

Abstract
Recent studies identified the SLC26A9 Cl− channel as a modifier and potential therapeutic target in cystic fibrosis (CF). However, understanding of the regulation of SLC26A9 in epithelia remains limited and cellular models with stable expression for biochemical and functional studies are missing. We, therefore, generated Fisher rat thyroid (FRT) epithelial cells with stable expression of HA-tagged SLC26A9 via retroviral transfection and characterized SLC26A9 expression and function using Western blotting, immunolocalization, whole cell patch-clamp, and transepithelial bioelectric studies in Ussing chambers. We demonstrate stable expression of SLC26A9 in transfected FRT (SLC26A9-FRT) cells on the mRNA and protein level. Immunolocalization and Western blotting detected SLC26A9 in different intracellular compartments and to a lesser extent at the cell surface. Whole cell patch-clamp recordings demonstrated significantly increased constitutive Cl− currents in SLC26A9-FRT compared with control-transduced FRT (Control-FRT) cells (P < 0.01). Similar, transepithelial measurements showed that the basal short circuit current was significantly increased in SLC26A9-FRT vs. Control-FRT cell monolayers (P < 0.01). SLC26A9-mediated Cl− currents were increased by cAMP-dependent stimulation (IBMX and forskolin) and inhibited by GlyH-101, niflumic acid, DIDS, and 5-nitro-2-(3-phenylpropylamino) benzoic acid (NPPB), as well as RNAi knockdown of WNK1 implicated in epithelial osmoregulation. Our results support that these novel epithelial cells with stable expression of SLC26A9 will be a useful model for studies of pharmacological regulation including the identification of activators of SLC26A9 Cl− channels that may compensate deficient cystic fibrosis transmembrane regulator (CFTR)-mediated Cl− secretion and serve as an alternative therapeutic target in patients with CF and potentially other muco-obstructive lung diseases.
Keywords: Cl− channels, SLC26A9, cell model, epithelial ion transport, cystic fibrosis
the solute carrier 26 gene family member A9 (SLC26A9) encodes a Cl− channel that is abundantly expressed in epithelia of the airways and the upper gastrointestinal (GI) tract (6, 13, 14, 41). In human bronchial epithelial cells, SLC26A9 was shown to act as a constitutively active Cl− channel in the apical cell membrane where it interacts functionally with the cystic fibrosis (CF) transmembrane regulator (CFTR) (3, 5). Recent evidence from genetic studies demonstrated that single nucleotide polymorphisms (SNP) in SLC26A9 are associated with the risk of developing meconium ileus, exocrine pancreatic damage, and diabetes in patients with CF indicating that SLC26A9 Cl− channels may compensate for deficient CFTR-mediated Cl− secretion in a variety of organs affected by CF multiorgan disease (12, 21, 34, 36). Furthermore, recent functional studies demonstrated that SLC26A9-mediated Cl− secretion is essential for preventing airway mucus obstruction due to mucin hypersecretion in type-2 airway inflammation in mice and that a functional SNP in the 3′-untranslated region of SLC26A9 (rs2282430) that reduced protein expression in vitro is associated with asthma (2). Finally, missense variants of SLC26A9 that abrogate its Cl− channel function were also found in patients with diffuse bronchiectasis (4). Collectively, these studies suggest SLC26A9 as a disease modifier and novel therapeutic target that may compensate for impaired CFTR-mediated Cl− secretion and improve mucus transport in patients with CF and potentially other muco-obstructive airways diseases (19).
Despite these persuasive results from recent mouse and human studies, cell models and reagents including antibodies for studies of SLC26A9 function and regulation and the identification of activator compounds for therapeutics development remain limited. The aim of this study was, therefore, to generate and characterize an epithelial cell model with stable expression of SLC26A9. To achieve this goal, we selected Fisher rat thyroid (FRT) epithelial cells as a model as they have been shown to be suitable for studies of other epithelial ion channels including CFTR and epithelial Na+ channels at the level of single cells, as well as cell monolayers suitable for integrated studies of transepithelial ion transport (32, 33). FRT cell lines transduced with CFTR mutants were also used successfully for functional high throughput screening assays that led to the identification of the clinical CFTR modulators ivacaftor and lumacaftor (9, 16, 23, 37–39, 43). Additionally, FRT cells have not been reported to express either SLC26A9 or CFTR endogenously and thus provide a cellular environment for studies of SLC26A9 Cl− channels in the absence of functional CFTR, i.e., mimicking the pathophysiological condition present in most patients with CF (7, 20, 26). To overcome limitations related to the lack of antibodies for detection of the native SLC26A9 protein in this model, we transduced FRT cells with NH2 and COOH terminally HA-tagged versions of SLC26A9 that enable biochemical studies including immunolocalization and immunoblotting with anti-HA antibodies (24). FRT cell lines with stable expression of HA-tagged SLC26A9 after retroviral transduction were characterized by immunolocalization and immunoblotting studies and functional studies in single cells and monolayers using whole cell patch-clamp and transepithelial Ussing chamber measurements. These SLC26A9-expressing FRT cell lines will provide a useful model for studies of the regulation and identification of activators of SLC26A9 Cl− channels that may serve as a novel therapeutic target in CF and potentially other muco-obstructive lung diseases.
MATERIALS AND METHODS
Materials.
Cell culture plastics were obtained from Greiner BioOne (Frickenhausen, Germany), with the exception of Corning Snapwell permeable filter inserts (no. 3407; Corning, Corning, NY). IBMX, forskolin (FSK), niflumic acid (NFA), and DIDS were obtained from Sigma-Aldrich (Taufkirchen, Germany). Cyclopiazonic acid (CPA), GlyH-101, and CFTRinh-172 were purchased from Calbiochem (Merck Millipore, Darmstadt, Germany), and 5-nitro-2-(3-phenylpropylamino) benzoic acid (NPPB) was from Tocris (R&D Systems, Wiesbaden, Germany). All chemicals were of the highest grade of purity available.
Cell culture.
FRT epithelial cells were kindly provided by Dr. E. Sorscher (University of Alabama at Birmingham School of Medicine, Birmingham, AL). FRT cells were grown in Coon's F12 modified medium (F0855; Biochrom, Berlin, Germany), supplemented with 5% FBS, 100 U/ml penicillin, and 100 μM/ml streptomycin. Cells were cultured in humidified atmosphere at 37°C in 5% CO2. The medium was changed every 2–3 days. Cell monolayers were grown on Snapwell filter membranes and considered confluent and differentiated after 5–8 days. Transepithelial electrical resistance values of FRT monolayers were in the range of 4,000 Ω/cm2. For patch-clamp studies, flask-grown cells were dissociated and plated at low density to poly-l-lysine-coated glass coverslip chips and single cells were patched 12–24 h after plating.
Retroviral transfection of epithelial cells.
The 2.6-kb cDNA coding sequence of human SLC26A9 containing a 3× HA tag at the COOH terminus (SLC26A9-3HA) or NH2 terminus (3HA-SLC26A9) in pReceiver-M07 and pReceiver-M06 expression vectors (GeneCopoeia, Rockville, MD), respectively, was kindly provided by Dr. L. Galietta (Istituto Giannina Gaslini, Genova, Italy) and subcloned into the retroviral expression vector pRIJ derived from the moloney murine leukemia retrovirus based plasmid pQCXIP (BD Biosciences, Heidelberg, Germany) with an extended multiple cloning site. The entire construct was sequenced for verification. Retroviral particles were generated as described previously (29). In brief, the packaging cell line phoenix-gp was transfected with 4.5 μg VSV-G and the plasmids 3HA-SLC26A9.pRIJ and SLC26A9-3HA.pRIJ or the control vector .pRIJ (each 13.5 μg) using the calcium phosphate transfection technique. Replication-defective retroviral particles were recovered in the supernatant of phoenix-gp cells and harvested every 24 h up to 6 days after transfection. FRT epithelial cells were infected twice with 500 μl of filtered retroviral supernatant in the presence of 4 μg/ml polybrene to enhance retroviral transduction efficiency. Three days after transduction, selection was initiated by culturing cells in the presence of puromycin at a concentration of 1 μg/ml. Cell lines with stable expression of SLC26A9 were identified by PCR, immunocytochemistry, and bioelectric studies over a time of 15 passages.
Semiquantitave real-time RT-PCR.
Messenger RNA (mRNA) was isolated using an RNeasy Mini kit (Qiagen, Hilden, Germany) according to the manufacturer's protocol and the concentration of RNA was determined by UV absorption at 260 nm (NanoDrop ND 1000 spectrophotometer; Thermo Scientific, Schwerte, Germany). Real-time RT-PCR was performed with an Applied Biosystems 7500 Real-Time PCR System (Life Technologies, Darmstadt, Germany) using FAM-MGB labeled TaqMan gene expression assays for SLC26A9 (Hs00369451_m1), WNK1 (Rn00671521_m1), WNK4 (Rn00598070_m1), STK39 (SPAK, Rn00571210_m1), and β-actin (Rn00667869_m1 and Hs01060665_g1). Relative fold changes in target gene expression were calculated from the efficiency of the PCR reaction and the crossing point deviation between samples from 16HBE14o- and Control-FRT vs. SLC26A9-FRT cells and determined by normalization to expression of the reference gene β-actin.
Surface biotinylation, deglycosylation, and immunoblot assays.
For surface biotinylation studies, sulfo-NHS-biotin (0.5 mg/ml; Pierce, Thermo Scientific) was applied to the apical or basolateral side of confluent filter-grown FRT cell monolayers and incubated at 4°C for 30 min. To prevent biotinylation of apical or basolateral membrane proteins, BSA (1%) in PBS supplemented with 2.0 mM Ca2+ was applied to the respective opposite membrane. Subsequently, cell monolayers were incubated with 1% BSA in PBS in both compartments to stop the biotinylation reaction. Following cell lysis, protein concentration of the whole cell lysates was determined by Pierce BCA Protein Assay Kit (Thermo Scientific) and equal amounts of protein were loaded onto streptavidin-agarose beads (Sigma-Aldrich) for overnight incubation at 4°C. Then, biotinylated membrane proteins were washed and resuspended in 4× NuPAGE LDS Sample Buffer.
Total protein extracts were obtained from epithelial cell monolayers grown on 6-well plates. Cell lysis was performed on ice in RIPA buffer (150 mM NaCl, 1% NP-40, 0.1% SDS, 10 mM Tris·Cl pH 8.0, and 1 mM EDTA) supplemented with cOmplete Protease Inhibitor (Roche Diagnostics, Mannheim, Germany) for 30 min. After centrifugation, supernatants were collected and protein concentrations determined using Pierce BCA Protein Assay Kit (Thermo Scientific). Reducing 4× NuPAGE LDS Sample Buffer (Life Technologies) was added and proteins were denatured for 30 min at 37°C.
Glycosylation assays were performed according to manufacturer's instructions (New England Biolabs, Frankfurt, Germany). In brief, 10 μg of cell lysates were denatured by incubation with 1× Glycoprotein Denaturing Buffer (0.5% SDS, 40 mM DTT) for 30 min. G5 reaction buffer was added to the samples in the case of endoglycosidase H (EndoH) treatment and G7 reaction buffer and 10% NP-40 in the case of peptide-I-glycosidase F (PNGaseF) treatment, respectively. Then, protein samples were incubated with 500 U of either EndoH or PNGaseF for 1 h at 37°C. The deglycosylation reactions were stopped by the addition of 4× NuPAGE LDS Sample Buffer.
Protein samples were separated by electrophoresis using NuPAGE Novex Bis-Tris 4 - 12% gels (Life Technologies). After electrophoresis, transfer onto nitrocellulose membranes (Bio-Rad, München, Germany) was performed overnight at 4°C. Membranes were then blocked in 5% dry milk powder in PBS followed by incubation with anti-HA tag (05-094; Merck Millipore), anti-Na+/K+-ATPase (06–520; Merck Millipore), or β-actin (Clone AC-15, A1978; Sigma-Aldrich) as primary antibodies at a dilution of 1:2,000 and 1:10,000, respectively. Membranes were washed with PBS and subsequently incubated with the relevant anti-mouse IgG peroxidase-conjugated secondary antibody (Sigma-Aldrich) at a concentration of 1:10,000. After additional washes, peroxidase activity was detected with Western Lightning Plus Enhanced Cheminluminescence Substrate (Perkin Elmer, Rodgau, Germany) on a Fusion Fx7 system (Peqlab, Erlangen, Germany). Protein abundance was determined by densitometry using a ChemiDoc MP documentation system (Bio-Rad). After being immunoblotted for HA-tagged SLC26A9, blots were stripped and analyzed for β-actin as loading control. The relative abundance of the 95- and 120-kDa SLC26A9 bands in SLC26A9-3HA-FRT and 3HA-SLC26A9-FRT cells, and their doublet ratios, were determined from the total surface area of the respective bands normalized to β-actin.
Immunofluorescence microscopy.
Cell monolayers were grown for 5 days, fixed with 4% (wt/vol) paraformaldehyde for 10 min, and permeabilized for 8 min with 0.1% (wt/vol) Triton X-100. Cells were incubated for 60 min with anti-HA tag (1: 200), anti-ZO-1 (1: 200; no. 61–7300; Invitrogen, Life Technologies), anti-Giantin (1:200; ab24586; Abcam, Cambridge, UK), anti-Calnexin (1:200; ADI-SPA-860-D; Enzo Life Science, Lörrach, Germany), and anti-EEA1 (1:100; no. 3288; Cell Signaling Technology, Leiden, The Netherlands) primary antibody, rinsed three times with PBS, and incubated with a 1:300 dilution of the respective Alexa Fluor-labeled F(ab')2 fragment (Invitrogen). Hoechst (1: 2,000) was used to counterstain cell nuclei. After 30 min of incubation, the specimens were again rinsed three times and embedded in FluorSave anti-fade medium (Merck Millipore). Images were obtained using a 60× oil-immersion objective on a confocal laser scanning microscope (Leica SP8, Wetzlar, Germany) with the instrument's settings adjusted so that no positive signal was observed in the channel corresponding to the fluorescence of the relevant isotypic controls (data not shown). Most studies were performed using automated XY stage scanning (adjusted to the cell nuclei). In some studies, XZ (vertical) images were generated taking single Z-series stacks through the epithelial cells at 0.05-μm steps (15 steps). All images were processed with the software Fiji based on ImageJ or Adobe Illustrator version CS2 (Adobe Systems, München, Germany), respectively.
Whole cell patch-clamp recordings.
All whole cell patch-clamp experiments were performed using a 200B Axopatch amplifier controlled by pCLAMP 10 software through a Digidata 1440A data acquisition system (Molecular Devices, Sunnyvale, CA). Experiments were performed as previously described (5). Briefly, cells were voltage clamped at a holding potential of −40 mV, and membrane current (Im) was recorded continuously as GlyH-101 (50 μM) and NFA (500 μM) were applied. The current-voltage (I/V) relationship was measured after each addition by stepping the holding potential from −100 to +100 mV in 20-mV steps. Bath solutions ± the agonists and inhibitors were continuously perfused and warmed to 37°C with an inline solution heater. Patch-clamp experiments were carried out in NMDG-Cl solution to isolate Cl− currents. The bath solution (140 mM NMDG-Cl, 10 mM HEPES, 5 mM d-glucose, 1 mM MgCl2, and 1.5 mM CaCl2, pH 7.3) and pipette solution (140 mM NMDG-Cl, 10 mM HEPES, 5 mM d-glucose, 1 mM MgCl2, 1 mM EGTA, 1 mM Mg-ATP, and 100 μM Na2-GTP, pH 7.2) were prepared fresh on the day of experiment.
Transepithelial Ussing chamber measurements.
Bioelectrical experiments were performed in EasyMount Ussing chambers (Physiologic Instruments, San Diego, CA) using the short circuit current technique. Corrections for electrode offset and bath solution resistances were performed before the experiments. Chambers and bathing solutions were maintained at 37°C. The transepithelial voltage (Vte) was clamped to 0 mV using a standard four Ag/AgCl electrode voltage clamp (VCC MC6; Physiologic Instruments) and the short-circuit current (Isc) was measured. Voltage pulses of 2 mV were applied every 60 s for 1 s for monitoring the transepithelial resistance (Rte) of cell monolayers. The Isc was continuously recorded using LabChart7 for Windows (ADInstruments, Oxfordshire, UK). FRT cell monolayers grown on Snapwell cell culture inserts were mounted into Ussing chambers. The basolateral compartment was perfused with a standard bath solution of the following composition: 145 mM NaCl, 0.4 mM KH2PO4, 1.6 mM K2HPO4, 5 mM d-glucose, 1 mM MgCl2, and 1.3 mM calcium-gluconate. The apical compartment was perfused with a Cl− free solution (0 mM Cl−) prepared by replacing NaCl with equivalent amounts of Na-gluconate to increase the driving force for Cl− transport across the epithelium. To determine effects of hypotonic and hypertonic conditions, the osmolality was adjusted to 220 mol/kgH2O by reducing the Na+ concentration by 40 mM and to 480 mol/kgH2O by adding mannitol (200 mM), respectively. IBMX (100 μM, both sides) and FSK (1 μM, both sides) were added to increase intracellular cAMP levels, and CPA (50 μM, apical), an inhibitor of Ca2+-ATPase in intracellular Ca2+ stores, was added to increase intracellular Ca2+ (18). Furthermore, individual effects of the following Cl− channel blockers were tested: CFTR inhibitor CFTRinh-172 (20 μM, apical), GlyH-101 (50 μM, apical), NFA (500 μM, basolateral), NPPB (100 μM, apical), and DIDS (100 μM, apical). In a subset of experiments, CFTRinh-172, GlyH-101, and NFA were added sequentially to assess for additivity of effects.
RNAi studies.
Transduced epithelial cells were seeded on permeable Snapwell filter supports at a density of 250,000 cells/cm2 and left to attach for 16–18 h at 37°C in Coon's F12 modified medium without antibiotics. Dharmafect transfection reagent (Dharmacon, GE Healthcare, Freiburg, Germany) and ON-TARGETplus SMARTpool of WNK1-, WNK4-, and SPAK-siRNA (150 nM; Dharmacon) or scrambled control (Dharmacon) were mixed, incubated for 20 min and added to the respective wells. After at least 6 h of transfection, the medium was replaced and bioelectric measurements were performed after 48 h.
Statistical analysis.
Data were analyzed with SigmaStat version 3.1 (Systat Software, Erkrath, Germany) and are shown as mean ± SE. Statistical analysis was performed using unpaired and paired, two-tailed Student's t-test and one-way ANOVA as appropriate, and P < 0.05 were accepted to indicate statistical significance. All data were obtained from at least three independent experiments.
RESULTS
Generation of FRT epithelial cell lines with stable expression of SLC26A9.
To generate FRT epithelial cell lines with stable expression of human SLC26A9 for biochemical and functional studies, FRT cells were transduced with a retroviral plasmid containing either COOH or NH2 terminally 3×HA-tagged SLC26A9 constructs (SLC26A9-3HA-FRT and 3HA-SLC26A9-FRT, respectively) or empty retroviral plasmid (pRIJ) (referred to as Control-FRT). Transcript analysis demonstrated that Control-FRT cells do not express SLC26A9 endogenously and that SLC26A9 mRNA levels were ∼1,500-fold increased in SLC26A9-3HA-FRT and 3HA-SLC26A9-FRT cells compared with endogenous transcript levels in human normal (16HBE14o-) and CF (CFBE41o-) bronchial epithelial cell lines (Fig. 1A). In SLC26A9-3HA-FRT cells, SLC26A9 mRNA levels tended to be slightly higher (∼1.4-fold) than in 3HA-SLC26A9-FRT cells, but this difference did not reach statistical significance (Fig. 1A). Western blotting using anti-HA antibodies detected bands at 95 and 120 kDa, as expected for SLC26A9 (8, 11) in 3HA-SLC26A9-FRT and SLC26A9-3HA-FRT cells but not in Control-FRT cells (Fig. 1B). Densitometric analysis revealed similar levels of the mature 120 kDa but 2.6-fold higher levels of the 95-kDa band in SLC26A9-3HA-FRT vs. 3HA-SLC26A9-FRT cells (P < 0.05; Fig. 1C). Cell surface biotinylation assays detected mature SLC26A9 protein (120 kDa) in the apical membrane and to a lesser extent also in the basolateral membrane of SLC26A9-3HA-FRT cells. Detection of Na+-K+-ATPase was used as control to confirm purity of biotinylated samples and was found in the basolateral fraction only (Fig. 1D). Furthermore, deglycosylation assays were carried out to study the glycosylation state of SLC26A9 in this epithelial cell model. Cell lysates were treated with EndoH to characterize basic N-glycosylation in the endoplasmic reticulum (ER) and PNGaseF to test for further N-linked glycosylation modifications and compared with untreated samples, respectively. In the presence of EndoH, two protein bands were detectable and the lower band was slightly shifted to a lower product size of ∼90 kDa compared with the lower 95-kDa band in untreated SLC26A9-expressing FRT cells (Fig. 1D). In the case of PNGaseF, the mobility of the bands was increased and only one protein band was detected (Fig. 1E). The size of this band was similar to the predicted molecular size of SLC26A9 of 87 kDa consistent with expression of the N-glycosylated form of SLC26A9 in FRT cells (8, 11).
Fig. 1.
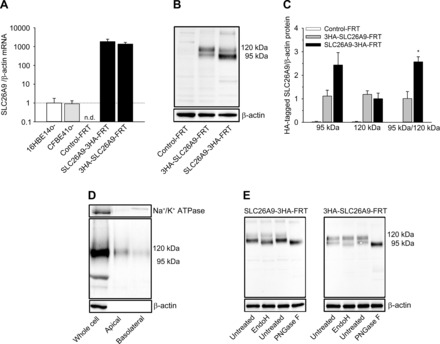
Expression of SLC26A9 in transduced Fisher rat thyroid (FRT) epithelial cell lines. A: mRNA transcript levels of SLC26A9 in FRT cells transduced with SLC26A9 constructs containing HA-tags at the COOH and NH2 termini (SLC26A9-3HA-FRT and 3HA-SLC26A9-FRT, respectively) compared with nontransduced FRT (Control-FRT) cells and endogenous expression in human normal (16HBE14o-) and cystic fibrosis (CF) (CFBE41o-) bronchial epithelial cells (n = 4 per group). B and C: representative Western blot (B) and densitometric analyses (C) of SLC26A9 protein bands detected by anti-HA antibody in 3HA-SLC26A9-FRT, SLC26A9-3HA-FRT, and Control-FRT cells. Relative abundance of the 95- and 120-kDa SLC26A9 bands was determined from the total surface area of the respective bands normalized to β-actin as loading control, and doublet ratios of the 2 bands were calculated for 3HA-SLC26A9-FRT and SLC26A9-3HA-FRT cells. *P < 0.05, compared with 3HA-SLC26A9-FRT; n = 4. D: representative Western blot of whole cell lysates, and apically and basolaterally biotinylated surface protein fractions of SLC26A9-3HA-FRT cells. β-Actin and Na+-K+-ATPase were used as controls. E: glycosylation studies of SLC26A9 in SLC26A9-3HA-FRT and 3HA-SLC26A9-FRT cells using endoglycosidase H (EndoH) or peptide-N-glycosidase F (PNGaseF) treatment. β-actin was used as a loading control.
Next, we performed immunolocalization studies to determine the cellular distribution of HA-tagged SLC26A9 in FRT cells. SLC26A9-3HA-FRT cells exhibited strong immunoreactive signals in intracellular compartments, whereas only little signal was observed at the cell surface (Fig. 2). SLC26A9 staining showed colocalization with the ER marker calnexin, the Golgi-matrix protein giantin, and the endosome marker EEA1 in SLC26A9-3HA-FRT cells and was not observed in Control-FRT cells (Fig. 2).
Fig. 2.
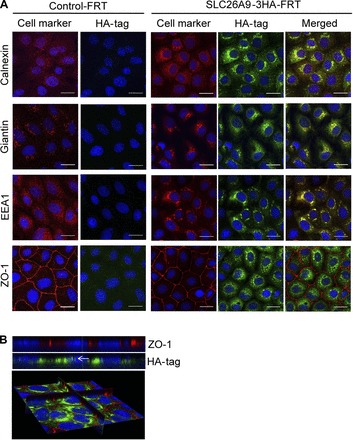
Immunolocalization of HA-tagged SLC26A9 protein in FRT epithelial cells. A: XY images of coimmunolocalization of SLC26A9 (green) with cell markers (red) in Control-FRT (left) and SLC26A9-3HA-FRT (right) cells. Cells were costained with the endoplasmic reticulum (ER) marker calnexin, the Golgi matrix protein giantin, the endosome marker EEA1, and the tight-junction marker ZO-1 and representative images are shown as individual immunostainings and the merged overlay. B: XZ images (top) and 3D reconstruction (bottom) of SLC26A9-3HA-FRT cells costained for SLC26A9 (green) and ZO-1 (red). The arrow indicates the vicinity of the luminal membrane. Nuclei were stained with Hoechst (blue). Scale bar = 20 μm.
Characterization of SLC26A9 Cl− channel function in single transduced FRT epithelial cells.
For functional characterization of SLC26A9 transduced FRT cells, we performed patch-clamp experiments and assessed whole cell Cl− currents in SLC26A9-3HA-FRT, 3HA-SLC26A9-FRT, and Control-FRT cells. For these experiments, cells were cultured and studied in a nonpolarized configuration and experimental solutions were used that limit conductive ions to Cl−. Consistent with previous reports (28), Control-FRT cells expressed small outwardly rectifying Cl− currents (Fig. 3A). In agreement with previous studies in HEK cells with transient expression of SLC26A9 (5), SLC26A9-3HA-FRT cells demonstrated a linear current-voltage (I–V) relationship, whereas membrane currents (Im) in 3HA-SLC26A9-FRT cells were slightly outwardly rectifying (Fig. 3A). Under voltage clamp at a holding potential of −40 mV, a negative current was measured in all cells (Cl− equilibrium potential ≅ 0 mV). In SLC26A9-3HA-FRT and 3HA-SLC26A9-FRT cells, whole cell Cl− currents were significantly increased compared with Control-FRT cells (Fig. 3A). SLC26A9-mediated Cl− currents were significantly inhibited by GlyH-101 and NFA, respectively (Fig. 3B).
Fig. 3.
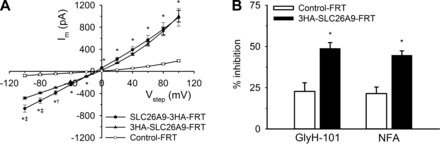
Characterization of Cl− currents in SLC26A9 transduced FRT epithelial cell lines by whole cell patch-clamp measurements. A: summary current-voltage (I–V) curves of SLC26A9-expressing FRT epithelial cells (COOH and NH2 terminus tagged protein: SLC26A9-3HA-FRT and 3HA-SLC26A9-FRT, respectively) and Control-FRT cells. *P < 0.01 vs. Control-FRT. †P < 0.05, ‡P < 0.01 vs. 3HA-SLC26A9-FRT; n = 4–7. B: effects of inhibitors GlyH-101 (50 μM) and niflumic acid (NFA; 500 μM) on whole cell currents in 3HA-SLC26A9-FRT cells (Vhold = −40 mV) expressed as %inhibition of total current. *P < 0.01 vs. Control-FRT; n = 6–8.
Characterization of SLC26A9-mediated Cl− currents in FRT epithelial cell monolayers.
To determine transepithelial SLC26A9-mediated Cl− currents across polarized FRT cell monolayer, we performed Ussing chamber measurements in SLC26A9-3HA-FRT, 3HA-SLC26A9-FRT, and Control-FRT cells in the presence of a basolateral-to-apical Cl− gradient to augment the electrochemical driving force for Cl− secretion across the epithelium (35). Under basal conditions, Control-FRT cells exhibited a low Isc of ∼4 μA/cm2 that was significantly increased to similar levels (∼3- to 4-fold) in SLC26A9-3HA-FRT and 3HA-SLC26A9-FRT monolayers (Fig. 4, A and B). This basal Isc was abolished when measurements were performed under Cl−-free conditions demonstrating that SLC26A9-mediated currents were carried by Cl− (Fig. 4B). Stimulation of monolayers with IBMX and FSK to increase intracellular cAMP levels increased Isc by ∼20 to 30% in both Control-FRT (ΔIsc = 0.7 ± 0.2 μA/cm2; n = 16; P < 0.15) and SLC26A9-3HA-FRT (ΔIsc = 4.5 ± 0.3 μA/cm2; n = 21; P < 0.004) cell monolayers (Fig. 4C). Treatment with CPA to increase intracellular Ca2+ had no effect on Isc in Control-FRT or SLC26A9-3HA-FRT monolayers (Fig. 4C). Testing of a series of Cl− channel blockers showed that SLC26A9-mediated currents across FRT monolayers were significantly inhibited by GlyH-101 (ΔIsc = 1.7 ± 0.2 μA/cm2; n = 8; P < 0.01) and NFA (2.7 ± 0.6 μA/cm2; n = 15; P < 0.05) that were previously reported to inhibit SLC26A9 Cl− channels (5, 9, 22). Furthermore, SLC26A9-mediated Isc was significantly inhibited by the broad spectrum Cl− channel blockers DIDS (ΔIsc = 5.1 ± 0.3 μA/cm2; n = 6; P < 0.01) and NPPB (ΔIsc = 2.7 ± 0.6 μA/cm2; n = 5; P < 0.01) but not by the CFTR inhibitor CFTRinh-172 (Fig. 4C). In Control-FRT cells, the endogenous Isc was slightly inhibited by GlyH-101 and DIDS, whereas CFTRinh-172, NFA, and NPPB had no effects (Fig. 4C). When CFTRinh-172, GlyH-101, and NFA were added sequentially, SLC26A9-mediated Isc was further inhibited when NFA was added in the presence of GlyH-101 indicating additivity of inhibitory effects on SLC26A9 Cl− channels (Fig. 4, D and E).
Fig. 4.
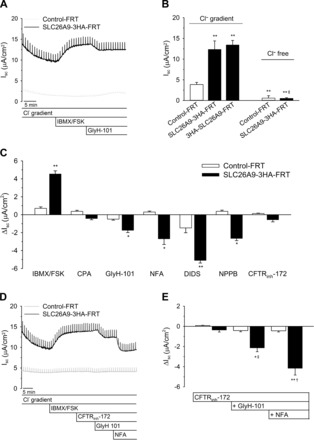
Characterization of transepithelial Cl− currents in SLC26A9 transduced FRT epithelial cell lines. A: representative original recordings of Ussing chamber measurements of SLC26A9-expressing FRT (SLC26A9-3HA-FRT) and Control-FRT monolayers sequentially exposed to IBMX/forskolin (FSK) and the Cl− channel inhibitor NFA in the presence of a Cl− gradient (0 mM Cl− apical/145 mM Cl− basolateral). B: summary of baseline short-circuit current (Isc) values across monolayers of COOH and NH2 terminally tagged SLC26A9-expressing FRT (3HA-SLC26A9-FRT and SLC26A9-3HA-FRT) and Control-FRT monolayers in the presence of a basolateral-to-apical Cl− gradient compared with Cl− free conditions. **P < 0.01 vs. Control-FRT (Cl− gradient); ‡P < 0.01 vs. basal Isc in SLC26A9-3HA-FRT cells (Cl− gradient). C: summary of individual effects of IBMX/FSK, cyclopiazonic acid (CPA), GlyH-101, NFA, DIDS, 5-nitro-2-(3-phenylpropylamino) benzoic acid (NPPB), and cystic fibrosis transmembrane regulator inhibitor (CFTRinh-172) in SLC26A9-3HA-FRT compared with Control-FRT cell monolayers in the presence of a Cl− gradient. *P < 0.05, **P < 0.01 vs. Control-FRT for each inhibitor; n = 9–21. D and E: representative original recordings (D) and summary (E) of Ussing chamber measurements of SLC26A9-expressing FRT (SLC26A9-3HA-FRT) and Control-FRT monolayers sequentially exposed to IBMX/FSK and the Cl− channel inhibitors CFTRinh-172, GlyH-101, and NFA in the presence of a Cl− gradient. *P < 0.05, **P < 0.01 vs. Control-FRT for each inhibitor; †P < 0.05, ‡P < 0.01 vs. SLC26A9-3HA-FRT under previous condition; n = 9.
Osmoregulation of SLC26A9 Cl− channels in FRT epithelial cell monolayers.
Next, we used SLC26A9-expressing FRT cell lines for initial studies of SLC26A9 Cl− channel regulation. Based on previous studies in Xenopus laevis oocytes and HEK293 cells, we hypothesized that SLC26A9 Cl− channels may contribute to epithelial volume regulation following osmotic challenge and that osmosensitive kinases such as WNK1, WNK4, and SPAK may be implicated in this process (8, 42). We, therefore, determined the effects of siRNA knockdown of these kinases, as well as hypotonic and hypertonic challenge conditions, on SLC26A9-mediated Cl− currents in FRT monolayers. Transcript analyses demonstrated that WNK1 and WNK4 were expressed at similar levels in Control-FRT and SLC26A9-3HA-FRT monolayers (Fig. 5A). Transcript levels of SPAK were reduced by ∼50% in SLC26A9-3HA-FRT cells (Fig. 5A). To test if these kinases were implicated in the regulation of SLC26A9-mediated Cl− secretion, we used siRNA knockdown in SLC26A9-3HA-FRT monolayers and determined the effects on transepithelial Isc in the presence of cAMP stimulation with IBMX and FSK. Knockdown efficiencies for WNK1, WNK4, and SPAK were determined by real time RT-PCR and ranged between ∼30 and 50% for specific siRNA, whereas scrambled control had no effect (Fig. 5A). Knockdown of WNK1 led to a significant reduction of SLC26A9-mediated Isc compared with mock-transfected SLC26A9-3HA-FRT cells, whereas treatment with WNK4- or SPAK-siRNA had no effects on Cl− transport in SLC26A9-3HA-FRT monolayers (Fig. 5B).
Fig. 5.
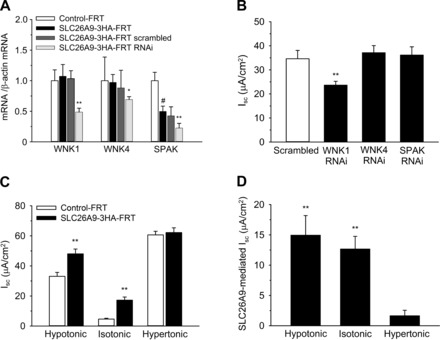
Osmoregulation of SLC26A9 in FRT epithelial cells. A: summary of transcript levels of WNK1, WNK4, and SPAK kinases in Control-FRT vs. SLC26A9-3HA-FRT cells after treatment with siRNA targeting the respective kinases or scrambled control. *P < 0.05; **P < 0.01 vs. SLC26A9-3HA-FRT; #P < 0.01 vs. Control-FRT; n = 4. B: effects of siRNA knockdown of WNK1, WNK4, and SPAK or scrambled control on transepithelial Isc across SLC26A9-3HA-FRT cell monolayers in the presence of cAMP stimulation with IBMX and FSK. **P < 0.01 vs. scrambled; n = 6–12. C: effect of hypotonic (220 mol/kgH2O) and hypertonic (480 mol/kgH2O) challenge on total Isc in Control-FRT and SLC26A9-3HA-FRT cell monolayers in the presence of IBMX and FSK. **P < 0.01 vs. Control-FRT; n = 6–12. D: comparison of SLC26A9-mediated Isc in SLC26A9-3HA-FRT cell monolayers under hypotonic, isotonic, and hypertonic conditions. Values reflect the difference of total Isc in the presence of IBMX and FSK between Control-FRT and SLC26A9-3HA-FRT cells. **P < 0.01 vs. hypertonic conditions; n = 6–12.
Next, we determined the effects of hypotonic (220 mol/kgH2O) and hypertonic (480 mol/kgH2O) solutions on SLC26A9-mediated Cl− transport in FRT cell monolayers. Exposure to both hypotonic and hypertonic solutions induced a large basal Isc in Control-FRT as well as SLC26A9-3HA-FRT monolayers (Fig. 5C). To determine the effects of osmotic challenge on SLC26A9-mediated Cl− conductance, experiments were performed in a paired fashion and Isc values obtained in Control-FRT monolayers were subtracted from Isc values obtained in SLC26A9-3HA-FRT monolayers. SLC26A9-mediated currents were largely abrogated under hypertonic conditions, whereas hypotonic conditions had no effect on SLC26A9-mediated Isc in FRT monolayers compared with studies in isotonic bath solutions (Fig. 5, C and D).
DISCUSSION
Emerging evidence suggests that the epithelial Cl− channel SLC26A9 may serve as a promising therapeutic target to compensate for deficient CFTR-mediated Cl− secretion in the airways and potentially other affected organs such as the pancreas and GI tract in patients with CF (2–6, 12, 15, 21, 34, 36). However, epithelial cell model systems with stable expression of SLC26A9 for studies of pharmacological regulation and drug discovery assays to identify SLC26A9 activators are lacking. In this study, we used retroviral transduction to generate FRT epithelial cells lines with stable expression of HA-tagged human SLC26A9. Furthermore, we performed a detailed biochemical and functional characterization of these novel SLC26A9-expressing FRT cell lines that will be important for future studies using this model to search for compounds that activate SCL26A9 Cl− channels and determine their mechanism of action at the molecular and cellular level.
Our biochemical studies demonstrated that both NH2 and COOH terminally tagged SLC26A9 FRT cell lines express two distinct protein bands reflecting the fully glycosylated form (∼120 kDa) and the immature form (∼95 kDa) of SLC26A9 (8, 11). In COOH terminally tagged SLC26A9 FRT cells, SLC26A9 transcript levels tended to be ∼1.4-fold higher and the 95-kDa band of the protein was increased compared with the NH2 terminally tagged SLC26A9 FRT cells. These data suggest that higher levels of SLC26A9 expression may lead to higher levels of immature protein in these cells. However, the levels of the 120-kDa band reflecting the mature SLC26A9 Cl− channel were similar in the two SLC26A9-expressing FRT cell lines (Fig. 1, A and C). Immunolocalization studies detected prominent intracellular localization of SLC26A9 with only little signal at the cell membrane (Fig. 2). However, surface biotinylation studies demonstrated that a fraction of mature SLC26A9 protein was inserted into the apical plasma membrane (Fig. 1D). SLC26A9 colocalized with the ER marker calnexin, the Golgi marker giantin, and the endosome marker EEA1, which is consistent with processing and the trafficking of the protein to these compartments (Fig. 2). Due to the lack of sensitive antibodies, the cellular localization of SLC26A9 in native human epithelial cells remains unknown. However, recent studies in mice demonstrated that type 2 airway inflammation results in induction of SLC26A9-mediated Cl− secretion but not SLC26A9 transcript levels in native murine airway epithelia (1, 2). These results are in line with posttranscriptional regulation of SLC26A9 in native mammalian epithelial cells and suggest that SLC26A9 Cl− channel function may be activated therapeutically by compounds that increase trafficking to the apical plasma membrane or compounds that activate SLC26A9 Cl− channels that are constitutively inserted into the plasma membrane.
Consistent with biochemical detection in the plasma membrane (Fig. 1), our functional studies demonstrated a robust constitutive Cl− channel function in SLC26A9-expressing FRT cells (Figs. 3 and 4). At the single cell level, Control-FRT cells exhibited a small outwardly rectifying Cl− conductance that may reflect endogenous expression of anoctamins (ANO6, ANO8, and ANO10) that were reported to be expressed in FRT cells (28). Both NH2 and COOH terminally tagged SLC26A9-FRT cells expressed constitutive Cl− conductances that were substantially greater in magnitude and inhibited by GlyH-101 and NFA (Fig. 3), an inhibitor profile consistent with previous studies of transient expression of SLC26A9 in different heterologous cells (5, 8). SLC26A9-3HA-FRT cells demonstrated a linear I–V relationship that is similar to previous results in HEK cells expressing SLC26A9 transiently (5). In 3HA-SLC26A9-FRT cells, the I–V curves showed somewhat outwardly rectifying membrane currents (Im) indicating that the position of the HA-tag may affect channel behavior (Fig. 3A). In epithelial monolayers, Control-FRT cells also exhibited a small constitutive Isc that was abolished under Cl− free conditions. Similar to patch-clamp studies in single cells, basal transepithelial Cl− currents were substantially greater (∼3 to 4-fold) in both NH2 and COOH terminally tagged SLC26A9-FRT monolayers compared with FRT control cells. Stimulation of cAMP signaling increased basal Cl− currents by 20–30% in Control-FRT and SLC26A9-expressing FRT monolayers (Fig. 4C) but not in single cell patch-clamp studies (data not shown). On the other hand, treatment of monolayers with CPA, a reversible inhibitor of the Ca2+-ATPase that blocks Ca2+ uptake into intracellular stores thereby increasing intracellular Ca2+ levels, had no effect on basal Cl− currents in control or SLC26A9-expressing FRT monolayers. When viewed in combination, we speculate that augmentation of SLC26A9-mediated Cl− currents by cAMP-dependent activation in FRT monolayers was mediated by activation of cAMP-dependent K+ channels that increased the electrochemical driving force for Cl− secretion (10, 17), rather than direct activation of SLC26A9 Cl− channels. Similar to SLC26A9 Cl− currents in single FRT cells, SLC26A9-mediated Cl− transport across FRT monolayers was inhibited by GlyH-101 and NFA. Interestingly, sequential addition of GlyH-101 and NFA showed additive inhibitory effects on SLC26A9-mediated Isc suggesting that these compounds may inhibit SLC26A9 Cl− channels via different binding sites (Fig. 4, D and E). In addition, SLC26A9-mediated Cl− transport in FRT monolayers was inhibited by the broad spectrum Cl− channel inhibitors DIDS and NPPB but not by CFTRinh-172. Although these blockers inhibit other Cl− channels including CFTR (30, 31), this inhibitor profile will be useful for future studies of SLC26A9 Cl− channel regulation in these novel FRT cell lines.
Previous reports implicated osmosensing kinases such as WNK1, WNK4, and SPAK in the regulation of surface expression and function of SLC26A9 and other transporters participating in electrolyte homeostasis including CFTR and the Na+-K+-2Cl− cotranspoter (NKCC) (9, 27, 42). Based on these results, we performed siRNA knockdown studies and determined effects of hypo- and hypertonic challenge on SLC26A9-mediated Cl− transport in FRT monolayers. Collectively, the results of these studies provide initial evidence that osmosensing kinases, in particular WNK1, and osmotic stress conditions participate in SLC26A9 regulation in FRT monolayers (Fig. 5).
Together, these novel SLC26A9-expressing FRT cells will be a useful model for further elucidation of the regulation of SLC26A9 Cl− channels in single cell as well as in the context of an epithelial monolayer, including intracellular trafficking and regulation of SLC26A9 Cl− channels inserted into the plasma membrane. Similar to FRT cells expressing mutant CFTR, these SLC26A9-expressing FRT cells may be used for functional high throughput screening assays for efficient testing of large chemical libraries. As exemplified by the rapid and highly successful development of clinical CFTR correctors and potentiators (25, 37, 38, 40), such FRT-based screening assays for SLC26A9 modulators should have a high potential to lead to the identification of compounds that activate SCL26A9 Cl− channel trafficking and/or open probability. SLC26A9 modulators may compensate for deficient CFTR function in the airways and other CF target tissues such as the GI tract and the pancreas and may be beneficial for all patients with CF independent of their CFTR genotypes. Furthermore, recent studies in mice with type 2 airway inflammation indicate that improvement of airway surface dehydration by activation of SLC26A9-mediated Cl− secretion may also be beneficial in other lung diseases associated with airway mucus hypersecretion and obstruction such as asthma and chronic obstructive pulmonary disease (2).
In summary, we generated FRT cell lines with stable expression of SLC26A9 Cl− channels. Furthermore, we performed detailed biochemical and functional analyses supporting that this novel cell model will be a useful for studies of pharmacological regulation and the identification of compounds that improve SLC26A9-mediated Cl− secretion in epithelial cells devoid of functional CFTR. Activation of SLC26A9 Cl− channels may compensate for deficient CFTR-mediated Cl− transport and improve mucosal surface hydration, which may be beneficial for patients with CF and potentially other muco-obstructive lung diseases.
GRANTS
This project was supported in part by the German Federal Ministry of Education and Research (01GM1110C, 82DZL00401, and 82DZL004A1 to M. A. Mall) and the Cystic Fibrosis Foundation (CFF BERTRA12G0 to C. A. Bertrand).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
J.J.S., S.S., X.W., J.F., C.A.B., and M.A.M. conception and design of research; J.J.S., S.S., X.W., and C.A.B. performed experiments; J.J.S., S.S., X.W., J.F., C.A.B., and M.A.M. analyzed data; J.J.S., S.S., X.W., J.F., C.A.B., and M.A.M. interpreted results of experiments; J.J.S., X.W., C.A.B., and M.A.M. prepared figures; J.J.S., J.F., and M.A.M. drafted manuscript; J.J.S., S.S., X.W., J.F., C.A.B., and M.A.M. edited and revised manuscript; J.J.S., S.S., X.W., J.F., C.A.B., and M.A.M. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank E. Sorscher (University of Alabama at Birmingham School of Medicine) for providing FRT epithelial cells, L. Galietta for providing the cDNA coding sequence of human HA-tagged SLC26A9, and H. Scheuermann (TLRC Cell Culture Laboratory), and M. Lampe (TLRC Microscopy Laboratory) for expert assistance.
REFERENCES
- 1.Anagnostopoulou P, Dai L, Schatterny J, Hirtz S, Duerr J, Mall MA. Allergic airway inflammation induces a pro-secretory epithelial ion transport phenotype in mice. Eur Respir J 36: 1436–1447, 2010. [DOI] [PubMed] [Google Scholar]
- 2.Anagnostopoulou P, Riederer B, Duerr J, Michel S, Binia A, Agrawal R, Liu X, Kalitzki K, Xiao F, Chen M, Schatterny J, Hartmann D, Thum T, Kabesch M, Soleimani M, Seidler U, Mall MA. SLC26A9-mediated chloride secretion prevents mucus obstruction in airway inflammation. J Clin Invest 122: 3629–3634, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Avella M, Loriol C, Boulukos K, Borgese F, Ehrenfeld J. SLC26A9 stimulates CFTR expression and function in human bronchial cell lines. J Cell Physiol 226: 212–223, 2011. [DOI] [PubMed] [Google Scholar]
- 4.Bakouh N, Bienvenu T, Thomas A, Ehrenfeld J, Liote H, Roussel D, Duquesnoy P, Farman N, Viel M, Cherif-Zahar B, Amselem S, Taam RA, Edelman A, Planelles G, Sermet-Gaudelus I. Characterization of SLC26A9 in patients with CF-like lung disease. Hum Mutat 34: 1404–1414, 2013. [DOI] [PubMed] [Google Scholar]
- 5.Bertrand CA, Zhang R, Pilewski JM, Frizzell RA. SLC26A9 is a constitutively active, CFTR-regulated anion conductance in human bronchial epithelia. J Gen Physiol 133: 421–438, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang MH, Plata C, Zandi-Nejad K, Sindić A, Sussman CR, Mercado A, Broumand V, Raghuram V, Mount DB, Romero MF. Slc26a9–anion exchanger, channel and Na+ transporter. J Membr Biol 228: 125–140, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, White GA, O'Riordan CR, Smith AE. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 63: 827–834, 1990. [DOI] [PubMed] [Google Scholar]
- 8.Dorwart MR, Shcheynikov N, Wang Y, Stippec S, Muallem S. SLC26A9 is a Cl− channel regulated by the WNK kinases. J Physiol 584: 333–345, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galietta LV, Jayaraman S, Verkman AS. Cell-based assay for high-throughput quantitative screening of CFTR chloride transport agonists. Am J Physiol Cell Physiol 281: C1734–C1742, 2001. [DOI] [PubMed] [Google Scholar]
- 10.Kunzelmann K, Mall M. Electrolyte transport in the mammalian colon: mechanisms and implications for disease. Physiol Rev 82: 245–289, 2002. [DOI] [PubMed] [Google Scholar]
- 11.Li J, Xia F, Reithmeier RA. N-glycosylation and topology of the human SLC26 family of anion transport membrane proteins. Am J Physiol Cell Physiol 306: C943–C960, 2014. [DOI] [PubMed] [Google Scholar]
- 12.Li W, Soave D, Miller MR, Keenan K, Lin F, Gong J, Chiang T, Stephenson AL, Durie P, Rommens J, Sun L, Strug LJ. Unraveling the complex genetic model for cystic fibrosis: pleiotropic effects of modifier genes on early cystic fibrosis-related morbidities. Hum Genet 133: 151–161, 2014. [DOI] [PubMed] [Google Scholar]
- 13.Liu X, Li T, Riederer B, Lenzen H, Ludolph L, Yeruva S, Tuo B, Soleimani M, Seidler U. Loss of Slc26a9 anion transporter alters intestinal electrolyte and HCO3− transport and reduces survival in CFTR-deficient mice. Pflügers Arch 467: 1261–1275, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lohi H, Kujala M, Makela S, Lehtonen E, Kestila M, Saarialho-Kere U, Markovich D, Kere J. Functional characterization of three novel tissue-specific anion exchangers SLC26A7, -A8, and -A9. J Biol Chem 277: 14246–14254, 2002. [DOI] [PubMed] [Google Scholar]
- 15.Loriol C, Dulong S, Avella M, Gabillat N, Boulukos K, Borgese F, Ehrenfeld J. Characterization of SLC26A9, facilitation of Cl− transport by bicarbonate. Cell Physiol Biochem 22: 15–30, 2008. [DOI] [PubMed] [Google Scholar]
- 16.Ma T, Vetrivel L, Yang H, Pedemonte N, Zegarra-Moran O, Galietta LJ, Verkman AS. High-affinity activators of cystic fibrosis transmembrane conductance regulator (CFTR) chloride conductance identified by high-throughput screening. J Biol Chem 277: 37235–37241, 2002. [DOI] [PubMed] [Google Scholar]
- 17.Mall M, Wissner A, Schreiber R, Kuehr J, Seydewitz HH, Brandis M, Greger R, Kunzelmann K. Role of K(V)LQT1 in cyclic adenosine monophosphate-mediated Cl− secretion in human airway epithelia. Am J Respir Cell Mol Biol 23: 283–289, 2000. [DOI] [PubMed] [Google Scholar]
- 18.Mall M, Gonska T, Thomas J, Hirtz S, Schreiber R, Kunzelmann K. Activation of ion secretion via proteinase-activated receptor-2 in human colon. Am J Physiol Gastrointest Liver Physiol 282: G200–G210, 2002. [DOI] [PubMed] [Google Scholar]
- 19.Mall MA, Galietta LJ. Targeting ion channels in cystic fibrosis. J Cyst Fibros 14: 561–570, 2015. [DOI] [PubMed] [Google Scholar]
- 20.Mall MA, Hartl D. CFTR: cystic fibrosis and beyond. Eur Respir J 44: 1042–1054, 2014. [DOI] [PubMed] [Google Scholar]
- 21.Miller MR, Soave D, Li W, Gong J, Pace RG, Boëlle PY, Cutting GR, Drumm ML, Knowles MR, Sun L, Rommens JM, Accurso F, Durie PR, Corvol H, Levy H, Sontag MK, Strug LJ. Variants in solute carrier SLC26A9 modify prenatal exocrine pancreatic damage in cystic fibrosis. J Pediatr 166: 1152–1157, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muanprasat C, Sonawane ND, Salinas D, Taddei A, Galietta LJ, Verkman AS. Discovery of glycine hydrazide pore-occluding CFTR inhibitors: mechanism, structure-activity analysis, and in vivo efficacy. J Gen Physiol 124: 125–137, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Namkung W, Park J, Seo Y, Verkman AS. Novel amino-carbonitrile-pyrazole identified in a small molecule screen activates wild-type and ΔF508 cystic fibrosis transmembrane conductance regulator in the absence of a cAMP agonist. Mol Pharmacol 84: 384–392, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pedemonte N, Lukacs GL, Du K, Caci E, Zegarra-Moran O, Galietta LJ, Verkman AS. Small-molecule correctors of defective ΔF508-CFTR cellular processing identified by high-throughput screening. J Clin Invest 115: 2564–2571, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Drevinek P, Griese M, McKone EF, Wainwright CE, Konstan MW, Moss R, Ratjen F, Sermet-Gaudelus I, Rowe SM, Dong Q, Rodriguez S, Yen K, Ordonez C, Elborn JS; VX08-770-102 Study Group. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med 365: 1663–1672, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rich DP, Anderson MP, Gregory RJ, Cheng SH, Paul S, Jefferson DM, McCann JD, Klinger KW, Smith AE, Welsh MJ. Expression of cystic fibrosis transmembrane conductance regulator corrects defective chloride channel regulation in cystic fibrosis airway epithelial cells. Nature 347: 358–363, 1990. [DOI] [PubMed] [Google Scholar]
- 27.Richardson C, Sakamoto K, de los Heros P, Deak M, Campbell DG, Prescott AR, Alessi DR. Regulation of the NKCC2 ion cotransporter by SPAK-OSR1-dependent and -independent pathways. J Cell Sci 124: 789–800, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schreiber R, Uliyakina I, Kongsuphol P, Warth R, Mirza M, Martins JM, Kunzelmann K. Expression and function of epithelial anoctamins. J Biol Chem 285: 7838–7845, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schuck S, Manninen A, Honsho M, Füllekrug J, Simons K. Generation of single and double knockdowns in polarized epithelial cells by retrovirus-mediated RNA interference. Proc Natl Acad Sci USA 101: 4912–4917, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scott-Ward TS, Li H, Schmidt A, Cai Z, Sheppard DN. Direct block of the cystic fibrosis transmembrane conductance regulator Cl− channel by niflumic acid. Mol Membr Biol 21: 27–38, 2004. [DOI] [PubMed] [Google Scholar]
- 31.Sheppard DN. CFTR channel pharmacology: novel pore blockers identified by high-throughput screening. J Gen Physiol 124: 109–113, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sheppard DN, Carson MR, Ostedgaard LS, Denning GM, Welsh MJ. Expression of cystic fibrosis transmembrane conductance regulator in a model epithelium. Am J Physiol Lung Cell Mol Physiol 266: L405–L413, 1994. [DOI] [PubMed] [Google Scholar]
- 33.Snyder PM. Liddle's syndrome mutations disrupt cAMP-mediated translocation of the epithelial Na+ channel to the cell surface. J Clin Invest 105: 45–53, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Soave D, Miller MR, Keenan K, Li W, Gong J, Ip W, Accurso F, Sun L, Rommens JM, Sontag M, Durie PR, Strug LJ. Evidence for a causal relationship between early exocrine pancreatic disease and cystic fibrosis-related diabetes: a Mendelian randomization study. Diabetes 63: 2114–2119, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stutts MJ, Fitz JG, Paradiso AM, Boucher RC. Multiple modes of regulation of airway epithelial chloride secretion by extracellular ATP. Am J Physiol Cell Physiol 267: C1442–C1451, 1994. [DOI] [PubMed] [Google Scholar]
- 36.Sun L, Rommens JM, Corvol H, Li W, Li X, Chiang TA, Lin F, Dorfman R, Busson PF, Parekh RV, Zelenika D, Blackman SM, Corey M, Doshi VK, Henderson L, Naughton KM, O'Neal WK, Pace RG, Stonebraker JR, Wood SD, Wright FA, Zielenski J, Clement A, Drumm ML, Boëlle PY, Cutting GR, Knowles MR, Durie PR, Strug LJ. Multiple apical plasma membrane constituents are associated with susceptibility to meconium ileus in individuals with cystic fibrosis. Nat Genet 44: 562–569, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van Goor F, Hadida S, Grootenhuis PD, Burton B, Cao D, Neuberger T, Turnbull A, Singh A, Joubran J, Hazlewood A, Zhou J, McCartney J, Arumugam V, Decker C, Yang J, Young C, Olson ER, Wine JJ, Frizzell RA, Ashlock M, Negulescu P. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci USA 106: 18825–18830, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van Goor F, Hadida S, Grootenhuis PD, Burton B, Stack JH, Straley KS, Decker CJ, Miller M, McCartney J, Olson ER, Wine JJ, Frizzell RA, Ashlock M, Negulescu PA. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci USA 108: 18843–18888, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van Goor F, Yu H, Burton B, Hoffman BJ. Effect of ivacaftor on CFTR forms with missense mutations associated with defects in protein processing or function. J Cyst Fibros 13: 29–36, 2014. [DOI] [PubMed] [Google Scholar]
- 40.Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, Cipolli M, Colombo C, Davies JC, De Boeck K, Flume PA, Konstan MW, McColley SA, McCoy K, McKone EF, Munck A, Ratjen F, Rowe SM, Waltz D, Boyle MP; TRAFFIC Study Group; TRANSPORT Study Group . Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med 373: 220–231, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu J, Song P, Miller ML, Borgese F, Barone S, Riederer B, Wang Z, Alper SL, Forte JG, Shull GE, Ehrenfeld J, Seidler U, Soleimani M. Deletion of the chloride transporter Slc26a9 causes loss of tubulovesicles in parietal cells and impairs acid secretion in the stomach. Proc Natl Acad Sci USA 105: 17955–17960, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang CL, Liu X, Paliege A, Zhu X, Bachmann S, Dawson DC, Ellison DH. WNK1 and WNK4 modulate CFTR activity. Biochem Biophys Res Commun 353: 535–540, 2007. [DOI] [PubMed] [Google Scholar]
- 43.Yu H, Burton B, Huang CJ, Worley J, Cao D, Johnson JP Jr, Urrutia A, Joubran J, Seepersaud S, Sussky K, Hoffman BJ, Van Goor F. Ivacaftor potentiation of multiple CFTR channels with gating mutations. J Cyst Fibros 11: 237–245, 2012. [DOI] [PubMed] [Google Scholar]