. The Western blot shown in (A) is a representative of three independent experiments. Data presented in (C) and (D) are representative of two independent experiments.
E596 is located on the face of αC opposite to the one containing F594 and F595 (Figure 1E), and this surface is predicted to face JH1 [11,12]. The JH2 αC harbours other charged amino acids (Supplementary Figure S1); R588 and E592, located on the same helical face, but their substitution to alanine did not lead to any reduction of JAK2 V617F activity (Figure 1A). Further, all possible combinations of alanine mutants of E592, E596 and R588 were generated (Supplementary Figure S1) and only substitution of E596 to alanine reduces the activity of JAK2 V617F. Intriguingly, E596 is necessary for JAK2 V617F oncogenic activation but not for the wild-type activation process, as introduction of this mutation into the wild-type protein did not affect signalling downstream of the kinase (Supplementary Figure S2A).
To begin to determine the mechanism by which E596A affects V617F constitutive activity, we determined the 1.54 Å (1 Å=0.1 nm) crystal structure of V617F E596A JAK2 JH2 bound to Mg2+-ATP (Figure 1E and Supplementary Table S1) (PDB code: 5I4N). The overall structure of the double mutant domain is highly similar to that of the V617F JAK2 JH2 (PDB: 4FVR; RMSD 0.45 Å; Figure 1D). Remarkably, neither the conformation of F537, F595, F617 nor αC were affected by the E596A mutation. These results led us to surmise that E596A was suppressing the effects of V617F by modulating interactions with other domains of JAK2 such as JH1.
Replacement of residue 596 with a basic residue efficiently inhibits JAK2 V617F constitutive activity without impairing cytokine inducibility
To gain additional insight into the role played by E596 in V617F-mediated activation, we probed the chemical basis of the effects of E596 by mutating it to amino acids with different physical properties. We tested the effect of the E596X mutations in the context of the V617F mutant using transcriptional assays as above (Figure 2A). Unexpectedly, neither replacement of E596 with polar (Q) or large aliphatic (W, L) amino acids significantly decreased constitutive activity, suggesting that the E596A mutation is not disrupting a salt bridge. Of the mutants tested, besides alanine, only basic residue substitutions (E596K and E596R) efficiently inhibited JAK2 V617F without affecting Epo-inducible signalling (Figure 2B). Substitution with arginine was the most potent inhibitory mutation that restored the wild-type phenotype (Figures 2A and 2B). In contrast, the conservative E596D mutation substantially increased the constitutive activity of JAK2 V617F (85% increase) (Figure 2A). Of interest, this substitution is not activating on its own (Supplementary Figures S2B and S2C). As replacement of the glutamate by a shorter negatively charged residue, aspartate, strongly increased V617F activation, this caused us to hypothesize that a negative charge at position 596 is required to support JAK2 V617F-mediated activation. We then confirmed the behaviour of our mutants in a functional assay. BaF3-EpoR cells were transduced with the set of JAK2 V617F double mutants and sorted for similar levels of GFP. Cells were starved and allowed to grow in cytokine-free medium for 6 days. Only BaF3 cells expressing JAK2 V617F and JAK2 V617F/E596D were able to proliferate, whereas cells expressing V617F/E596A and V617F/E596R were not (Figure 2C). Next, we determined whether the side chain of the E596R mutant inhibits V617F activity by interacting with the nearby carboxylate of E592. This is not the case since the E596R substitution was also able to inhibit JAK2 V617F/E592A (Supplementary Figure S3). In conclusion, we found that besides the substitution of E596 to alanine, positive residues such as arginine or lysine could be placed at position 596 to specifically inhibit the activity of JAK2 V617F without altering the Epo-inducible activity.
Figure 2. Investigation of the nature of the interaction mediated by E596 in V617F activation.

(A) STAT5 transcriptional activity was measured in γ2A cells by luciferase assay. Mutation of E596 to either arginine or lysine induces a decrease in constitutive activity of JAK2 V617F up to 71% and 60% respectively whereas substitution to aspartate increases the activity of the mutant protein up to 86%. The activity of the wild-type JAK2 amounted to 25% of the activity of JAK2 V617F. Shown are means for three independent experiments each performed in triplicate and data were normalized by taking 100% as the basal activity of JAK2 V617F. (B) Substitution of E596 by protonated residues in JAK2 V617F displays an Epo-induced response similar to JAK2 WT. These two mutants behave like the wild-type JAK2. Shown are means±S.E.M. for three independent experiments done in triplicate. n.s, not significant. (C) BaF3-EpoR stably expressing JAK2 double mutants were starved for 4 h and allow to grow in a cytokine-free media. The number of living cells was evaluated by ATP measurement during 6 days. Only JAK2 V617F and JAK2 V617F/E596D transform the BaF3 into factor-independent cells. Although E596/K and A mutations prevent autonomous grow of JAK2 V617F.
Homologous E596R substitutions in JAK1 (E637R) and TYK2 (E657R) decrease constitutive signalling of mutant proteins
Other JAK family members are activated by mutations analogous to V617F, namely V658F in JAK1 and V678F in TYK2 [37]. In addition, the glutamate at position 596 is conserved in all the JAKs (Figure 3A). Substitutions similar to E596R in JAK2 were introduced into JAK1 and TYK2 in order to demonstrate the relevance of this inhibitory mechanism to these proteins. For the JAK1 V658F mutant, we used JAK1-deficient U4C cells [28], where the STAT5 reporter Spi_Luc was co-transfected along with the JAK1 variants and the chimeric receptor EpoR/gp130 (consisting of the EpoR extracellular domain and gp130 transmembrane and intracellular domain [38]). In U4C cells, co-transfection of EpoR/gp130 with JAK1 restores Epo-mediated activation of STAT5 (Figure 3B) whereas JAK1 V658F drives constitutive STAT5 signalling. Interestingly, JAK1 E637R mutation decreases the constitutive signalling of the mutant as we observed with JAK2 V617F. To test the activity of TYK2 V678F double mutant, we used TYK2-deficient 11.1 cells [26,27]. The STAT3 reporter, pGL3bPpr2_Luc [39] was used to assess the activity of the TYK2 variants co-transfected with the thrombopoietin receptor (TpoR) in 11.1 cells[37] (Figure 3C). Although TYK2 V678F leads to STAT3 signalling in the absence of cytokine, the double mutant V678F/E657R does not, consistent with what we observed for both JAK2 and JAK1. Placement of an arginine at the position analogous to E596 in the JH2 αC can significantly lower the hyperactivity of JAK1 and TYK2 V617F-like mutants to a basal level similar to wild-type protein without compromising cytokine-mediated signalling. Hence, this inhibition approach might also be relevant for JAK1 V678F-driven adult T-cell lymphoblastic leukaemia (T-ALL) [40,41].
Figure 3. Study of the applicability of the inhibitory substitution to other JAKs.
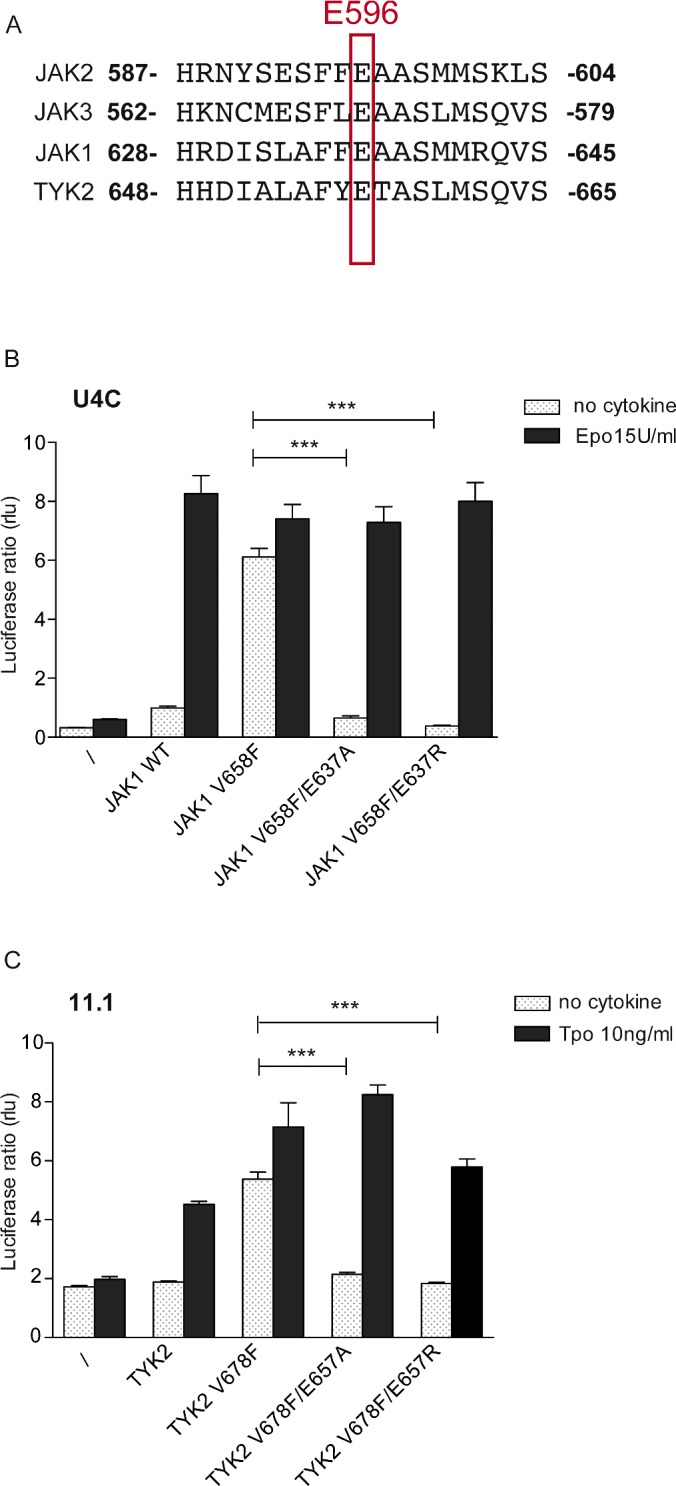
(A) Alignment of the JH2 αC sequence of JAKs. A glutamate at position 596 is conserved in other JAK family members. (B) JAK1 deficient cells, U4C, were transfected with the JAK1 variants and a chimaeric receptor EpoR/gp130, allowing stimulation by Epo and intracellular binding of two JAK1 molecules signalling in a homodimeric complex. STAT5 transcriptional activity was measured using Spi_Luc reporter. Similarly to JAK2 V617F, its homologous JAK1 V678F could be inhibited by replacement of the glutamate by either alanine or arginine, but still allow normal response to Epo. (C) TYK2 deficient cells, 11.1 were transfected with TpoR and the TYK2 variants. STAT3 transcriptional activity was monitored using pGL3bPpr2_Luc reporter. A profound decrease in constitutive activity is observed by substitution of E657 by alanine or arginine. Shown in (B) and (C) are average units+S.E.M. for three independent experiments performed in triplicate (***P<0.0001, Student's unpaired t test with unequal variance).
Interfacial placement of arginines along JH2 αC blocks V617F-mediated activation
Based on the preceding findings, we hypothesized that introducing positively charged residues in the vicinity of E596 would be similarly inhibitory. Residues 592, 596 and 600 are located on the same solvent-exposed face of αC, opposite to F595 and proposed to face JH1. After analyses of the available coordinates [11,12] and modelling to visualize where arginines would likely point toward the kinase domain, we also selected position 599. Arginine substitutions of the targeted residues were introduced into JAK2 V617F and the activity of these mutants was assessed by luciferase assays and Western blotting in JAK2 deficient cells (Figures 4A and 4B). Remarkably, arginines placed at positions 592 and 599, like the E596R mutation, could also drastically decrease the pathogenic activation of JAK2 V617F (Figure 4A), whereas the M600R substitution exhibited a more modest effect. Most importantly, all the double mutants were able to induce STAT5 transcriptional activity in response to Epo stimulation to the same extent as the wild-type JAK2 (Figure 4A). Arginine substitutions at positions 592, 599 and 600 also inhibited the hyper-phosphorylation of Y1007/1008 of JAK2 V617F in the absence of Epo (Figure 4B). Phosphorylation of the double mutants was only triggered after Epo stimulation, albeit to varying degrees. We then investigated the effect of arginine substitutions on the activity of the mutant JAK2 V617F in stably expressing BaF3-EpoR and examined the ability of these mutations to reduce the autonomous growth triggered by JAK2 V617F as above (Figure 4C). All the cell lines were able to grow in the presence IL-3 or Epo. After washing, cells were grown in cytokine-free medium for 5 days and counted each day. Cytokine withdrawal led to death of cells expressing either JAK2 WT or JAK2 double mutants, whereas JAK2 V617F expressing cells survived and proliferated (Figure 4C). In order to verify that the double mutants expressing BaF3 cells respond to Epo simulation, we measured the levels of phosphorylation of STAT5 (Y694) and JAK2 (Y1007/Y1008) before and after stimulation with 50 unit/ml of Epo by flow cytometry (Figure 4D). Although BaF3 JAK2 V617F exhibited phosphorylation of STAT5 and JAK2 in the absence of cytokine, BaF3 JAK2 WT and the E592R, E596R, S599R and M600R JAK2 double mutants did not. In contrast, these cells only displayed STAT5 and JAK2 phosphorylation after Epo stimulation.
Figure 4. Modulation of JAK2 V617F activity via arginine placement on the JH2 αC.

(A) Luciferase assay performed in JAK2 deficient cells, γ2A, reveals that arginines placed at either position 592, 596, 599 can achieve significant decrease in the activity of the mutant, whereas a modest decrease is observed with M600R. (B) Y2A cells were transiently transfected with EpoR and the JAK2 variants. The whole cell lysate was analysed by Western blot. Analysis of the phosphorylation at Y1007/1008 reveals that arginines placed at different interfacial location suppress V617F-mediated hyperactivation, whereas it allows phosphorylation of JAK2 activation loop after stimulation with Epo. (C) BaF3-EpoR cells stably expressing the JAK2 V617F with additional arginine mutation were no longer able to grow in the absence of cytokine. (D) Activation of JAK2 and STAT5 was monitored in the BaF3 cell lines by phospho-flow cytometry. JAK2 V617F cells exhibit activation of STAT5 and JAK2 in the absence of cytokine whereas this effect is suppressed by the arginine placement at positions 592, 596, 599 and 600. Cells expressing the JAK2 V617F inhibited forms respond nicely to Epo stimulation. Data presented in (A), (B) and (C) are representative of three independent experiments. Data presented in (D) are representative of two independent experiments.
JAK2 V617F activity is abolished through modulation of the SH2-JH2 linker C-terminus
The flexible linker between the SH2-like and JH2 domains is involved in the regulation of JAK2 activity [22,23], and was observed to be in the vicinity of V617 and αC by MD simulation [11]. Remodelling of this linker has also been observed in the crystal structure of JAK1 JH2-V658F, where F658 induces a phenylalanine-flipped rearrangement of the linker residue F575 (F537 in JAK2) [19]. Curiously, several residues located in this linker have been associated with clinically observed gain-of-function mutations, namely exon 12 mutations [42]. Hence, we extended our study to the SH2-JH2 linker and screened for residues that are critical for the activity of JAK2 V617F. We used a set of mutants where residues from 501 to 544 in groups of three were substituted with alanine [22] and introduced the mutation V617F. We examined the basal activity of these mutants by Western blot for phosphorylation at Y1007/1008 of JAK2 in transiently transfected γ2A cells (Figure 5A). An absence of phosphorylation was observed for alanine mutants in the N-terminal part of the linker, consistent with previous data obtained in wild-type JAK2 [22]. Indeed, the linker N-terminal fragment was shown to be essential for interaction with the EpoR [22] and it is known that cytokine receptor binding is required for constitutive activation of JAK2 V617F [43,44]. In contrast, the C-terminal part of the linker appears to be specifically required for V617F activity. Mutation of residues from Q534 to L545 significantly reduced JAK2 V617F phosphorylation but not wild type [22]. Next, using structure-guided mutagenesis, we individually mutated residues in the linker C-terminal segment to determine which amino acids are involved in V617F activation process (Supplementary Figure S4A). First, we confirmed that mutation F537A strongly impaired the constitutive activity of the mutant, as has been previously demonstrated [19], without impeding Epo normal response (Figures 5B and 5C). Secondly, we found that mutations K539E and E543/D544 to KK (EDtKK) can also uncouple V617F from cytokine-induced activation, similarly to E596R (Figures 5B and 5C). It was earlier demonstrated that the negatively charged ED motif (E543/D544) was involved in the auto-inhibitory regulation of JAK2 [22] and its deletion has been associated with MPN [42]. Unexpectedly, mutation of this ED motif can either increase JAK2 WT (Supplementary Figure S4B) or decrease JAK2 V617F basal activity (Figure 5B). Since F537A and K539E preserve the Epo inducibility of JAK2 V617F (Figures 5B and 5C), the inhibition of V617F is not due to a lack of interaction of the JAK with EpoR. Here again, with K539E, we show that a charge inversion can uncouple V617F from cytokine-induced activation of JAK2.
Figure 5. JAK2 V617F-mediated hyperactivation is suppressed by modulation of the SH2-JH2 linker.

(A) The sequence of the linker between the SH2-like and JH2 is represented above. JAK2 V617F constucts containing alanine-scanning mutations in the SH2-JH2 linker (from PKP/AAA to EDL/AAA) were expressed in JAK2 deficient cells and their levels of activity were assessed by Western blot using anti-phospho JAK2 (Y1007/Y1008) antibodies. JAK2 V617F basal activity is abolished by mutation of the linker N-terminal region and C-terminal region. Mutation of the middle portion sustains phosphorylation of JAK2 V617F. C-terminal region of the linker is important to maintain hyperactivity of JAK2 V617F and contains several charge residues that have been associated with diseases (red). (B) STAT5 transcriptional activity measurements by luciferase assay in γ2A show that F537A, K539E strongly decrease V617F, whereas E543/D544KK do so to a lower extent. All the three mutants preserved a normal Epo response. Shown are average units+S.E.M. for three independent experiments performed in triplicate (**P<0.01, Student's unpaired t test with unequal variance). (C) Western blot analysis of the three inhibited forms of JAK2 V617F. JAK2 V617F/K539E and JAK2 V617F/F537A were transfected in Y2A and were assessed for activity. Both proteins exhibit a similar decrease in constitutive activity then JAK2 V617F/E596R and a marked response to Epo. Western blots shown in (A) and (C) are representative of three independent experiments.
E596R inhibits activation induced by the V617F circuit but not other activating JAK2 mutations relevant for pathology
Our data lead us to hypothesize that the change of conformation of the JH2 αC induced by the mutation V617F rearranges E596, which in turn, triggers a series of activating events via the C-terminal end of the SH2-JH2 linker. Based on inspection of the recently described MD-based model of JAK2 JH1/JH2 [11], we identified several potential networks of residues that could enable inter-domain communication via the SH2-JH2 linker. One of these proposed activation circuits would be switched-on by F537, and would contain E543/D544 (SH2-JH2 linker C-terminal end), D569/Y570 (JH2 N-terminus, β2–β3 loop) and two kinase domain residues K857 (JH1 N-terminus, β1–β2 loop, G-loop) and K883 (JH1 N-terminus, strand-β3) (Figures 6A and 6B). In the context of JAK2 V617F, E596 clashes with the C-terminus of the SH2-JH2 linker, on one hand de-stabilizing the inhibitory JH2–JH1 interface via the E543/D544 and downstream β2–β3 JH2 N-terminus (D569/Y570) and on the other hand allowing F537 (liberated from its interaction with helix αC phenylalanine residues by the V617F mutation [19]) to clash with K857 of JH1 (Supplementary Figure S5C).
Figure 6. V617F-induced activation circuit.

(A) Schematic representation the V617F activation circuit. (B) Model of the interface between JH1 (blue) and JH2 (green) from MD simulation data (11). E596 (pink) interacts with the C-terminus of the SH2-JH2 linker (grey) and initiates a cascade of activating events involving residues F537 (purple), E543, D569, Y570, K883, K857 (red). (C) Y2A cells were transiently transfected with the JAK2 variants and STAT5 transcriptional reporter. E596R mutation decreases the activity of the different hyperactive JAK2 mutants that comprise the V617F activation circuit without impairing Epo normal response. (D) Y2A cells were transiently transfected with several JAK2 oncogenic mutants and basal transcriptional activity was measured with STAT5 transcriptional reporter, Spi_Luc. K539L, T875N and R683G all induce constitutive signalling of JAK2. No significant decrease in the constitutive activation of these mutants is observed when substituting E596 with either alanine or arginine. Shown in (C) and (D) are average units+S.E.M. for three independent experiments performed in triplicate (****P<0.0001, n.s, not significant; Dunn's multiple comparison test).
Were this activation circuit model to be correct, we would make several predictions: (i) the participants in this circuit, except for the switch F537 would, when appropriately mutated, induce constitutive activation; (ii) the E596R mutation would suppress this activation like it suppresses V617F; (iii) activating mutations in JAK2 unrelated to this activating circuit would not be inhibited by the E596R mutation. Indeed, we show that mutants E543A, D569A, Y570F, K857A and K883E predicted by our modelling to be in the V617F circuit, are all constitutively active (Figure 6C). Importantly, this hyperactivity is prevented by the E596R mutation, which is remarkable, especially for the JH1 mutation K857A, which is far away from helix αC (Figures 6B and 6C).
Next, we introduced the E596R mutation in other activated forms of JAK2 previously shown to be active and exhibit STAT5 constitutive signalling [45–47], but that are placed outside of the V617F circuit. These mutations include the exon 12 polycythaemia vera (PV) mutation K539L, located in the SH2-JH2 linker of JAK2 [45], as well as the JAK2 R683S mutation, located between the JH2 N- and C-lobes which is found in B-cell acute lymphoblastic leukaemia [47]. We also examined the JAK2 T875N mutant, associated with a megakaryoblastic leukaemia line, located in the β2–β3 loop of the kinase domain of JAK2 [46] (Figure 6B). All the mutants exhibited STAT5 signalling in the absence of Epo (Figure 6D). Remarkably, E596R exclusively impaired the constitutive activity of V617F and not the activity of the other mutants. Considering the proximity of K539L to V617F (15 Å) (Figure 6B), these results strongly support the specificity of the molecular process involving E596 in the activation process of JAK2 by the V617F mutation.
Overall, our results support the notion that V617F changes the conformation of the αC of JH2 repositioning E596 and leading to changes involving F537, E543/D544 (SH2-JH2 linker C-terminus), D569/Y570 (JH2 N-terminus) and eventually JH1 kinase residues K857/K883 (JH1 N-terminus), which could activate the kinase activity of JH1 (Supplementary Figures S5A–S5C).
DISCUSSION
The prevailing model is that JH2 maintains JAK2 in its auto-inhibited state by restraining JH1, whereas the oncogenic V617F JH2 mutation releases the structural hindrance on JH1 enabling the kinase domain to adopt an active conformation. A second activating event contributes to establish the high and persistent JAK2 V617F activation [11,16,48]. This activation process seems to be first initiated by modulation of the JH2 αC, where the main conformational changes can be detected in the X-ray crystal [16]. We addressed how the molecular events induced by V617F around JH2 helix C are transmitted to the kinase domain.
Our main findings are that: (i) a glutamic acid (E596) of the JH2 αC is an essential mediator of JAK2 V617F pathogenic activity. This is also the case for JAK1 V678F and TYK2 V658F, suggesting a common activation mechanism by V → F mutations in these three proteins. We suggest that E596 participates in the activation of V617F by making a new contact that is only present in the JAK2 V617F conformation, that is likely a steric clash with SH2-JH2 linker C-terminal amino acid backbones; (ii) the V617F mutation causes residues from αC to collaborate with the SH2-JH2 linker to trigger JH1 kinase domain activity and (iii) JAK2 V617F constitutive activity can be uncoupled from cytokine-induced signalling by either placement of positive charges along the JH2 αC or by charge inversion on the SH2-JH2 linker C-terminus, which contributes to activation.
By extensive mutagenesis of the SH2-JH2 linker and of the N-lobes of JH1 and JH2, we propose a defined circuit for JAK2 V617F activation where a sequential set of activating events propagates from JH2 αC to JH1 active site (Figure 6A). We propose that the JAK2 V617F activation derives from the combination of small activating events, which is relevant for the potency of this pathologic mutant. We show that mutations in the nodes of this circuit induce constitutive activation and, importantly, in all cases, the E596R mutation prevents activation. E596R appears to both restore an inhibitory JH2–JH1 interaction and prevent the final step of JH1 activation by V617F. In support of the former, we tested the effect of the E596R mutation on several newly described JH1 activating mutations which break the JH1/JH2 inhibitory interface [11]. E596R could significantly reduce the hyperactivity of such JAK2 mutants, namely K883E and R947E (Supplementary Figure S6A). Since only R947 appears to possibly be in the vicinity of E596 (7 Å), with the K883 residue on JH1 being far from the JH2 αC (28 Å), (Supplementary Figure S6B), the inhibitory effect of E596R is likely mediated by intermediate, possibly SH2-JH2 C-terminus linker residues. Importantly, other activating JAK2 mutations (K539L, R683G, T875N), which affect residues not involved in the V617F-circuit, are not inhibited by the E596R mutation.
Overall, our data indicate that V617F activation results from both loss of the basal auto-inhibition and from a positive regulatory interaction directly mediated by JH1. Indeed, the circuit we identify only partially overlaps with residues that belong to the inhibitory interface previously identified, such as the pair pY570-K883 [11]. In addition, activation involves residue K857 via a different mechanism. We hypothesize that the conformation of K857 in JH1 is influenced by that of the SH2-JH2 linker residue F537. In our model the side chain of F537 comes in close proximity of K857 (Supplementary Figure S5C). This is possible because F537 is liberated from its interaction with F595 by the novel aromatic stacking interaction between F617 and F594/F595 in the V617F context [19]. Mutation of K857 itself leads to activation (Figure 6C). This particular residue is predicted to contribute to the stabilization of the G-loop in an inactive conformation (as is observed in the DFG-out conformation of JH1, PDB: 3UGC; Supplementary Figure S5B). Perturbation of K857 by the linker residue F537 would destabilize the G-loop and favour an active conformation (as is observed in the DFG-in conformation of JH1, PDB: 2B7A; Supplementary Figure S5B). Figure 7 outlines our model of V617F activation circuit. The change of conformation induced in the αC of JH2 by V617F will affect the position of the first oncogenic switch E596, which will clash with the SH2-JH2 linker C-term. The destabilized linker will then trigger two sets of molecular events. The first one will be initiated by a second oncogenic switch, the linker residue F537 (previously described in [19]) that likely contacts K857 in JH1. The second set of events will be initiated by E543 that is predicted to destabilize the position of the JH2 β2–β3 and the inhibitory salt bridge between pY570 and K883 [11] allowing formation of the key salt bridge between K882 (β3 lysine) and the αC glutamate E898 in JH1. Collectively, these conformational changes are suggested to favour an active conformation of JH1.
Figure 7. Schematic representation of the V617F-induced activation circuit.

Activation of JAK2 V617F is suggested to result from the combination of several activating events that are triggered by the mutation V617F and transmitted through two switch residues E596 and F537. These activating events are part of two different molecular mechanisms that, when combined, will result in the strong activity of the mutant. The first mechanism is the disruption of one of the inhibitory interactions meditated between JH2 and JH1 (pY570-K883) and the second one, is the positive regulatory interaction mediated by F537 (SH2-JH2 linker C-term) on K857 (JH1 N-term).
The αC of JH2 is a typical amphipathic helix where amino acids with hydrophilic side chains extend from one side (R588, E592, E596) whereas hydrophobic side chains of F594 and F595 extend from the opposite side. Since placement of arginines on the same helical face specifically reduced the constitutive activity of JAK2 V617F, it implies that this particular helical interface plays a specific role in mediating V617F activation. Furthermore, the negative E596 seems to play a central role in supporting the activation by V617F as its presence is required for the mutant activity. In JAK2 V617F, the F594/F595/F617 stacking participates in the stabilization of the JH2 αC. However, MD simulations showed that these contacts are only transient [16], so stabilization of this helix seems to require other contacts that might involve SH2-JH2 linker C-terminus residues or inter-molecular interactions between two pseudokinase domains as was observed in the non-crystallographic dimer of JAK1-JH2 crystal structure [19].
Our data and others [11,22] established the importance of the SH2-JH2 linker in both JAK2 WT and V617F basal activity. E596A/R mutations are predicted to re-build one of the autoinhibitory JH2/JH1 contacts and to directly suppress the positive regulatory interaction between F537 and K857. Whether the autoinhibitory JH2/JH1 interaction occurs in cis or in trans has been debated for the past years and lines of evidence supporting both models have been provided [11–14]. Our study supports the cis model in which JH2 requires close proximity with the kinase domain to exert an intrinsic auto-inhibition. Nevertheless, we cannot exclude that both inhibitory interfaces might co-exist and represent two levels of auto-inhibition in the JAK2-receptor complex.
One surprising result is that our inhibitory mutations in the JH2 αC do not affect cytokine-induced signalling, which offers intriguing possibilities from a therapeutic perspective. Efforts have been made into the development of allosteric inhibitors [49], however ATP mimicking small molecules prevail in the treatment of cancer. But to date, none of these compounds were able to achieve uncoupling of the oncogenic from the normal activities of the protein kinases such as JAK2. Our results indicate that targeting the region around residue E596 could lead to such effect, thereby avoiding the problematic side effects related to the loss of the normal function of the kinase. Molecules such as α-helix mimetics could be designed to target the JH2 αC face with E596, and introduce a positive charge in the vicinity of the linker and JH1. These compounds would destabilize the activated state of V617F, but would have negligible effects on the wild-type protein. This strategy is not only relevant for JAK2 V617F, but also for the JAK1 V658F mutant associated with adult T-ALL. Such molecules would not affect the wild-type JAK2 allowing to use these molecules at higher doses compared with what is used now in the clinic.
Acknowledgments
We thank Dr O. Silvennoinen and D. Ungureanu from the University of Tampere, Finland, for providing the pFAST-Bac-JH2 plasmid. Lidvine Genet and Céline Mouton for expert technical assistance. Nicolas Dauguet for expert cell sorting. We thank the Advanced Photon Source NE-CAT (supported by NIH P41 GM103403 and S10 RR029205) for assistance with X-ray data collection.
Abbreviations
- DMEM
Dulbecco's modified Eagle's medium
- EpoR
erythropoietin receptor
- FACS
fluorescence activated cell sorting
- IRES
internal ribosomal entry site
- JAK
Janus kinase
- JH
JAK homology
- MD
molecular dynamics
- MPN
myeloproliferative neoplasms
- SH
Src homology
- STAT
signal transducers and activators of transcription
- TCEP
Tris (2-carboxyethyl) phosphine
- TpoR
thrombopoietin receptor
- TYK2
tyrosine kinase 2
AUTHOR CONTRIBUTION
Stefan Constantinescu and Emilie Leroy conceived and designed the study, interpreted the data and wrote the paper. Emilie Leroy, Alexandra Dusa, Didier Colau and Céline Mouton performed the experiments. Andrew Shiau and Amir Motamedi determined the structure. Emilie Leroy, Stefan Constantinescu, Lily Huang and Andrew Shiau analysed the data. Xavier Cahu performed flow cytometry and phospho flow analysis.
FUNDING
This work was supported by the Ludwig Institute for Cancer Research, FRS-FNRS [grant number 3.4596.12], the Fondation Salus Sanguinis, Belgium; the Fondation contre le cancer [grant number F/2014/266]; the Actions de Recherche Concertées of the communauté Française de Belgique [grant number ARC10/15-027]; the PAI program Belgian Medical Genetics Initiative [grant number IAP-P7/43]; the FRIA PhD fellowship (to E.L.); and the National Institutes of Health [grant number L089966 (to L.J.H.)].
References
- 1.Akada H., Akada S., Hutchison R.E., Sakamoto K., Wagner K.U., Mohi G. Critical role of Jak2 in the maintenance and function of adult hematopoietic stem cells. Stem Cells. 2014;32:1878–1889. doi: 10.1002/stem.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ghoreschi K., Laurence A., O'Shea J.J. Janus kinases in immune cell signaling. Immunol. Rev. 2009;228:273–287. doi: 10.1111/j.1600-065X.2008.00754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.James C., Ugo V., Le Couedic J.P., Staerk J., Delhommeau F., Lacout C., Garçon L., Raslova H., Berger R., Bennaceur-Griscelli A., et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–1148. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 4.Baxter E.J., Scott L.M., Campbell P.J., East C., Fourouclas N., Swanton S., Vassiliou G.S., Bench A.J., Boyd E.M., Curtin N., et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–1061. doi: 10.1016/S0140-6736(05)74230-6. [DOI] [PubMed] [Google Scholar]
- 5.Kralovics R., Passamonti F., Buser A.S., Teo S.S., Tiedt R., Passweg J.R., Tichelli A., Cazzola M., Skoda R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005;352:1779–1790. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 6.Levine R.L., Wadleigh M., Cools J., Ebert B.L., Wernig G., Huntly B.J., Boggon T.J., Wlodarska I., Clark J.J., Moore S., et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387–397. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 7.Verstovsek S., Mesa R.A., Gotlib J., Levy R.S., Gupta V., DiPersio J.F., Catalano J.V., Deininger M., Miller C., Silver R.T., et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N. Engl. J. Med. 2012;366:799–807. doi: 10.1056/NEJMoa1110557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harrison C., Kiladjian J.J., Al-Ali H.K., Gisslinger H., Waltzman R., Stalbovskaya V., McQuitty M., Hunter D.S., Levy R., Knoops L., et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N. Engl. J. Med. 2012;366:787–798. doi: 10.1056/NEJMoa1110556. [DOI] [PubMed] [Google Scholar]
- 9.Vannucchi A.M., Kiladjian J.J., Griesshammer M., Masszi T., Durrant S., Passamonti F., Harrison C.N., Pane F., Zachee P., Mesa R., et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N. Engl. J. Med. 2015;372:426–435. doi: 10.1056/NEJMoa1409002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saharinen P., Silvennoinen O. The pseudokinase domain is required for suppression of basal activity of Jak2 and Jak3 tyrosine kinases and for cytokine-inducible activation of signal transduction. J. Biol. Chem. 2002;277:47954–47963. doi: 10.1074/jbc.M205156200. [DOI] [PubMed] [Google Scholar]
- 11.Shan Y., Gnanasambandan K., Ungureanu D., Kim E.T., Hammaren H., Yamashita K., Silvennoinen O., Shaw D.E., Hubbard S.R. Molecular basis for pseudokinase-dependent autoinhibition of JAK2 tyrosine kinase. Nat. Struct. Mol. Biol. 2014;21:579–584. doi: 10.1038/nsmb.2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lupardus P.J., Ultsch M., Wallweber H., Bir Kohli P., Johnson A.R., Eigenbrot C. Structure of the pseudokinase-kinase domains from protein kinase TYK2 reveals a mechanism for Janus kinase (JAK) autoinhibition. Proc. Natl. Acad. Sci. U.S.A. 2014;111:8025–8030. doi: 10.1073/pnas.1401180111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brooks A.J., Dai W., O'Mara M.L., Abankwa D., Chhabra Y., Pelekanos R.A., Gardon O., Tunny K.A., Blucher K.M., Morton C.J., et al. Mechanism of activation of protein kinase JAK2 by the growth hormone receptor. Science. 2014;344:1249783. doi: 10.1126/science.1249783. [DOI] [PubMed] [Google Scholar]
- 14.Varghese Leila N., Ungureanu D., Liau Nicholas P.D., Young Samuel N., Laktyushin A., Hammaren H., Lucet I.S., Nicola N.A., Silvennoinen O., Babon J.J., Murphy J.M. Mechanistic insights into activation and SOCS3-mediated inhibition of myeloproliferative neoplasm-associated JAK2 mutants from biochemical and structural analyses. Biochem. J. 2014;458:395–405. doi: 10.1042/BJ20131516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Springuel L., Hornakova T., Losdyck E., Lambert F., Leroy E., Constantinescu S.N., Flex E., Tartaglia M., Knoops L., Renauld J.C. Cooperating JAK1 and JAK3 mutants increase resistance to JAK inhibitors. Blood. 2014;124:3924–3931. doi: 10.1182/blood-2014-05-576652. [DOI] [PubMed] [Google Scholar]
- 16.Bandaranayake R.M., Ungureanu D., Shan Y., Shaw D.E., Silvennoinen O., Hubbard S.R. Crystal structures of the JAK2 pseudokinase domain and the pathogenic mutant V617F. Nat. Struct. Mol. Biol. 2012;19:754–759. doi: 10.1038/nsmb.2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lucet I.S., Fantino E., Styles M., Bamert R., Patel O., Broughton S.E., Walter M., Burns C.J., Treutlein H., Wilks A.F., Rossjohn J. The structural basis of Janus kinase 2 inhibition by a potent and specific pan-Janus kinase inhibitor. Blood. 2006;107:176–183. doi: 10.1182/blood-2005-06-2413. [DOI] [PubMed] [Google Scholar]
- 18.Kulagowski J.J., Blair W., Bull R.J., Chang C., Deshmukh G., Dyke H.J., Eigenbrot C., Ghilardi N., Gibbons P., Harrison T.K., et al. Identification of imidazo-pyrrolopyridines as novel and potent JAK1 inhibitors. J. Med. Chem. 2012;55:5901–5921. doi: 10.1021/jm300438j. [DOI] [PubMed] [Google Scholar]
- 19.Toms A.V., Deshpande A., McNally R., Jeong Y., Rogers J.M., Kim C.U., Gruner S.M., Ficarro S.B., Marto J.A., Sattler M., et al. Structure of a pseudokinase-domain switch that controls oncogenic activation of Jak kinases. Nat. Struct. Mol. Biol. 2013;20:1221–1223. doi: 10.1038/nsmb.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wallweber H.J., Tam C., Franke Y., Starovasnik M.A., Lupardus P.J. Structural basis of recognition of interferon-alpha receptor by tyrosine kinase 2. Nat. Struct. Mol. Biol. 2014;21:443–448. doi: 10.1038/nsmb.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dusa A., Mouton C., Pecquet C., Herman M. Constantinescu SN. JAK2 V617F constitutive activation requires JH2 residue F595: a pseudokinase domain target for specific inhibitors. PLoS One. 2010;5:e11157. doi: 10.1371/journal.pone.0011157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao L., Dong H., Zhang C.C., Kinch L., Osawa M., Iacovino M., Grishin N.V., Kyba M., Huang L.J. A JAK2 interdomain linker relays Epo receptor engagement signals to kinase activation. J. Biol. Chem. 2009;284:26988–26998. doi: 10.1074/jbc.M109.011387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sanz Sanz A., Niranjan Y., Hammaren H., Ungureanu D., Ruijtenbeek R., Touw I.P., Silvennoinen O., Hilhorst R. The JH2 domain and SH2-JH2 linker regulate JAK2 activity: a detailed kinetic analysis of wild type and V617F mutant kinase domains. Biochim. Biophys. Acta. 2014;1844:1835–1841. doi: 10.1016/j.bbapap.2014.07.003. [DOI] [PubMed] [Google Scholar]
- 24.Liu X., Constantinescu S.N., Sun Y., Bogan J.S., Hirsch D., Weinberg R.A., Lodish H.F. Generation of mammalian cells stably expressing multiple genes at predetermined levels. Anal. Biochem. 2000;280:20–28. doi: 10.1006/abio.2000.4478. [DOI] [PubMed] [Google Scholar]
- 25.Kohlhuber F., Rogers N.C., Watling D., Feng J., Guschin D., Briscoe J., Witthuhn B.A., Kotenko S.V., Pestka S., Stark G.R., et al. A JAK1/JAK2 chimera can sustain alpha and gamma interferon responses. Mol. Cell. Biol. 1997;17:695–706. doi: 10.1128/MCB.17.2.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pellegrini S., John J., Shearer M., Kerr I.M., Stark G.R. Use of a selectable marker regulated by alpha interferon to obtain mutations in the signaling pathway. Mol. Cell. Biol. 1989;9:4605–4612. doi: 10.1128/MCB.9.11.4605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Velazquez L., Fellous M., Stark G.R., Pellegrini S. A protein tyrosine kinase in the interferon alpha/beta signaling pathway. Cell. 1992;70:313–322. doi: 10.1016/0092-8674(92)90105-L. [DOI] [PubMed] [Google Scholar]
- 28.Muller M., Briscoe J., Laxton C., Guschin D., Ziemiecki A., Silvennoinen O., Harpur A.G., Barbieri G., Witthuhn B.A., Schindler C., et al. The protein tyrosine kinase JAK1 complements defects in interferon-alpha/beta and -gamma signal transduction. Nature. 1993;366:129–135. doi: 10.1038/366129a0. [DOI] [PubMed] [Google Scholar]
- 29.Seubert N., Royer Y., Staerk J., Kubatzky K.F., Moucadel V., Krishnakumar S., Smith S.O., Constantinescu S.N. Active and inactive orientations of the transmembrane and cytosolic domains of the erythropoietin receptor dimer. Mol. Cell. 2003;12:1239–1250. doi: 10.1016/S1097-2765(03)00389-7. [DOI] [PubMed] [Google Scholar]
- 30.Sliva D., Wood T.J., Schindler C., Lobie P.E., Norstedt G. Growth hormone specifically regulates serine protease inhibitor gene transcription via gamma-activated sequence-like DNA elements. J. Biol. Chem. 1994;269:26208–26214. [PubMed] [Google Scholar]
- 31.Kabsch W. Xds. Acta Crystallogr. D Biol. Crystallogr. 2010;66(Pt 2):125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G., McCoy A., et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011;67(Pt 4):235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40(Pt 4):658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W., et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66(Pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Müller K., Amman H.J., Doran D.M., Gerber P.R., Gubernator K., Schrepfer G. MOLOC: a molecular modeling program. Bull. Soc. Chim. Belg. 1988;97:655–667. [Google Scholar]
- 36.Gnanasambandan K., Magis A., Sayeski P.P. The constitutive activation of Jak2-V617F is mediated by a pi stacking mechanism involving phenylalanines 595 and 617. Biochemistry. 2010;49:9972–9984. doi: 10.1021/bi1014858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Staerk J., Kallin A., Demoulin J-B., Vainchenker W., Constantinescu S.N. JAK1 and Tyk2 activation by the homologous Polycythemia Vera JAK2 V617F mutation: cross-talk with IGF1 receptor. J. Biol. Chem. 2005;280:41893–41899. doi: 10.1074/jbc.C500358200. [DOI] [PubMed] [Google Scholar]
- 38.Hemmann U., Gerhartz C., Heesel B., Sasse J., Kurapkat G., Grotzinger J., Wollmer A., Zhong Z., Darnell J.E., Jr, Graeve L., et al. Differential activation of acute phase response factor/Stat3 and Stat1 via the cytoplasmic domain of the interleukin 6 signal transducer gp130. II. Src homology SH2 domains define the specificity of stat factor activation. J. Biol. Chem. 1996;271:12999–13007. doi: 10.1074/jbc.271.22.12999. [DOI] [PubMed] [Google Scholar]
- 39.Eyckerman S., Waelput W., Verhee A., Broekaert D., Vandekerckhove J., Tavernier J. Analysis of Tyr to Phe and fa/fa leptin receptor mutations in the PC12 cell line. Eur. Cytokine Netw. 1999;10:549–556. [PubMed] [Google Scholar]
- 40.Jeong E.G., Kim M.S., Nam H.K., Min C.K., Lee S., Chung Y.J., Yoo N.J., Lee S.H. Somatic mutations of JAK1 and JAK3 in acute leukemias and solid cancers. Clin Cancer Res. 2008;14:3716–3721. doi: 10.1158/1078-0432.CCR-07-4839. [DOI] [PubMed] [Google Scholar]
- 41.Flex E., Petrangeli V., Stella L., Chiaretti S., Hornakova T., Knoops L., Ariola C., Fodale V., Clappier E., Paoloni F., et al. Somatically acquired JAK1 mutations in adult acute lymphoblastic leukemia. J. Exp. Med. 2008;205:751–758. doi: 10.1084/jem.20072182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma W., Kantarjian H., Zhang X., Yeh C.H., Zhang Z.J., Verstovsek S, Albitar M. Mutation profile of JAK2 transcripts in patients with chronic myeloproliferative neoplasias. J. Mol. Diagn. 2009;11:49–53. doi: 10.2353/jmoldx.2009.080114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu X., Levine R., Tong W., Wernig G., Pikman Y., Zarnegar S., Gilliland D.G., Lodish H. Expression of a homodimeric type I cytokine receptor is required for JAK2V617F-mediated transformation. Proc. Natl. Acad. Sci. U.S.A. 2005;102:18962–18967. doi: 10.1073/pnas.0509714102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu X., Huang L.J., Lodish H.F. Dimerization by a cytokine receptor is necessary for constitutive activation of JAK2V617F. J. Biol. Chem. 2008;283:5258–5266. doi: 10.1074/jbc.M707125200. [DOI] [PubMed] [Google Scholar]
- 45.Scott L.M., Tong W., Levine R.L., Scott M.A., Beer P.A., Stratton M.R., Futreal P.A., Erber W.N., McMullin M.F., Harrison C.N., et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N. Engl. J. Med. 2007;356:459–468. doi: 10.1056/NEJMoa065202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mercher T., Wernig G., Moore S.A., Levine R.L., Gu T.L., Frohling S., Cullen D., Polakiewicz R.D., Bernard O.A., Boggon T.J., et al. JAK2T875N is a novel activating mutation that results in myeloproliferative disease with features of megakaryoblastic leukemia in a murine bone marrow transplantation model. Blood. 2006;108:2770–2779. doi: 10.1182/blood-2006-04-014712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mullighan C.G., Zhang J., Harvey R.C., Collins-Underwood J.R., Schulman B.A., Phillips L.A., Tasian S.K., Loh M.L., Su X., Liu W., et al. JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. U.S.A. 2009;106:9414–9418. doi: 10.1073/pnas.0811761106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Constantinescu S.N., Leroy E., Gryshkova V., Pecquet C., Dusa A. Activating Janus kinase pseudokinase domain mutations in myeloproliferative and other blood cancers. Biochem. Soc. Trans. 2013;41:1048–1054. doi: 10.1042/BST20130084. [DOI] [PubMed] [Google Scholar]
- 49.Cowan-Jacob S.W., Jahnke W., Knapp S. Novel approaches for targeting kinases: allosteric inhibition, allosteric activation and pseudokinases. Future Med. Chem. 2014;6:541–561. doi: 10.4155/fmc.13.216. [DOI] [PubMed] [Google Scholar]