

Abstract
Individuals with long-standing spinal cord injury (SCI) often present with extreme muscle atrophy and impaired glucose metabolism at both the skeletal muscle and whole body level. Persistent inflammation and increased levels of proinflammatory cytokines in the skeletal muscle are potential contributors to dysregulation of glucose metabolism and atrophy; however, to date no study has assessed the effects of long-standing SCI on their expression or intracellular signaling in the paralyzed muscle. In the present study, we assessed the expression of genes (TNFαR, TNFα, IL-6R, IL-6, TWEAK, TWEAK R, atrogin-1, and MuRF1) and abundance of intracellular signaling proteins (TWEAK, TWEAK R, NF-κB, and p-p65/p-50/105) that are known to mediate inflammation and atrophy in skeletal muscle. In addition, based on the effects of muscle inflammation on promotion of skeletal muscle fibrosis, we assessed the degree of fibrosis between myofibers and fascicles in both groups. For further insight into the distribution and variability of muscle fiber size, we also analyzed the frequency distribution of SCI fiber size. Resting vastus lateralis (VL) muscle biopsy samples were taken from 11 men with long-standing SCI (≈22 yr) and compared with VL samples from 11 able-bodied men of similar age. Our results demonstrated that chronic SCI muscle has heightened TNFαR and TWEAK R gene expression and NF-κB signaling (higher TWEAK R and phospho-NF-κB p65) and fibrosis, along with substantial myofiber size heterogeneity, compared with able-bodied individuals. Our data suggest that the TWEAK/TWEAK R/NF-κB signaling pathway may be an important mediator of chronic inflammation and fibrotic adaptation in SCI muscle.
Keywords: TNF-like weak inducer of apoptosis, nuclear factor κ-light-chain-enhancer of activated B cells
chronic spinal cord injury (SCI) is associated with severe skeletal muscle atrophy as well as many atrophy and physical inactivity-related comorbidity factors such as diabetes, obesity, and cardiometabolic diseases (4–6, 19, 45). Our previous findings and findings from other SCI studies have shown repeatedly that individuals with long-standing SCI have impaired glucose metabolism at the whole body (4–6) and skeletal muscle level (44). Persistent inflammation and increased levels of proinflammatory cytokines in the skeletal muscle have been emerging as potential contributors to muscle insulin resistance (13, 14), impaired oxidative metabolism (35, 36), increased fibrosis (39), and impaired regenerative capacity (27, 32). Given that paralyzed muscle presents with extreme atrophy (11), impaired muscle metabolism (16, 26), and impaired regenerative capacity (1) shortly after SCI, it is important to consider the impact of skeletal muscle inflammation on these reported muscle pathologies.
Skeletal muscle inflammation involves several candidate cytokines and molecular pathways that appear to be shared across multiple chronic disease conditions. Tumor necrosis factor (TNF)-α and interleukin (IL)-6 are two major regulators of chronic inflammation in the muscle (38). TNFα exerts its action on cells (via TNF receptor binding) primarily by upregulating transcriptional activity under the control of the transcription factor nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB), whereas activation of the IL-6 receptor by IL-6 binding initiates a JAK/STAT signaling cascade that upregulates the transcriptionally regulated program governed by STAT3. Both of these transcription factors upregulate many genes that encode proinflammatory cytokines and other inflammatory mediators (8, 17, 24, 27).
TNF-like weak inducer of apoptosis (TWEAK), a member of the TNF superfamily, has also been identified as an important mediator of inflammation. Chronic elevation of TWEAK signaling in muscle promotes atrophy, metabolic dysfunction, and fibrogenesis (9, 12, 28, 29, 35, 36). The TWEAK-TWEAK receptor (R) system mediates the activation of the NF-κB signaling pathway, which leads to the increased expression of muscle RING finger 1 (MuRF1) and muscle atrophy F-box (atrogin-1), degradation of muscle proteins, including MyHC (39), and repression of peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) levels in skeletal muscle (35, 36). This leads to muscle atrophy (in catabolic conditions, disuse, unloading, and aging), reduced expression of several mitochondrial genes, reduced proportion of type I fibers, and increased proportion of type II fibers. Although TWEAK signaling has never been studied in the SCI muscle, the catabolic signaling targets MURF1 and atrogin-1 are shown to be upregulated in quadriceps muscle shortly after injury and downregulated [compared with able-bodied (AB) men] in the chronic stages of SCI (23).
The TWEAK-TWEAK R system is also an important regulator of fibrosis in skeletal muscle (39, 40). The TWEAK-TWEAK R axis promotes fibrogenic activities through its direct action on stromal cell types, thereby causing the accumulation of myofibroblasts. Moreover, the TWEAK-TWEAK R pathway regulates collagen gene expression. For instance, TWEAK R-knockout (KO) mice demonstrated significantly reduced levels of collagen I, III, and IV in the skeletal muscle. Despite the knowledge that TNFα, IL-6, and TWEAK-TWEAK R-NF-κB signaling pathways are influential in regulating skeletal muscle mass, metabolism, and fibrosis, surprisingly, no study has assessed their expression or associated intracellular signaling in the paralyzed muscles of individuals with long-standing SCI. Therefore, in the present study, we assessed skeletal muscle candidate genes and signaling proteins that are known to mediate inflammation and atrophy in resting vastus lateralis (VL) muscle biopsy samples taken from 11 men with long-standing SCI (≈22 yr), with direct comparisons to VL samples from 11 healthy, AB men of similar age. In addition, based on the effects of muscle inflammation on promotion of skeletal muscle fibrosis, we assessed the degree of fibrosis between myofibers and fascicles in both groups. For further insight into the distribution and variability of muscle fiber size, we also analyzed the frequency distribution of SCI fiber size.
METHODS
Human subjects and tissue collection.
Resting VL muscle biopsy samples from 11 men with SCI (injury levels: C5-T8; American Spinal Injury Association A and B; 22.4 ± 10 yr postinjury, 49.6 ± 10.0 yr of age) and 11 AB men (41.2 ± 10.6 yr of age) were utilized for quantitative PCR, immunoblotting, and determination of fibrosis. Subjects were excluded for any medical or other health conditions that would be expected to affect testing (e.g., frank diabetes) or for which testing might be contraindicated (e.g., dystrophinopathy). The study was approved by the University of Alabama at Birmingham (UAB) Institutional Review Board. All participants gave written informed consent after hearing a thorough explanation of study procedures and risks and after having an opportunity to ask questions. Medications were recorded, and the only medication that was used in either group was anti-spasticity medication (Baclofen) in two of the SCI individuals. All muscle biopsies were performed before noon following an overnight fast.
Muscle biopsy.
Muscle samples were collected from the VL before noon after an overnight fast. Briefly, biopsies were performed under local anesthetic (1% lidocaine) using a 5-mm Bergstrom-type biopsy needle under suction, as described previously (2, 27, 41). Approximately 50–70 mg of muscle for immunohistochemistry was mounted cross-sectionally and frozen in liquid nitrogen-cooled isopentane. The remaining tissue was snap-frozen in ∼30-mg portions for biochemical assays.
Muscle protein and RNA isolation.
Mixed-muscle protein lysate was prepared, utilizing established methods in our laboratory (2, 25). Briefly, muscle samples (30 mg) were homogenized after a 15-min preincubation in 6 μl/mg muscle of ice-cold lysis buffer with protease and phosphatase inhibitors and then centrifuged at 15,000 g for 40 min at 4°C. The supernatant was stored at −80°C until it was assayed for protein content using the bicinchoninic acid technique with BSA as a standard. Total RNA was isolated and further purified from frozen muscle samples (30 mg) using Tri-Reagent (Molecular Research Center, Cincinnati, OH) and RNeasy Mini Kits (Qiagen, Valencia, CA), respectively, following the manufacturer's instructions. RNA quantity and quality were determined using a spectrophotometer (NanoDrop ND-1000; Thermo Scientific, Rockford, IL) to measure absorbance at 260 nm and the 260:280 ratio.
Quantitative RT-PCR.
Skeletal muscle transcript levels for eight target genes known to be involved in muscle inflammation and/or protein breakdown were measured using quantitative RT-PCR (StepOne System; Applied Biosystems, Foster City, CA). cDNA was synthesized via reverse transcription using the SuperScript VILO cDNA Synthesis kit (Invitrogen, Carlsbad, CA). Specific mRNAs of interest quantified via Taqman Gene Expression Assays (Applied Biosystems) included the following: IL-6 (Hs00985639_m1), IL-6R (Hs00794121_m1), TNFA (TNFα; Hs00174128_m1), TNFRSF1B (TNF-αR Hs00153550_ m1), TNFSF12 (TWEAK; Hs00356411_m1), TNFRSF12A (TWEAK R′ Fn14; Hs0017993_m1), TRIM63 (MURF1, Hs00822397_m1), and FBXO32 (ATROGIN-1, Hs01041408_m1). GAPDH (Hs02758991_g1) expression served as internal control; its expression was not significantly different between groups. All samples were run in triplicate. Relative amounts of target mRNA (i.e., ΔCT values) were determined using the comparative threshold cycle method (37) via StepOne software version 2.2.2 (Applied Biosystems), and the results are shown as the relative fold difference (i.e., 2−ΔΔCT) compared with AB individuals.
Immunoblotting.
Based on gene expression results (see Fig. 1), immunoblotting of muscle protein lysate was performed to examine protein signaling associated with the TNF and TWEAK pathways. Thirty-five micrograms of mixed-muscle protein lysate were resolved on 4–12% SDS-PAGE gels and transferred to PVDF membranes, as we have described previously (25, 41). Equal loading was verified on Ponceau S stained membranes before any antibody (Ab) treatments. All gels contained samples from both AB and SCI subjects loaded in series. Primary Abs were purchased from Cell Signaling Technology (Danvers, MA) and used at 1:1,000 dilution in 5% goat serum (monoclonal Abs) or 2% milk + 2% BSA (polyclonal Abs) against the following: NF-κB p50/p105 (no. 3035), phosphorylated (Ser536) NF-κB p65 (no. 3031), and TNF receptor-associated factor (TRAF)6 (no. 8028). In addition, 1:500 dilution in 2% milk + 2% BSA against TWEAK R/Fn 14 (no. 4403) was utilized. Horseradish peroxidase-conjugated secondary Abs were used at 1:50,000, followed by chemiluminescent detection (SuperSignal West Femto Chemiluminescent Substrate; Thermo Scientific), in a Bio-Rad ChemiDoc imaging system, with band densitometry performed using Bio-Rad Quantity One software (version 4.5.1). Parameters for image development in the ChemiDoc were consistent across all membranes using predefined saturation criteria for the charge-coupled device camera, as described previously (2).
Fig. 1.
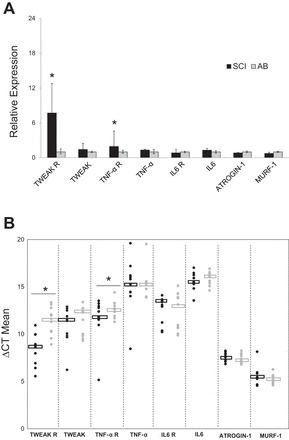
Effects of long-standing spinal cord injury (SCI) on skeletal muscle proinflammatory/atrophy gene expression. A: shown are relative fold differences (i.e., 2−ΔΔCT) in SCI compared with able-bodied (AB) vastus lateralis (VL) muscle at baseline, with error bars representing relative quantification (2−ΔΔCT) minimum and maximum; n = 11 in each group. B: ΔCT mean values in SCI vs. AB VL muscle. Dot plot represents the complete data set, and median values are indicated by central rectangles; n = 11. *P < 0.05, different from AB. TNFαR, TNFα receptor; IL-6R, IL-6 receptor; TWEAK, TNF-like weak inducer of apoptosis; TWEAK R, TWEAK receptor; MuRF1, muscle RING finger 1; ATROGIN-1, muscle atrophy F-box before MuRF1.
Determination of skeletal muscle fibrosis.
The degree of fibrosis between myofibers and fascicles was assessed as we described recently (20) using a lectin [wheat germ agglutinin (WGA) conjugated to Texas Red; Invitrogen W21405]. Texas Red WGA binds to sialic acid and N-acetylglucosaminyl residues and, therefore, reveals primarily collagen content in the extracellular matrix. Briefly, 6-μm sections were fixed for 20 min at room temperature in 3% neutral-buffered formalin, washed for 3 × 5 min in 1× PBS, incubated in Texas Red WGA (1:50 in 1× PBS) for 1 h at room temperature, washed again (3 × 5 min in 1× PBS), mounted, and stored protected from light at −20°C. Adobe Photoshop CC 2014 (Adobe Systems Incorporated, San Jose, CA) was utilized for measuring the areas positive for WGA. A stitched grid of ×10 pictures encompassing the entire sample was analyzed (total myofiber no./sample SCI = 862 ± 140, AB = 1,021 ± 70).
Determination of type-specific muscle fiber size.
Our laboratory routinely assesses myofiber type distribution and type-specific myofiber size via myosin heavy chain isoform immunohistochemistry and has published these methods previously (21, 22). The relative distributions of myofiber types I, IIa, and IIax/IIx were determined immunohistochemically and published elsewhere (44). As in the previous report, because the IIax hybrid population was very high in SCI muscle, we collapsed IIax and IIx myofibers into one group here. For cross-sectional area (CSA) measurements, at least 50 myofibers of each type were randomly selected and manually traced along their laminin-stained borders. SCI samples contained an insufficient number of type I myofibers for accurate fiber size determination. Mean fiber area was computed as the weighted average CSA (weighted by %distribution of each type). In addition, within-subjects myofiber size heterogeneity for type IIa and IIax/IIx myofibers was expressed as coefficient of variation (CV%).
Statistical analysis.
Results are expressed as means ± SE for muscle signaling and fibrosis and mean ± RQ (2−ΔΔCT) minimum and maximum for gene expression. Differences between SCI and AB individuals were compared using the two-group t-test. Relationships between variables were examined using Pearson correlation analysis. Analysis of histogram data for CSAs of type IIa and IIax/IIx myofibers, i.e., testing whether the distribution of a variable is the same across different groups (SCI and AB), was performed using the two-group Kolmogorov-Smirnov test. All statistical tests were two-tailed and were performed using a significance level of 5%. Statistical analyses were performed using SAS version 9.4.
RESULTS
Chronic SCI muscle shows heightened TNFα and TWEAK-R gene expression.
Skeletal muscle expression of target genes [IL-6, IL-6R, TNFα, TNFα receptor 1B, TWEAK, TWEAK R (Fn14), atrogin-1, and MuRF1] is shown in Fig. 1. Muscle TNFα receptor mRNA was approximately twofold and TWEAK-R eightfold higher in SCI vs. AB (P < 0.05). For the remaining targets, muscle mRNA levels were not different between groups.
Chronic SCI muscle shows heightened NF-κB signaling and fibrosis.
The total levels of TWEAK-R, NF-κB p50 and p105, TRAF6, and phosphorylated levels of NF-κB p65 and representative blots are shown in Fig. 2. TWEAK-R protein was 54% higher and NF-κB p65 phosphorylation 73% higher in SCI vs. AB muscle (P < 0.05). No other protein levels were different between groups. In addition, there was no significant correlation between TWEAK-R protein level and NF-κB p-p65 for the SCI (r = −0.25, P = 0.47; n = 11) or AB group (r = −0.16, P = 0.65; n = 11). As shown in Fig. 3, we found a significant difference in muscle fibrosis (i.e., %area positive for WGA) among groups. SCI muscle showed ∼50% more fibrosis (P < 0.05); however, there was no significant relationship between levels of fibrosis and levels of genes of interest or between levels of fibrosis and levels of proteins of interest (P > 0.15 for all correlations) for SCI or AB (P > 0.07 for all correlastions).
Fig. 2.
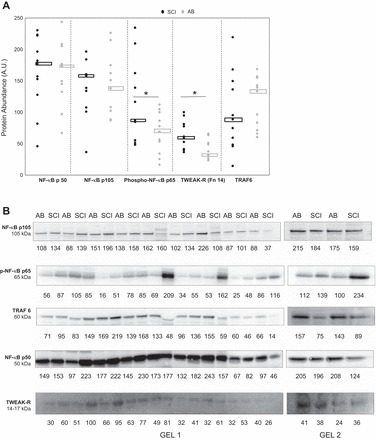
Effects of long-standing SCI on skeletal muscle TWEAK-Fn14-NF-κB signaling. A: shown are the abundance of total levels of TWEAK R, NF-κB p50 and p105, TNF receptor-associated factor 6 (TRAF6), and phosphorylated levels of NF-κB p65. Dot plot represents the complete data set, and median values are indicated by central rectangles; n = 11. B: representative immunoblots for studied proteins in VL muscle of 11 SCI vs. AB individuals. Quantification of band intensity is printed below each band. AU, arbitrary units; NF-κB, nuclear factor κ-light-chain-enhancer of activated B cells.
Fig. 3.
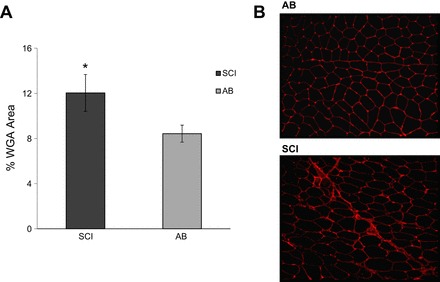
Effects of long-standing SCI on the degree of fibrosis between myofibers and fascicles. Data are means ± SE. A: percentage of wheat germ agglutinin (WGA)-positive areas in 11 SCI vs. AB individuals. B: representative immunohistochemical images of WGA stain; WGA-positive areas are in red.
Greater myofiber size heterogeneity in SCI muscle.
Surprisingly, we found no significant group differences in CSAs of type IIa or IIax/IIx myofibers (IIa: 4,955 vs. 5,960 μm2; IIax/IIx: 4,706 vs. 4,920 μm2 in SCI vs. AB, respectively). It was not possible to compare type I CSA between groups due to an insufficient number of type I myofibers in SCI muscle [see Yarar-Fisher et al. (44)]. Mean fiber area, which takes into account this small number of type I myofibers in SCI, was also not different among groups. On the other hand, greater within-subjects myofiber size heterogeneity (P < 0.05) was noted in SCI vs. AB for both type IIa (49 vs. 32 CV%) and type IIax/IIx (56 vs. 39 CV%) myofibers. Myofiber size histograms for the two groups shown in Fig. 4 represent all of the myofibers assessed for CSA within each group. Comparisons of the histograms for type IIa CSA and for type IIax/IIx CSA (SCI and AB) myofibers did not yield statistically significant results (P > 0.20 for all comparisons); however, the histograms show a leftward shift in SCI for all type IIa and type IIax/IIx myofibers <8,000 μm2, which is indicative of atrophy.
Fig. 4.
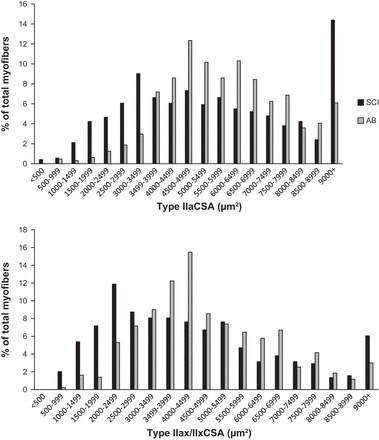
Muscle fiber size distribution for type IIa and IIax/IIx fibers in 11 SCI vs. AB individuals. CSA, cross-sectional area.
DISCUSSION
Comparing skeletal muscle proinflammatory/atrophy gene expression, signaling, and the degree of fibrosis in males with chronic SCI vs. healthy, AB men of similar age, the major findings of this study include a marked upregulation of muscle TWEAK R and TNFαR gene expression and TWEAK R protein content coupled with greater TWEAK-TWEAK R-NF-κB signaling, fibrosis, and myofiber size heterogeneity in SCI compared with AB controls. The widely disparate myofiber sizes in SCI resulted in no statistically detected mean myofiber atrophy compared with AB, but a leftward shift in the size histograms for all myofibers <8,000 μm2 suggests primarily atrophy counteracted by compensatory hypertrophy of a minority pool of myofibers.
TWEAK is a member of the TNF superfamily of cytokines and an important stimulus for the activation of NF-κB signaling in skeletal muscle (39, 40). TWEAK R, the smallest member of the TNF superfamily, has been identified as the unique TWEAK R (28, 31, 35). TWEAK R contains a TRAF binding site (9), which leads to downstream signaling and NF-κB transcriptional regulation upon stimulation by TWEAK. TWEAK R is expressed at low levels in healthy tissues, and its expression significantly increases in response to injury, aging, disuse (40), and acute resistance exercise (33). Short-term TWEAK-TWEAK R activation promotes tissue repair and regeneration following an acute injury; however, during chronic injury or disease conditions, sustained TWEAK-TWEAK R activation may drive these responses toward a pathological remodeling in the muscle in which healthy tissue is replaced by fibrotic tissue (15, 39, 40). TWEAK-TWEAK R signaling promotes fibrogenic activities via increasing accumulation of fibroblasts (31, 42) and/or increasing collagen gene expression (Col1a1 and Col1a2) (30, 31) in skeletal muscle through its activation of inflammatory signaling pathways. For example, ablation of TWEAK R decreases fibrosis and expression of collagens in skeletal muscle of mice during aging (40). Our findings of higher fibrosis (Fig. 3A) and upregulation of NF-κB signaling (via phosphorylation of NF-κB p65), in conjunction with higher TWEAK R protein in the SCI muscle (Fig. 2A), are consistent with the idea that TWEAK-TWEAK R-NF-κB signaling likely mediates fibrosis in chronically paralyzed muscle. In addition to the effects of TWEAK-TWEAK R on muscle fibrosis, TWEAK is an important regulator of skeletal muscle fiber type distribution. Previous studies have demonstrated that transgenic overexpression of TWEAK in mouse models leads to a marked reduction in the proportion of type I fibers with a concomitant increase in type II fibers in both soleus and extensor digitorum longus muscle (28, 36). In addition, TWEAK-KO mice were present with higher type I, IIa, and mitochondrial content (36). Our data support this finding. Previously, we (44) showed that individuals with SCI had a predominance of IIax/IIx myofibers and far fewer type I fibers than AB individuals, and here we report eightfold higher TWEAK R gene expression, as well as higher TWEAK R protein levels, in the SCI muscle.
In addition to TWEAK R, TNFαR gene expression was higher in SCI vs AB muscle. TNFα and TWEAK are known to exert affects via activation of both canonical and noncanonical NF-κB pathways in response to many stimuli, including aging and denervation (10, 24). Activation of NF-κB-regulated gene transcription causes muscle atrophy via increasing the components of the ubiquitin proteasome system, including the E3 ligases MuRF1 and atrogin-1 (7, 12, 24). Although higher NF-κB signaling was evident in the SCI group (Fig. 3), we did not find any differences in either MuRF1 or atrogin-1 expression among groups (Fig. 1). This may be due to a plateau in skeletal muscle atrophy in this chronic (≥20 yr) SCI group such that a heightened compensatory anabolic state may exist for off-setting elevated protein degradation (3, 23). Our previous work partly supports this hypothesis, as we found that pathways (via phosphorylation of p70 S6 kinase and ribosomal protein S6) that favor net protein synthesis/hypertrophy were hyperactivated even 22 yr after the injury (43). TWEAK also inhibits the activity of the phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathway, which further bolsters the robust catabolic action of TWEAK on skeletal muscle; however, we previously showed in the same set of subjects that SCI muscle sustains the ability to activate the PI3K/Akt pathway in response to muscle contractions (44). In fact, there is a higher basal activation of Akt in SCI vs. AB muscle. This compensatory state may have neutralized some of the effects of NF-κB on muscle atrophy and shifted the effects of upregulation more toward fibrosis.
In addition, our previous findings (44) from the same set of subjects demonstrated markedly impaired whole body insulin sensitivity (despite similar fasting glucose), impaired muscle signaling for glucose utilization, and markedly low glucose transporter 4 levels in SCI. This impairment, when combined with higher inflammation, fibrosis, and the transformation of skeletal muscles from a slow oxidative to a fast glycolytic phenotype, may play an important role in muscle insulin resistance and type 2 diabetes associated with SCI. Therefore, our studies are underway to determine the potential role of heightened muscle inflammation in metabolic disturbances among individuals with SCI. Higher TWEAK R in SCI muscle may also cause impaired oxidative metabolism, as recent animal studies (18, 36) have demonstrated that TWEAK inhibits skeletal muscle oxidative metabolism via activating the NF-κB signaling pathway, which represses PGC-1α levels in skeletal muscle. For example, TWEAK KO mice present with higher skeletal muscle mitochondrial content and oxidative phosphorylation capacity via increasing PGC-1α levels compared with wild-type mice. Although we did not measure muscle oxidative metabolism in the present study, previous work in human chronic SCI muscle has shown that oxidative metabolism is reduced to a similar extent as seen in people with mitochondrial myopathies and heart failure (26), which we now suggest may partly result from higher TWEAK R.
Surprisingly, we did not find significant group differences in fiber type-specific (IIa and IIax/IIx) myofiber size. Our findings are not consistent with previous work (11, 34) that has shown significant myofiber atrophy in the SCI vs. AB muscle. For further insight into the distribution and variability of muscle fiber size, we analyzed the frequency distribution of fiber size in both groups. Intervals of 500 μm2 were defined, and the percentage of muscle fibers in each interval was determined for the IIa and IIax/IIx myofibers separately. We were surprised to find populations of extremely large myofibers (>8,000 μm2) in the SCI group. This apparent compensatory hypertrophy in subpopulations of type IIa and IIax/IIx myofibers cannot be explained but likely negated the ability to detect marked atrophy since there was generally a leftward shift in the myofiber size histograms (in SCI vs. AB) for myofibers (<8,000 μm2). The population of enlarged myofibers in SCI warrants further investigation. In conclusion, we found that individuals with long-standing SCI have heightened muscle inflammatory signaling and fibrosis, along with substantial myofiber size heterogeneity, compared with AB individuals. Our collective data suggest that the TWEAK-TWEAK R-NF-κB signaling pathway may be an important mediator of the fibrotic adaptation in paralyzed muscle.
GRANTS
This work was supported by a Veterans Affairs Merit Award (M. M. Bamman), the UAB Center for Exercise Medicine, Department of Physical Medicine and Rehabilitation, 5T32-DK-62710, and the UAB Center for Clinical and Translational Science (UL1-TR-000165).
DISCLOSURES
None of the authors have any conflicts of interest to disclose, financial or otherwise.
AUTHOR CONTRIBUTIONS
C.Y.-F., C.S.B., and M.M.B. conception and design of research; C.Y.-F. and S.T.W. performed experiments; C.Y.-F., R.A.O., and M.M.B. analyzed data; C.Y.-F., C.S.B., N.A.K., M.J.S., A.B.M., R.A.O., and M.M.B. interpreted results of experiments; C.Y.-F. and M.M.B. prepared figures; C.Y.-F. drafted manuscript; C.Y.-F., C.S.B., and M.M.B. edited and revised manuscript; C.Y.-F., C.S.B., N.A.K., M.J.S., S.T.W., A.B.M., R.A.O., and M.M.B. approved final version of manuscript.
ACKNOWLEDGMENTS
We sincerely thank the participants for their tireless dedication and S. C. Tuggle for assistance with project coordination.
REFERENCES
- 1.Akiyama K, Noguchi J, Hirose M, Kajita S, Katayama K, Khalaj M, Tsuji T, Fairfield H, Byers C, Reinholdt L, Ogura A, Kunieda T. A mutation in the nuclear pore complex gene Tmem48 causes gametogenesis defects in skeletal fusions with sterility (sks) mice. J Biol Chem 288: 31830–31841, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bamman MM, Ragan RC, Kim JS, Cross JM, Hill VJ, Tuggle SC, Allman RM. Myogenic protein expression before and after resistance loading in 26- and 64-yr-old men and women. J Appl Physiol 97: 1329–1337, 2004. [DOI] [PubMed] [Google Scholar]
- 3.Batt J, Bain J, Goncalves J, Michalski B, Plant P, Fahnestock M, Woodgett J. Differential gene expression profiling of short and long term denervated muscle. FASEB J 20: 115–117, 2006. [DOI] [PubMed] [Google Scholar]
- 4.Bauman WA, Adkins RH, Spungen AM, Waters RL. The effect of residual neurological deficit on oral glucose tolerance in persons with chronic spinal cord injury. Spinal Cord 37: 765–771, 1999. [DOI] [PubMed] [Google Scholar]
- 5.Bauman WA, Spungen AM. Carbohydrate and lipid metabolism in chronic spinal cord injury. J Spinal Cord Med 24: 266–277, 2001. [DOI] [PubMed] [Google Scholar]
- 6.Bauman WA, Spungen AM. Disorders of carbohydrate and lipid metabolism in veterans with paraplegia or quadriplegia: a model of premature aging. Metabolism 43: 749–756, 1994. [DOI] [PubMed] [Google Scholar]
- 7.Bhatnagar S, Mittal A, Gupta SK, Kumar A. TWEAK causes myotube atrophy through coordinated activation of ubiquitin-proteasome system, autophagy, and caspases. J Cell Physiol 227: 1042–1051, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bodell PW, Kodesh E, Haddad F, Zaldivar FP, Cooper DM, Adams GR. Skeletal muscle growth in young rats is inhibited by chronic exposure to IL-6 but preserved by concurrent voluntary endurance exercise. J Appl Physiol 106: 443–53, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown SA, Richards CM, Hanscom HN, Feng SL, Winkles JA. The Fn14 cytoplasmic tail binds tumour-necrosis-factor-receptor-associated factors 1, 2, 3 and 5 and mediates nuclear factor-kappaB activation. Biochem J 371: 395–403, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cai D, Frantz JD, Tawa NE Jr, Melendez PA, Oh BC, Lidov HG, Hasselgren PO, Frontera WR, Lee J, Glass DJ, Shoelson SE. IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell 119: 285–298, 2004. [DOI] [PubMed] [Google Scholar]
- 11.Castro MJ, Apple DF Jr, Hillegass EA, Dudley GA. Influence of complete spinal cord injury on skeletal muscle cross-sectional area within the first 6 months of injury. Eur J Appl Physiol Occup Physiol 80: 373–378, 1999. [DOI] [PubMed] [Google Scholar]
- 12.Dogra C, Changotra H, Wedhas N, Qin X, Wergedal JE, Kumar A. TNF-related weak inducer of apoptosis (TWEAK) is a potent skeletal muscle-wasting cytokine. FASEB J 21: 1857–1869, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Esser N, Legrand-Poels S, Piette J, Scheen AJ, Paquot N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res Clin Pract 105: 141–50, 2014. [DOI] [PubMed] [Google Scholar]
- 14.Fink LN, Oberbach A, Costford SR, Chan KL, Sams A, Blüher M, Klip A. Expression of anti-inflammatory macrophage genes within skeletal muscle correlates with insulin sensitivity in human obesity and type 2 diabetes. Diabetologia 56: 1623–1628, 2013. [DOI] [PubMed] [Google Scholar]
- 15.Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci 69, Suppl 1: S4–S9, 2014. [DOI] [PubMed] [Google Scholar]
- 16.Gorgey AS, Dolbow DR, Dolbow JD, Khalil RK, Castillo C, Gater DR. Effects of spinal cord injury on body composition and metabolic profile - part I. J Spinal Cord Med 37: 693–702, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haddad F, Zaldivar F, Cooper DM, Adams GR. IL-6-induced skeletal muscle atrophy. J Appl Physiol 98: 911–917, 2005. [DOI] [PubMed] [Google Scholar]
- 18.Hindi SM, Mishra V, Bhatnagar S, Tajrishi MM, Ogura Y, Yan Z, Burkly LC, Zheng TS, Kumar A. Regulatory circuitry of TWEAK-Fn14 system and PGC-1alpha in skeletal muscle atrophy program. FASEB J 28: 1398–1411, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jensen MP, Molton IR, Groah SL, Campbell ML, Charlifue S, Chiodo A, Forchheimer M, Krause JS, Tate D. Secondary health conditions in individuals aging with SCI: terminology, concepts and analytic approaches. Spinal Cord 50: 373–378, 2012. [DOI] [PubMed] [Google Scholar]
- 20.Kelly NA, Ford MP, Standaert DG, Watts RL, Bickel CS, Moellering DR, Tuggle SC, Williams JY, Lieb L, Windham ST, Bamman MM. Novel, high-intensity exercise prescription improves muscle mass, mitochondrial function, and physical capacity in individuals with Parkinson's disease. J Appl Physiol 116: 582–592, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim JS, Kosek DJ, Petrella JK, Cross JM, Bamman MM. Resting and load-induced levels of myogenic gene transcripts differ between older adults with demonstrable sarcopenia and young men and women. J Appl Physiol 99: 2149–2158, 2005. [DOI] [PubMed] [Google Scholar]
- 22.Kosek DJ, Kim JS, Petrella JK, Cross JM, Bamman MM. Efficacy of 3 days/wk resistance training on myofiber hypertrophy and myogenic mechanisms in young vs. older adults. J Appl Physiol 101: 531–544, 2006. [DOI] [PubMed] [Google Scholar]
- 23.Léger B, Senese R, Al-Khodairy AW, Dériaz O, Gobelet C, Giacobino JP, Russell AP. Atrogin-1, MuRF1, and FoXO, as well as phosphorylated GSK-3beta and 4E-BP1 are reduced in skeletal muscle of chronic spinal cord-injured patients. Muscle Nerve 40: 69–78, 2009. [DOI] [PubMed] [Google Scholar]
- 24.Li H, Malhotra S, Kumar A. Nuclear factor-kappa B signaling in skeletal muscle atrophy. J Mol Med 86: 1113–1126, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mayhew DL, Hornberger TA, Lincoln HC, Bamman MM. Eukaryotic initiation factor 2B epsilon induces cap-dependent translation and skeletal muscle hypertrophy. J Physiol 589: 3023–3037, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McCully KK, Mulcahy TK, Ryan TE, Zhao Q. Skeletal muscle metabolism in individuals with spinal cord injury. J Appl Physiol 111: 143–148, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Merritt EK, Stec MJ, Thalacker-Mercer A, Windham ST, Cross JM, Shelley DP, Craig Tuggle S, Kosek DJ, Kim JS, Bamman MM. Heightened muscle inflammation susceptibility may impair regenerative capacity in aging humans. J Appl Physiol 115: 937–948, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mittal A, Bhatnagar S, Kumar A, Lach-Trifilieff E, Wauters S, Li H, Makonchuk DY, Glass DJ, Kumar A. The TWEAK-Fn14 system is a critical regulator of denervation-induced skeletal muscle atrophy in mice. J Cell Biol 188: 833–849, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mittal A, Bhatnagar S, Kumar A, Paul PK, Kuang S, Kumar A. Genetic ablation of TWEAK augments regeneration and post-injury growth of skeletal muscle in mice. Am J Pathol 177: 1732–1742, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Novoyatleva T, Diehl F, van Amerongen MJ, Patra C, Ferrazzi F, Bellazzi R, Engel FB. TWEAK is a positive regulator of cardiomyocyte proliferation. Cardiovasc Res 85: 681–690, 2010. [DOI] [PubMed] [Google Scholar]
- 31.Novoyatleva T, Schymura Y, Janssen W, Strobl F, Swiercz JM, Patra C, Posern G, Wietelmann A, Zheng TS, Schermuly RT, Engel FB. Deletion of Fn14 receptor protects from right heart fibrosis and dysfunction. Basic Res Cardiol 108: 325, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ogura Y, Mishra V, Hindi SM, Kuang S, Kumar A. Proinflammatory cytokine tumor necrosis factor (TNF)-like weak inducer of apoptosis (TWEAK) suppresses satellite cell self-renewal through inversely modulating Notch and NF-kappaB signaling pathways. J Biol Chem 288: 35159–35169, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raue U, Jemiolo B, Yang Y, Trappe S. TWEAK-Fn14 pathway activation after exercise in human skeletal muscle: insights from two exercise modes and a time course investigation. J Appl Physiol 118: 569–578, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Round JM, Barr FM, Moffat B, Jones DA. Fibre areas and histochemical fibre types in the quadriceps muscle of paraplegic subjects. J Neurol Sci 116: 207–211, 1993. [DOI] [PubMed] [Google Scholar]
- 35.Sato S, Ogura Y, Kumar A. TWEAK/Fn14 Signaling Axis Mediates Skeletal Muscle Atrophy and Metabolic Dysfunction. Front Immunol 5: 18, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sato S, Ogura Y, Mishra V, Shin J, Bhatnagar S, Hill BG, Kumar A. TWEAK promotes exercise intolerance by decreasing skeletal muscle oxidative phosphorylation capacity. Skeletal Muscle 3: 18, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3: 1101–1108, 2008. [DOI] [PubMed] [Google Scholar]
- 38.Spate U, Schulze PC. Proinflammatory cytokines and skeletal muscle. Curr Opin Clin Nutr Metab Care 7: 265–269, 2004. [DOI] [PubMed] [Google Scholar]
- 39.Tajrishi MM, Sato S, Shin J, Zheng TS, Burkly LC, Kumar A. The TWEAK-Fn14 dyad is involved in age-associated pathological changes in skeletal muscle. Biochem Biophys Res Commun 446: 1219–1224, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tajrishi MM, Zheng TS, Burkly LC, Kumar A. The TWEAK-Fn14 pathway: a potent regulator of skeletal muscle biology in health and disease. Cytokine Growth Factor Rev 25: 215–225, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thalacker-Mercer AE, Dell'Italia LJ, Cui X, Cross JM, Bamman MM. Differential genomic responses in old vs. young humans despite similar levels of modest muscle damage after resistance loading. Physiol Genomics 40: 141–149, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ucero AC, Benito-Martin A, Fuentes-Calvo I, Santamaria B, Blanco J, Lopez-Novoa JM, Ruiz-Ortega M, Egido J, Burkly LC, Martinez-Salgado C, Ortiz A. TNF-related weak inducer of apoptosis (TWEAK) promotes kidney fibrosis and Ras-dependent proliferation of cultured renal fibroblast. Biochim Biophys Acta 1832: 1744–1755, 2013. [DOI] [PubMed] [Google Scholar]
- 43.Yarar-Fisher C, Bickel CS, Kelly NA, Windham ST, McLain AB, Bamman MM. Mechanosensitivity may be enhanced in skeletal muscles of spinal cord-injured versus able-bodied men. Muscle Nerve 50: 599–601, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yarar-Fisher C, Bickel CS, Windham ST, McLain AB, Bamman MM. Skeletal muscle signaling associated with impaired glucose tolerance in spinal cord-injured men and the effects of contractile activity. J Appl Physiol 115: 756–764, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yarar-Fisher C, Chen Y, Jackson AB, Hunter GR. Body mass index underestimates adiposity in women with spinal cord injury. Obesity 21: 1223–1225, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]