

Abstract
The early inflammatory response to influenza A virus infection contributes to severe lung disease and continues to pose a serious threat to human health. The mechanisms by which inflammatory cells invade the respiratory tract remain unclear. Uncontrolled inflammation and oxidative stress cause lung damage in response to influenza A infection. We have previously shown that the fatty acid binding protein 5 (FABP5) has anti-inflammatory properties. We speculate that, as a transporter of fatty acids, FABP5 plays an important protective role against oxidative damage to lipids during infection as well. Using FABP5-/- and wild-type (WT) mice infected with influenza A virus, we showed that FABP5-/- mice had increased cell infiltration of macrophages and neutrophils compared with WT mice. FABP5-/- mice presented lower viral burden but lost as much weight as WT mice. The adaptive immune response was also increased in FABP5-/- mice as illustrated by the accumulation of T and B cells in the lung tissues and increased levels of H1N1-specific IgG antibodies. FABP5 deficiency greatly enhanced oxidative damage and lipid peroxidation following influenza A infection and presented with sustained tissue inflammation. Interestingly, FABP5 expression decreased following influenza A infection in WT lung tissues that corresponded to a decrease in the anti-inflammatory molecule PPAR-γ activity. In conclusion, our results demonstrate a previously unknown contribution of FABP5 to influenza A virus pathogenesis by controlling excessive oxidative damage and inflammation. This property could be exploited for therapeutic purposes.
Keywords: FABP5, influenza A, lipid peroxidation, inflammation
although scientific research has been able to develop highly effective vaccines and treatments for many infectious diseases, influenza viruses remain a challenge because of their resilience and adaptability. New vaccines are required every year to combat the ever changing viral genome. Despite the administration of annual vaccinations, the U.S. still faces a burden caused by influenza that results in ∼36,000 deaths and more than 200,000 hospitalizations each year (43). Massive and rapid inflammatory cell infiltration is often associated with severe complications arising from pandemic influenza viruses (25). Fatal outcome of human influenza A has been shown to be associated with high viral titers and dysregulated cytokine and chemokine production (10). The airway epithelium and alveolar macrophages represent the first line of host defense where the virus is recognized and innate immune responses are initiated (21). Infection with sublethal doses of virus results in an acute infection since wild-type (WT) mice mount a vigorous immune response that limits viral spread and promotes complete clearance from the lungs within 7–10 days. Viral infections are known to predominantly drive a CD4 Th1 immune response, which promotes the activation of macrophages and CD8 T cells as well as B cell differentiation (12).
The generation of reactive oxygen species (ROS) represents an important component of the host's arsenal to combat invading microorganisms (14, 30, 35). In addition to their potent antimicrobial activity, ROS also possess a significant cell-signaling role in biological systems, capable of regulating the phenotype and function of immune cells (24, 26, 28). However, the extreme toxicity and lack of specificity means they are also capable of eliciting significant immunopathology to surrounding tissue (39). Influenza infections can cause high levels of ROS, leading to increased inflammation and lung tissue damage. Thus keeping ROS levels in check is necessary to prevent lung damage with influenza infection.
Fatty acids have been shown to alter the oxidative environment of cellular lipids (18). Fatty acid binding proteins (FABPs) are a family of small, highly conserved, cytoplasmic proteins that bind long-chain fatty acids and other hydrophobic ligands. FABPs are thought to be involved in fatty acid uptake, transport, and metabolism (47). The FABP5 gene encodes the epidermal fatty acid binding protein and was first found to be upregulated in psoriasis tissue (41). In the lung, FABP5 is mainly expressed by bronchial epithelial cells, alveolar type II epithelial cells, alveolar macrophages, and fibroblasts (15, 16, 33). FABP5 is upregulated in alveolar macrophages during acute lung rejection (20). We have recently shown that FABP5 acts as an anti-inflammatory mediator during bacterial infections of the lung. Peroxisome proliferator-activated receptor-γ (PPAR-γ) has been shown to act as an anti-inflammatory protein in the lung. Specifically, the presence of FABP5 increases PPAR-γ activity and results in less inflammation (13). Furthermore, a covalent modification of FABP5 by 4-hydroxynonenal suggests an antioxidant role for FABP5 (4). However, FABP5 function and regulation during viral infection remain unknown. We hypothesize that FABP5 plays an important protective role against inflammation and oxidative lung injury during H1N1 influenza A infection.
In this study we sought to identify the role of FABP5 in the outcome of H1N1 influenza A virus infection and to explore the immune response elicited in FABP5-deficient mice.
MATERIALS AND METHODS
Viral infection in mice.
FABP5-/- mice and littermate controls, on a C57BL/6J background, were kindly provided by Dr. Gokhan Hotamisligil at Harvard University (Boston, MA) and bred in our Biological Resources Center. All experimental animals used in this study were covered under protocols approved by the Institutional Animal Care and Use Committee of National Jewish Health.
Mice at 8–12 wk of age were anesthetized by isoflurane and then inoculated intranasally with 50 μl of 1 × 105 focus formation units of H1N1 (PR8) influenza A. As a control, mice were inoculated with 50 μl of saline. On days 1, 3, 7, 10, and 14 postinfection, mice were euthanized to examine bronchoalveolar lavage (BAL) cell profiles, lung tissue histopathology, lung viral loads, body weight, and FABP5 expression. Four to six mice were harvested per group per time point.
BAL processing.
Mice were euthanized with pentobarbital sodium (200 mg/kg) by intraperitoneal injection and tracheotomized. The lungs were lavaged with 1 ml of phosphate-buffered saline (PBS). BAL cell cytospins were stained with the Diff-Quick Stain Kit (IMEB) for cell differential counts.
ELISA.
Virus-specific antibody levels in sera were determined by ELISA. Samples were added to virus-coated plates. Bound antibody was detected with alkaline phosphatase-conjugated goat anti-mouse IgG (Southern Biotechnology).
Real-time RT-PCR.
Real-time RT-PCR was used to measure viral titers and the mRNA expression of FABP5 gene. Briefly, lungs were homogenized in 500 μl of RLT buffer with a tissue homogenizer (IKA T25 digital ULTRA-TURRAX). Total RNA of lung tissue was extracted by use of a RNeasy Mini Kit (Qiagen). One nanogram of total RNA was used for reverse transcription by using a TaqMan RNA-to-Ct 1-Step kit with a primer made against the nuclear protein of segment 5 of the PR8 virus (PrimerDesign) or FABP5 and GAPDH TaqMan probes (Applied Biosystems). Influenza titers were quantified by using an absolute quantification that involves comparing the threshold cycle values of lung tissues to those of known influenza quantity plotted on a standard curve. Standards were amplified in parallel with samples. GAPDH was used as the housekeeping gene. The threshold cycle was recorded for each sample to reflect the mRNA expression levels. The comparative threshold cycle method was used to demonstrate the relative expression level of the gene of interest and data were normalized as described in the figure legends.
CD3 and B220 immunohistochemistry.
Immunohistochemistry was used to localize T and B cells in lung tissues of FABP5-/- and WT mice. Paraffin-embedded lung tissue sections of 5-μm thickness were deparaffinized in xylene and rehydrated through graded ethanol. Citrate buffer (pH 6.0) was used for antigen retrieval at subboiling temperature. Sections were then treated with 0.3% hydrogen peroxide in 0.05 M Tris-buffered saline (TBS; pH 7.6) for 30 min to inhibit endogenous peroxidase. After blocking with 1% normal goat serum (Vector Laboratories), the slides were incubated overnight with rabbit anti-CD3 (1:1,000 dilution) (DakoCytomation), biotin anti-B220 (1:3,000 dilution) (BioLegend), or a rabbit IgG control at 4°C, followed by incubation with biotinylated secondary antibody for the anti-CD3 antibody. Avidin-biotin-peroxidase complex (Vector Laboratories) was added to the slides for 30 min at room temperature. Thereafter, 0.03% aminoethylcarbazole solution with hydrogen peroxide was used for chromogen reaction. The number of positively stained T or B cells was then enumerated blindly in pictures taken at ×10 magnification. The total number of positively stained T or B cells in the lung was then calculated for each strain of mice.
Glutathione and 8-isoprostane assays.
Glutathione and 8-isoprostane kits (Cayman Chemical, Ann Arbor, MI) were used per manufacturer's instructions to quantify reduced glutathione (GSH) and oxidized glutathione (GSSG) as well as 8-isoprostane in lung tissue samples. Briefly, fresh lung tissues were homogenized in 2 ml of phosphate buffer, pH 7, containing 1 mM EDTA using a tissue homogenizer (IKA T25 digital ULTRA-TURRAX). The samples were then processed per manufacturer's instructions.
Lung histopathology.
Lungs were fixed in 10% phosphate-buffered formalin, dehydrated, embedded in paraffin, and cut at thickness of 4 μm. Hematoxylin and eosin (H&E)-stained lung sections were evaluated in a double-blinded fashion using light microscopy. Microphotographs were taken with a digital camera interfaced with a software system (ImagePro Plus, Media Cybernetics, Rockville, MD).
Cell culture and immunoprecipitation.
The human bronchial epithelial cell line BEAS-2B was purchased from American Type Culture Collection. BEAS-2B cells were cultured in 75-cm2 tissue culture plates coated with a mixture of 0.01 mg/ml fibronectin, 0.03 mg/ml bovine collagen type I, and 0.01 mg/ml bovine serum albumin dissolved in BEBM medium. Cells were maintained in BEBM serum-free medium (Lonza) supplemented with the BEGM kit additives (Lonza) and penicillin-streptomycin (50 U/ml) in a 5% CO2 incubator at 37°C; then 2 × 106 cells were plated in six-well plates and subjected to H3N2 infection at an multiplicity of infection of 0.1.
At the indicated times, cells were trypsinized, washed with PBS, resuspended in 0.05% Triton-X and protease and phosphatase inhibitors, and sonicated. Protein concentrations were measured by BCA assay and normalized to 100 μg protein per sample. Lysate was precleared by adding 1 μg IgG anti-rat or anti-rabbit (Jackson Laboratories) and 20 μl Protein A/G (Santa Cruz: sc-2003) for 30 min at 4°C on an orbital rocker. Beads were pelleted and cell lysate supernatant was transferred to a new 1.5-ml tube. Primary antibody (rat anti-human FABP5, R&D or rabbit anti-human PPAR-γ, Abcam) was added and incubated for 1 h at 4°C on an orbital rocker, and 20 μl of Protein A/G were added and incubated with agitation overnight at 4°C on an orbital rocker. Beads were washed five times with cold Triton-X for 1 min at 10,000 g, and 40 μl Laemmli buffer with β-mercaptoethanol was added to the beads. Samples were boiled for 5 min and 20 μl of sample were loaded per well on a polyacrylamide gel. Western blot was then carried out with the opposite antibody. Briefly, 20 μl of samples were electrophoresed on 12% SDS-PAGE, transferred onto nitrocellulose membrane, blocked with 5% milk, and then incubated with rat anti-human FABP5 (R&D) or rabbit anti-human PPAR-γ (Abcam) overnight at 4°C. After washes in PBS with 0.1% Tween-20, the membranes were incubated with anti-rat or anti-rabbit IgG conjugated to horseradish peroxidase for FABP5 and PPAR-γ protein detection.
PPAR-γ activity assay.
Cells were lysed to extract nuclear proteins following manufacturer's instructions (Active Motif). Nuclear proteins (10 μg per sample) were used to perform the PPAR-γ ELISA (Active Motif), which measures PPAR-γ activation levels as previously described (13).
Immunohistofluorescence.
We used immunohistofluorescence to compare FABP5 expression in different cell types obtained from lung tissue of control wild-type mice and infected with PR8 virus in vivo. Immunohistofluorescence was performed for lung tissue sections fixed in 4% paraformaldehyde, embedded in paraffin, and cut at 4 μm. These sections were deparaffinized and rehydrated. Antigen retrieval was carried out and consisted of boiling slides in a microwave pressure cooker (Tender Cooker; NordicWare) for 10 min in 0.01 M citrate buffer (pH 6.0). After blocking with 3% normal donkey serum (Jackson ImmunoResearch; West Grove, PA) in PBS for 1 h, tissue sections were incubated with goat anti-FABP5, rabbit anti-pro-surfactant protein-C (SP-C) (Millipore), rabbit anti-club cell (Clara cell) secretory protein (CCSP) (Millipore), rabbit anti-CD68 (Santa Cruz), or rabbit anti-aquaporin 5 (Alpha Diagnostic International) antibodies. The secondary antibodies Alexa Fluor 594 anti-goat IgG and Alexa Fluor 488 anti-rabbit IgG (both from Invitrogen, Carlsbad, CA) were applied for 1 h. Sections were mounted with Vectashield medium containing DAPI and cells were analyzed via a Zeiss Axioskop2 microscope and AxioVision software package (Carl Zeiss MicroImaging) and magnification 10 × 63. The number of specific lung cells staining positive for FABP5 was then enumerated. FABP5 is labeled with a red probe and the specific cell markers [club cell (Clara cell), alveolar type I cell, alveolar type II cell, and alveolar macrophage] were labeled with green probes. The percentage of cells expressing FABP5 was calculated by counting the number of positively double-stained cells (green and red) per total number of each cell type (green).
Statistical analysis.
Normally distributed data are presented as means ± SD. One-way ANOVA was used for multiple comparisons, and a Tukey's post hoc test was applied where appropriate. Student's t-test was used for normally distributed continuous variables. The Mann-Whitney test was used when data were not normally distributed (nonparametric distribution) A P value <0.05 was considered statistically significant.
RESULTS
FABP5-/- mice have increased BAL inflammation in response to H1N1 PR8 virus.
We analyzed the extent of cellular infiltration in the BAL of FABP5-/- and WT mice infected with H1N1 PR8 virus 0, 1, 3, 7, 10, and 14 days postinfection. At baseline, mock-infected FABP5-/- mice had twice the total cell numbers in the BAL compared with WT mice (Fig. 1A). One day following H1N1 infection, there was a fourfold increase in the cellular infiltrate in FABP5-/- mice compared with basal levels (Fig. 1A). WT mice also had a fourfold increase in leukocytes. Because FABP5-/- had increased cell numbers at baseline, the total number of cells was significantly greater in FABP5-/- mice compared with WT mice. By day 3, there was no difference in the total cell numbers between FABP5-/- and WT mice, but the cell numbers from the two groups were still significantly higher than baseline numbers (Fig. 1A). Leukocyte numbers were elevated in FABP5-/- mice compared with WT mice 7, 10, and 14 days postinfection (Fig. 1A).
Fig. 1.
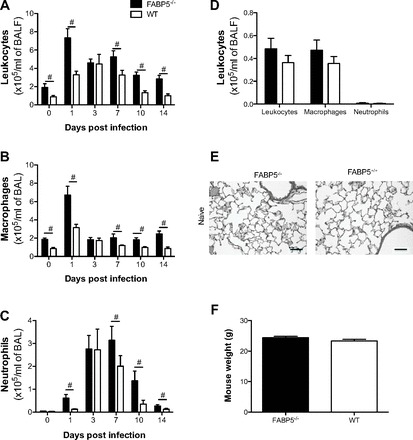
Effect of H1N1 PR8 influenza A virus infection on bronchoalveolar lavage fluid (BALF) cells in FABP5-/- and wild-type (WT) mice. Mice were treated with 1 × 105 focus formation units (FFU) of the H1N1 PR8 strain of influenza A virus or saline as a mock-infected control. Graphs represent number of total cells (A), macrophages (B), and neutrophils (C) counted in the BAL 0 (saline treated), 1, 3, 7, 10, and 14 days postinfection. Data are shown as means ± SD for 4–6 mice per group. #P < 0.05 vs. WT mice. D: total cells counted in the BALF of naive FABP5-/- and WT mice. Data are shown as means ± SD for 4 mice per group. E: representative hematoxylin and eosin-stained paraffin sections of lungs from naive FABP5-/- and WT mice. F: mouse body weight in grams at baseline. Data are shown as means ± SD for 31 mice per group.
Analysis of the infiltrating and resident populations by cell differential counting revealed that, except for day 3 postinfection, BAL from FABP5-/- mice contained significantly more macrophages (P < 0.05) than WT mice (Fig. 1B). Similarly, neutrophil numbers were significantly higher (P < 0.05) at all time points in FABP5-/- mice compared with WT mice, except for day 0 and day 3 postinfection (Fig. 1C).
Since FABP5-/- mice showed increased macrophages numbers when treated with intranasal saline compared with WT mice we had to confirm that FABP5-/- mice did not present any differences with WT mice at baseline. Thus we harvested the BAL of naive FABP5-/- and WT mice. Figure 1 shows that naive FABP5-/- and WT mice present similar leukocyte numbers (Fig. 1D) and similar lung histology (Fig. 1E). Furthermore, FABP5-/- mice used in this study presented similar body weight at baseline compared with WT mice (Fig. 1F).
Effect of FABP5 deletion on viral titers and body weight.
To determine whether increased inflammation of FABP5-/- mice correlated with increased levels of virus, we measured lung virus titers by measuring H1N1 PR8 mRNA levels by real-time PCR and determined absolute viral genome numbers. Lung virus titers were similar 1 day postinfection, but were significantly lower throughout the course of the infection in FABP5-/- mice compared with WT mice. Virus titers were undetectable by day 14 postinfection in both FABP5-/- and WT mice (Fig. 2A). We explored whether FABP5 deletion had an impact on H1N1-induced body weight loss. FABP5-/- and WT mice lost similar amounts of weight (∼20%) following infection (Fig. 2B).
Fig. 2.
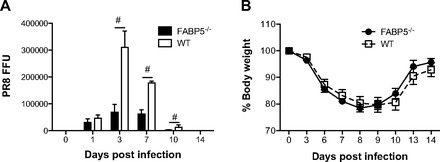
Effect of H1N1 (PR8) influenza A virus infection on viral titer and body weight. Mice were treated with 1 × 105 FFU of H1N1 PR8 influenza A virus. A: viral titers were determined at 0 (saline treated), 1, 3, 10, and 14 days postinfection. B: percent body weight loss was calculated throughout the course of the 14 days. Data are shown as means ± SD for 4–6 mice per group. #P < 0.05 vs. WT mice.
Effect of FABP5 deletion on adaptive immune cell responses.
Intranasal administration of influenza virus infects airway and alveolar epithelial cells and stays localized and confined to the respiratory tract. Infection with sublethal doses of virus results in an acute infection since WT mice mount a vigorous immune response that limits viral spread and promotes complete clearance from the lungs within 7–10 days. Viral infections are known to predominantly drive a CD4 Th1 immune response, which promotes the activation of macrophages and CD8 T cells as well as B cell differentiation (12). To determine whether T and B cells contribute to the increased viral clearance seen in FABP5-/- mice, we stained for T and B cells in lung tissue sections of FABP5-/- and WT mice infected with H1N1 for 10 days. Figure 3, A and B, indicate that T and B cells do accumulate in the lung tissues of FABP5-/- mice. We also measured H1N1-specific IgG antibodies in the serum of FABP5-/- and WT mice. As shown in Fig. 3C, FABP5-/- mice had increased H1N1-specific IgG antibodies in the serum compared with WT mice.
Fig. 3.
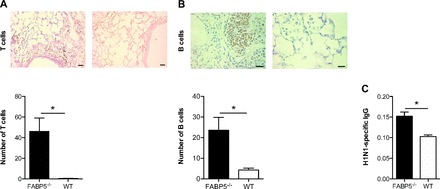
Effect of FABP5 deficiency on adaptive immune cell responses. A: anti-CD3 (T cell marker)-stained section with quantification of the number of positively stained T cells in each mouse strain. B: anti-B220 (B cell marker)-stained paraffin sections of lungs from FABP5-/- and WT mice obtained 10 days postinfection with quantification of the number of positively stained B cells in each mouse strain. Bars represent 50 and 20 μm, respectively. C: H1N1-specific IgG levels in serum of FABP5-/- and WT mice obtained 10 days postinfection. These data are representative of 4–6 mice per group.
Effect of FABP5 deficiency on influenza A-induced oxidative damage and lipid peroxidation.
Since an oxidative environment has been implicated in the pathogenesis of certain viral infections (5, 8), we measured the ratios of GSH to GSSG in the lungs of FABP5-/- and WT mice infected with H1N1 influenza virus. GSH protects the cell from oxidative damage by scavenging hydrogen peroxide. Thus a reduction in GSH/GSSG ratios suggests an increased oxidative environment. As shown in Fig. 4A, GSH/GSSG ratios were significantly reduced in H1N1-infected FABP5-/- lung tissues compared with saline-treated FABP5-/- lung tissues or H1N1-infected WT lung tissues. Ratios in FABP5-/- lungs were significantly lower than WT lungs 3, 7, 10, and 14 days postinfection. Interestingly the GSH/GSSG ratio remained low 14 days postinfection in FABP5-/- mice, whereas it came back to baseline levels in WT animals (Fig. 4A).
Fig. 4.
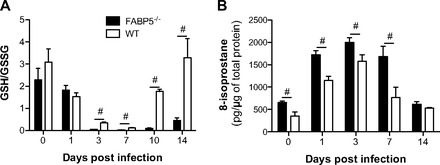
Effect of FABP5 deficiency on oxidative stress and lipid peroxidation in H1N1-infected mouse lungs. A: ratios of reduced glutathione (GSH) and oxidized glutathione (GSSG) of lung homogenates indicate a more oxidative environment in FABP5-/- mouse lung tissues following influenza A virus infection compared with WT mouse lung tissues. B: levels of 8-isoprostane are induced in FABP5-/- lung tissues following influenza A virus infection. Data are shown as means ± SD for 4–5 mice per group. #P < 0.05 vs. WT mice.
Since fatty acids have been shown to alter the oxidative environment of the cellular lipids (18), we sought to investigate whether FABP5 deficiency would alter lipid peroxidation. The isoprostanes are a family of eicosanoids of nonenzymatic origin produced by the random oxidation of tissue phospholipids by oxygen radicals. Isoprostanes appear in the plasma and urine under normal conditions and are elevated during periods of increased oxidant burden. 8-Isoprostane is a potent pulmonary vasoconstrictor (3) and has been implicated as a causative mediator in pulmonary oxygen toxicity (45). It has been proposed as a marker of antioxidant deficiency and oxidative stress, and elevated levels have been found in heavy smokers (31). Thus we quantified the levels of 8-isoprostanes in the lung tissues of H1N1-infected FABP5-/- and WT mice. Saline-treated FABP5-/- mice had more lipid oxidative damage than WT mice. H1N1 infection significantly increased 8-isoprostane levels in the lung tissues of FABP5-/- and WT mice compared with saline-treated lung tissues. 8-Isoprostane levels in FABP5-/- lung tissues were significantly higher than in WT lung tissues following H1N1 infection at all time points except 14 days postinfection (Fig. 4B).
H1N1-infected FABP5-/- mice have increased lung tissue inflammation compared with WT mice.
We examined the degree of airway inflammation caused by H1N1 in H&E-stained lung sections from FABP5-/- and WT mice euthanized 1, 3, 7, 10, and 14 days postinfection. We did not see any difference 1 day postinfection. By day 3 postinfection, some cell infiltration was evident in the lung tissues of FABP5-/- mice. At day 7 postinfection, which represents the peak of airways inflammation in WT mice as determined by our preliminary experiments, lungs from FABP5-/- mice had obvious peribronchial inflammation and prominent bronchial infiltrates consisting of mononuclear cells and neutrophils (Fig. 5, left). In contrast, histological examination of WT mice had considerably fewer bronchial and peribronchial infiltrates (Fig. 5, right). Therefore, we decided to examine the lungs at later time points.
Fig. 5.
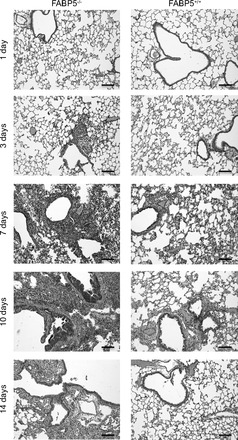
Lung histology in response to H1N1 (PR8) influenza A virus infection. Hematoxylin and eosin-stained paraffin sections of lungs from FABP5-/- and WT mice obtained 1, 3, 7, 10, and 14 days postinfection. Bars represent 100 μm. These data are representative of 4–6 mice per group.
Although WT mouse lung gradually recovered and the inflammation was resolved by days 10 and 14 postinfection, FABP5-/- mouse lung remained inflamed and damaged at 10 and 14 days postinfection (Fig. 5). Collectively, these data indicate that FABP5-/- mice undergo severe inflammation following influenza A infection that persists long after WT mice recovery.
FABP5 coimmunoprecipitates and activates PPAR-γ.
In a recent publication (13) we showed that FABP5 modulates PPAR-γ activation. PPAR-γ is a member of the nuclear hormone receptor superfamily of ligand-activated transcription factors. The PPAR-γ agonist, 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2), has been shown to reduce inflammatory cytokine and chemokine expression in the lungs of infected mice (9). Therefore, we next evaluated whether this pathway was involved in the increased and persistent inflammatory response in FABP5-/- mice. Since airway epithelial cells are the primary targets of influenza viruses, we infected the human airway epithelial cell line, Beas2B, with human influenza H3N2. Figure 6A shows that FABP5 coimmunoprecipitates with PPAR-γ in the absence of infection. However, the interaction between FABP5 and PPAR-γ is decreased following H3N2 infection. Similarly, PPAR-γ activation is decreased 24 and 48 h postinfection but increased 72 h postinfection when the interaction with FABP5 is reestablished (Fig. 6B).
Fig. 6.
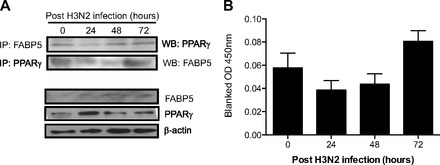
FABP5 binds to and activates the anti-inflammatory transcription factor peroxisome proliferator-activated receptor-γ (PPAR-γ) in airway epithelial cells. A: coimmunoprecipitation (IP) of FABP5 and PPAR-γ in Beas2B cells infected with influenza H3N2. Total FABP5, PPAR-γ, and β-actin proteins are shown as controls. B: PPAR-γ activity measured in the nuclear fraction of H3N2-infected Beas2B cells. Data are representative of 3 independent experiments.
Effect of H1N1 infection on FABP5 expression.
Given that FABP5-/- mice lost as much weight as WT mice, cleared the virus earlier than WT mice, but presented increased inflammation, we examined FABP5 mRNA expression in WT lungs following H1N1 infection. As indicated in Fig. 7A, levels of FABP5 mRNA were greatly reduced 1 day postinfection and remained significantly lower than baseline levels at all time points examined.
Fig. 7.
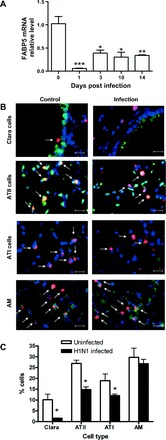
Effect of H1N1 (PR8) influenza A virus infection on FABP5 expression. A: FABP5 mRNA expression in WT mice is dramatically decreased 1 day postinfluenza A virus infection and remains low up to 14 days postinfection. Data are normalized to the relative abundance value obtained at day 0. Data are shown as means ± SD for 4–6 mice per group. *P < 0.05, **P < 0.01, ***P < 0.001. B: immunohistofluorescence of FABP5 in WT lung tissue sections in uninfected and H1N1-infected mice. DAPI stain is shown in blue, FABP5 in red, and CD68 [alveolar macrophages (AM)], club cell (Clara cell) secretory protein (CCSP) [club cells (Clara cells)], pro-surfactant protein-C (SP-C) [alveolar type II cells (ATII)] or aquaporin 5 [alveolar type I cells (ATI)] in green. Magnification bar equals 100 μm. C: quantification of FABP5 protein in specific lung cells. The percentage of cells expressing FABP5 was calculated in fixed lung tissues in WT mice without infection and after H1N1 infection. Data are representative of 4–6 mice per group.
To identify which cell type was responsible for influenza-induced decrease in FABP5 expression, we performed coimmunostaining of FABP5 with different lung cell types. We found decreased FABP5 protein expression in airway and alveolar epithelial cells 3 days postinfection. Specifically, decreases were observed in club, alveolar type I (ATI), and alveolar type II (ATII) cells (Fig. 7, B and C). Alveolar macrophages did not present any change in FABP5 expression (Fig. 7, B and C).
DISCUSSION
The early inflammatory response to influenza virus infection contributes to severe lung disease and continues to pose a serious threat to human health. Although FABP5 has been shown to play an important role in obesity and insulin resistance (27), little is known about its role in lung disease and infection. We have recently shown that FABP5 exerts anti-inflammatory properties in airway epithelial cells (13). In this study, we compared the immune response induced by influenza A virus in FABP5-/- and WT mice. We demonstrated that FABP5-/- mice had increased cell infiltration (i.e., macrophages and neutrophils) in BAL and increased adaptive immune response compared with WT mice. FABP5-/- mice presented lower viral burden but lost as much weight as WT mice. FABP5 deficiency greatly enhanced lipid peroxidation and overall lung oxidation following influenza A infection. Inflammation in H1N1-infected lung tissues was markedly enhanced in FABP5-/- mice compared with WT mice. Fourteen days postinfection FABP5-/- mice still had not resolved the lung inflammation while WT mice had. Interestingly, FABP5 was downregulated following influenza A infection in WT airway and alveolar epithelial cells and so was PPAR-γ activity.
A robust innate immune response is the first line of lung defense against respiratory viral infection. However, excessive responses may also cause additional tissue damage and inflammation. FABP5-/- mice presented with increased immune cell infiltration, which reduced the influenza virus titer but resulted in increased lung damage.
Interestingly, the increased number of macrophages seen in FABP5-/- mice may actually contribute to their protection and to the control of influenza virus replication at early time points of infection. Indeed, it has been shown that depletion of macrophages, prior to influenza infection, resulted in an uncontrolled replication of the virus and in increased mortality (44). Thus massive recruitment of macrophages at day 1 postinfection could account for control of viral titers and their subsequent reduction by day 3 postinfection. Neutrophils have also been linked to the initial control of influenza infection (44), but their presence is also associated with lung injury (32).
Studies of mouse models of influenza A virus infection have enhanced our understanding of the innate and adaptive antiviral mechanisms that contribute to viral clearance and recovery. The initial phase of influenza virus replication in epithelial cells, local macrophages, and dendritic cells triggers the rapid release of a range of cytokines and chemokines with antiviral and proinflammatory activities (12, 25). In addition to limiting viral replication in the respiratory tract, these processes are critical for the optimal activation of antigen-specific B and CD4 T cells and the development of an adaptive immunity (22). The elimination of infectious virus from the respiratory tract is ultimately dependent on increasing antibody levels from B cells (46) and cytotoxic activity of CD8 T cells. In our model, FABP5-/- mice presented increased numbers of T and B cells in the lung tissues compared with WT mice and increased H1N1-specific IgG levels. These data are in accordance with the fact that FABP5-/- mice cleared the virus earlier than WT mice.
In addition to inflammatory cytokines, innate immune cells produce ROS in response to pathogens. It has long been known that ROS play an important part in the innate immune system as an antimicrobial compound produced by NADPH oxidases in phagosomes of macrophages and neutrophils (36). More recently, it has been demonstrated that ROS are also involved in activation and regulation of a wider range of processes in the innate immune system, including autophagy, signal transduction, gene expression, activation of the inflammasome, and programmed necrosis (6, 7, 11, 29, 42). However, when produced at high concentration, ROS can have damaging effects on tissues at the site of infection (1). Our data demonstrate that FABP5-/- infected lung tissues had decreased GSH levels and produced increased amounts of 8-isoprostane, indicating increased oxidative stress in FABP5-/- lung tissues compared with WT lung tissues. This may explain why FABP5-/- lung tissues displayed more damage than WT tissues even though viral load was lower in FABP5-/- mice. Our results are in agreement with previous work where FABP5 has been described as a target of covalent 4-hydroxynonenal modification and therefore likely scavenges toxic reactive lipids (4).
To understand the mechanism behind FABP5 immunomodulatory function, we questioned whether the peroxisome proliferator-activated receptor-γ (PPAR-γ) was implicated. Indeed, PPAR-γ not only regulates fatty acid storage and glucose metabolism but also exerts anti-inflammatory activity (23, 37). Furthermore, FABP5 has recently emerged as a regulator of PPAR-γ activity (2). We have shown that in primary NHBE cells infected with bacteria, FABP5 downregulation resulted in reduced PPAR-γ activity, whereas FABP5 upregulation resulted in increased PPAR-γ activity (13). This contradicts the observation by Babaev et al. (2) that macrophage FABP5 expression suppresses PPAR-γ activity, but differences may be explained by the use of different cell types. Furthermore, inhibition of FABP5 by influenza infection resulted in decreased PPAR-γ activity, thus elucidating a potential immunomodulatory role for FABP5/PPAR in airway epithelial cells.
The reduction of FABP5 mRNA levels in lung homogenates following influenza A infection was surprising given that we previously showed upregulation of FABP5 following Mycoplasma pneumoniae or Pseudomonas aeruginosa infection in bronchial epithelial cells (13, 15). Similar upregulation was observed in alveolar macrophages following acute lung rejection (20). As mentioned previously, FABP5 is highly expressed in bronchial epithelial cells, alveolar type II epithelial cells, lung fibroblasts, and alveolar macrophages (15, 16, 33). Given this large array of expression in different lung cell types, we performed coimmunostaining of FABP5 with alveolar macrophages, airway epithelial cells, and type I and type II alveolar epithelial cells. We determined that airway and alveolar epithelial cells are responsible for FABP5 downregulation following H1N1 infection. Since FABP5 was severely downregulated in WT mice in response to H1N1 infection, repeating this experiment using FABP5 overexpressing mice would be important in determining how the presence of FABP5 affects the pathogenesis of H1N1 influenza. Furthermore, given that the alveolar macrophages were not affected, the generation of bone marrow chimeric mice may help elucidate the importance of FABP5 expression in the hematopoietic compartment. The downregulation of FABP5 following H1N1 infection and its effect on PPAR-γ activity let us speculate that airway epithelial cells downregulate FABP5 to initiate a robust immune response that is necessary to clear the virus. The inflammation is then resolved when FABP5 is reexpressed and PPAR-γ activity is increased. In the absence of FABP5, the virus is cleared rapidly but tissue inflammation remains.
As mentioned earlier, there are several FABP proteins. Functional redundancy might explain some of the modest effects observed in FABP5-/- mice in response to H1N1 infection compared with WT mice. For instance, FABP4 knockout animals have increased expression of FABP5 in the adipose tissue (40) and relatively minor metabolic alterations due to compensations. The interdependency of expression for the various FABPs is clearly evident for FABP5. In transgenic mice overexpressing the FABP5 gene, the adipocyte levels of FABP5 were 10-fold higher than in WT animals, whereas the levels of FABP4 were reduced by half (19). Furthermore, FABP5 deletion in the liver of FABP5-/- mice was compensated by overexpression of FABP3 during perinatal development (34). Thus more robust and convincing differences might be observed in mice lacking multiple FABP proteins.
In conclusion, influenza A virus induces a robust innate immune response in both FABP5-/- and WT mice. Although FABP5-/- mice presented increased numbers of inflammatory cells and lung damage, viral loads were lower in FABP5-/- mice compared with WT mice. We further demonstrated that FABP5 deficiency causes an enhanced oxidative environment, which could be responsible for the observed increased inflammation and lung tissue damage. Further investigations are needed to fully understand the signaling pathway involved in the absence of FABP5 that paradoxically leads to both antiviral responses and overly inflamed lung tissues in response to viral infection.
GRANTS
This study was supported by the Flight Attendant Medical Research Institute (F. Gally and B. Kosmider).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
F.G., B.K., and R.E.O.-D. conception and design of research; F.G., B.K., M.R.W., K.M.P., and R.E.O.-D. performed experiments; F.G., B.K., and R.E.O.-D. analyzed data; F.G., B.K., and R.E.O.-D. interpreted results of experiments; F.G. and B.K. prepared figures; F.G. drafted manuscript; F.G., B.K., K.M.P., K.L.H., and R.E.O.-D. edited and revised manuscript; F.G., B.K., M.R.W., K.M.P., K.L.H., and R.E.O.-D. approved final version of manuscript.
REFERENCES
- 1. Akaike T. Role of free radicals in viral pathogenesis and mutation. Rev Med Virol 11: 87–101, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Babaev VR, Runner RP, Fan D, Ding L, Zhang Y, Tao H, Erbay E, Gorgun CZ, Fazio S, Hotamisligil GS, Linton MF. Macrophage Mal1 deficiency suppresses atherosclerosis in low-density lipoprotein receptor-null mice by activating peroxisome proliferator-activated receptor-gamma-regulated genes. Arterioscler Thromb Vasc Biol 31: 1283–1290, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Banerjee M, Kang KH, Morrow JD, Roberts LJ, Newman JH. Effects of a novel prostaglandin, 8-epi-PGF2α, in rabbit lung in situ. Am J Physiol Heart Circ Physiol 263: H660–H663, 1992. [DOI] [PubMed] [Google Scholar]
- 4. Bennaars-Eiden A, Higgins L, Hertzel AV, Kapphahn RJ, Ferrington DA, Bernlohr DA. Covalent modification of epithelial fatty acid-binding protein by 4-hydroxynonenal in vitro and in vivo. Evidence for a role in antioxidant biology. J Biol Chem 277: 50693–50702, 2002. [DOI] [PubMed] [Google Scholar]
- 5. Buffinton GD, Christen S, Peterhans E, Stocker R. Oxidative stress in lungs of mice infected with influenza A virus. Free Radic Res Commun 16: 99–110, 1992. [DOI] [PubMed] [Google Scholar]
- 6. Chiang E, Dang O, Anderson K, Matsuzawa A, Ichijo H, David M. Cutting edge: apoptosis-regulating signal kinase 1 is required for reactive oxygen species-mediated activation of IFN regulatory factor 3 by lipopolysaccharide. J Immunol 176: 5720–5724, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137: 1112–1123, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Choi AM, Knobil K, Otterbein SL, Eastman DA, Jacoby DB. Oxidant stress responses in influenza virus pneumonia: gene expression and transcription factor activation. Am J Physiol Lung Cell Mol Physiol 271: L383–L391, 1996. [DOI] [PubMed] [Google Scholar]
- 9. Cloutier A, Marois I, Cloutier D, Verreault C, Cantin AM, Richter MV. The prostanoid 15-deoxy-Δ12,14-prostaglandin-j2 reduces lung inflammation and protects mice against lethal influenza infection. J Infect Dis 205: 621–630, 2012. [DOI] [PubMed] [Google Scholar]
- 10. de Jong MD, Simmons CP, Thanh TT, Hien VM, Smith GJ, Chau TN, Hoang DM, Chau NV, Khanh TH, Dong VC, Qui PT, Cam BV, Ha do Q, Guan Y, Peiris JS, Chinh NT, Hien TT, Farrar J. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat Med 12: 1203–1207, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320: 674–677, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fukuyama S, Kawaoka Y. The pathogenesis of influenza virus infections: the contributions of virus and host factors. Curr Opin Immunol 23: 481–486, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gally F, Chu HW, Bowler RP. Cigarette smoke decreases airway epithelial FABP5 expression and promotes Pseudomonas aeruginosa infection. PLoS One 8: e51784, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gao XP, Standiford TJ, Rahman A, Newstead M, Holland SM, Dinauer MC, Liu QH, Malik AB. Role of NADPH oxidase in the mechanism of lung neutrophil sequestration and microvessel injury induced by Gram-negative sepsis: studies in p47phox-/- and gp91phox-/- mice. J Immunol 168: 3974–3982, 2002. [DOI] [PubMed] [Google Scholar]
- 15. Green RM, Gally F, Keeney JG, Alper S, Gao B, Han M, Martin RJ, Weinberger AR, Case SR, Minor MN, Chu HW. Impact of cigarette smoke exposure on innate immunity: a Caenorhabditis elegans model. PLoS One 4: e6860, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guthmann F, Hohoff C, Fechner H, Humbert P, Borchers T, Spener F, Rustow B. Expression of fatty-acid-binding proteins in cells involved in lung-specific lipid metabolism. Eur J Biochem 253: 430–436, 1998. [DOI] [PubMed] [Google Scholar]
- 17. Hama Y, Kurokawa M, Imakita M, Yoshida Y, Shimizu T, Watanabe W, Shiraki K. Interleukin 12 is a primary cytokine responding to influenza virus infection in the respiratory tract of mice. Acta Virol 53: 233–240, 2009. [DOI] [PubMed] [Google Scholar]
- 18. Hart CM, Tolson JK, Block ER. Supplemental fatty acids alter lipid peroxidation and oxidant injury in endothelial cells. Am J Physiol Lung Cell Mol Physiol 260: L481–L488, 1991. [DOI] [PubMed] [Google Scholar]
- 19. Hertzel AV, Bennaars-Eiden A, Bernlohr DA. Increased lipolysis in transgenic animals overexpressing the epithelial fatty acid binding protein in adipose cells. J Lipid Res 43: 2105–2111, 2002. [DOI] [PubMed] [Google Scholar]
- 20. Holler J, Zakrzewicz A, Garn H, Hirschburger M, Kummer W, Padberg W, Grau V. Increased expression of epidermal fatty acid-binding protein by alveolar macrophages during acute rejection of rat lungs. APMIS 118: 791–800, 2010. [DOI] [PubMed] [Google Scholar]
- 21. Ichinohe T, Iwasaki A, Hasegawa H. Innate sensors of influenza virus: clues to developing better intranasal vaccines. Expert Rev Vaccines 7: 1435–1445, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Iwasaki A, Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science 327: 291–295, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jiang C, Ting AT, Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature 391: 82–86, 1998. [DOI] [PubMed] [Google Scholar]
- 24. Kasahara Y, Iwai K, Yachie A, Ohta K, Konno A, Seki H, Miyawaki T, Taniguchi N. Involvement of reactive oxygen intermediates in spontaneous and CD95 (Fas/APO-1)-mediated apoptosis of neutrophils. Blood 89: 1748–1753, 1997. [PubMed] [Google Scholar]
- 25. La Gruta NL, Kedzierska K, Stambas J, Doherty PC. A question of self-preservation: immunopathology in influenza virus infection. Immunol Cell Biol 85: 85–92, 2007. [DOI] [PubMed] [Google Scholar]
- 26. Lundqvist-Gustafsson H, Bengtsson T. Activation of the granule pool of the NADPH oxidase accelerates apoptosis in human neutrophils. J Leukoc Biol 65: 196–204, 1999. [DOI] [PubMed] [Google Scholar]
- 27. Maeda K, Uysal KT, Makowski L, Gorgun CZ, Atsumi G, Parker RA, Bruning J, Hertzel AV, Bernlohr DA, Hotamisligil GS. Role of the fatty acid binding protein mal1 in obesity and insulin resistance. Diabetes 52: 300–307, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mellqvist UH, Hansson M, Brune M, Dahlgren C, Hermodsson S, Hellstrand K. Natural killer cell dysfunction and apoptosis induced by chronic myelogenous leukemia cells: role of reactive oxygen species and regulation by histamine. Blood 96: 1961–1968, 2000. [PubMed] [Google Scholar]
- 29. Mogensen TH, Melchjorsen J, Hollsberg P, Paludan SR. Activation of NF-kappa B in virus-infected macrophages is dependent on mitochondrial oxidative stress and intracellular calcium: downstream involvement of the kinases TGF-beta-activated kinase 1, mitogen-activated kinase/extracellular signal-regulated kinase kinase 1, and I kappa B kinase. J Immunol 170: 6224–6233, 2003. [DOI] [PubMed] [Google Scholar]
- 30. Morgenstern DE, Gifford MA, Li LL, Doerschuk CM, Dinauer MC. Absence of respiratory burst in X-linked chronic granulomatous disease mice leads to abnormalities in both host defense and inflammatory response to Aspergillus fumigatus. J Exp Med 185: 207–218, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Morrow JD, Frei B, Longmire AW, Gaziano JM, Lynch SM, Shyr Y, Strauss WE, Oates JA, Roberts LJ., 2nd Increase in circulating products of lipid peroxidation (F2-isoprostanes) in smokers. Smoking as a cause of oxidative damage. N Engl J Med 332: 1198–1203, 1995. [DOI] [PubMed] [Google Scholar]
- 32. Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew AA, Phoon MC, van Rooijen N, Chow VT. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am J Pathol 179: 199–210, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Owada Y, Abdelwahab SA, Suzuki R, Iwasa H, Sakagami H, Spener F, Kondo H. Localization of epidermal-type fatty acid binding protein in alveolar macrophages and some alveolar type II epithelial cells in mouse lung. Histochem J 33: 453–457, 2001. [DOI] [PubMed] [Google Scholar]
- 34. Owada Y, Suzuki I, Noda T, Kondo H. Analysis on the phenotype of E-FABP-gene knockout mice. Mol Cell Biochem 239: 83–86, 2002. [PubMed] [Google Scholar]
- 35. Pollock JD, Williams DA, Gifford MA, Li LL, Du X, Fisherman J, Orkin SH, Doerschuk CM, Dinauer MC. Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat Genetics 9: 202–209, 1995. [DOI] [PubMed] [Google Scholar]
- 36. Rada B, Leto TL. Oxidative innate immune defenses by Nox/Duox family NADPH oxidases. Contrib Microbiol 15: 164–187, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 391: 79–82, 1998. [DOI] [PubMed] [Google Scholar]
- 38. Schmitz N, Kurrer M, Bachmann MF, Kopf M. Interleukin-1 is responsible for acute lung immunopathology but increases survival of respiratory influenza virus infection. J Virol 79: 6441–6448, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schwarz KB. Oxidative stress during viral infection: a review. Free Radic Biol Med 21: 641–649, 1996. [DOI] [PubMed] [Google Scholar]
- 40. Shaughnessy S, Smith ER, Kodukula S, Storch J, Fried SK. Adipocyte metabolism in adipocyte fatty acid binding protein knockout mice (aP2-/-) after short-term high-fat feeding: functional compensation by the keratinocyte [correction of keritinocyte] fatty acid binding protein. Diabetes 49: 904–911, 2000. [DOI] [PubMed] [Google Scholar]
- 41. Siegenthaler G, Hotz R, Chatellard-Gruaz D, Didierjean L, Hellman U, Saurat JH. Purification and characterization of the human epidermal fatty acid-binding protein: localization during epidermal cell differentiation in vivo and in vitro. Biochem J 302: 363–371, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tal MC, Sasai M, Lee HK, Yordy B, Shadel GS, Iwasaki A. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc Natl Acad Sci USA 106: 2770–2775, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Thompson WW, Shay DK, Weintraub E, Brammer L, Cox N, Anderson LJ, Fukuda K. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA 289: 179–186, 2003. [DOI] [PubMed] [Google Scholar]
- 44. Tumpey TM, Garcia-Sastre A, Taubenberger JK, Palese P, Swayne DE, Pantin-Jackwood MJ, Schultz-Cherry S, Solorzano A, Van Rooijen N, Katz JM, Basler CF. Pathogenicity of influenza viruses with genes from the 1918 pandemic virus: functional roles of alveolar macrophages and neutrophils in limiting virus replication and mortality in mice. J Virol 79: 14933–14944, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vacchiano CA, Tempel GE. Role of nonenzymatically generated prostanoid, 8-iso-PGF2α, in pulmonary oxygen toxicity. J Appl Physiol 77: 2912–2917, 1994. [DOI] [PubMed] [Google Scholar]
- 46. Waffarn EE, Baumgarth N. Protective B cell responses to flu—no fluke! J Immunol 186: 3823–3829, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zimmerman AW, Veerkamp JH. Fatty-acid-binding proteins do not protect against induced cytotoxicity in a kidney cell model. Biochem J 360: 159–165, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]