

Abstract
Hermansky-Pudlak syndrome (HPS) is a rare autosomal recessive disorder, and some patients with HPS develop pulmonary fibrosis, known as HPS-associated interstitial pneumonia (HPSIP). We have previously reported that HPSIP is associated with severe surfactant accumulation, lysosomal stress, and alveolar epithelial cell type II (AECII) apoptosis. Here, we hypothesized that defective autophagy might result in excessive lysosomal stress in HPSIP. Key autophagy proteins, including LC3B lipidation and p62, were increased in HPS1/2 mice lungs. Electron microscopy demonstrated a preferable binding of LC3B to the interior of lamellar bodies in the AECII of HPS1/2 mice, whereas in wild-type mice it was present on the limiting membrane in addition to the interior of the lamellar bodies. Similar observations were noted in human HPS1 lung sections. In vitro knockdown of HPS1 revealed increased LC3B lipidation and p62 accumulation, associated with an increase in proapoptotic caspases. Overexpression of LC3B decreased the HPS1 knockdown-induced p62 accumulation, whereas rapamycin treatment did not show the same effect. We conclude that loss of HPS1 protein results in impaired autophagy that is restored by exogenous LC3B and that defective autophagy might therefore play a critical role in the development and progression of HPSIP.
Keywords: autophagy, Hermansky-Pudlak syndrome, alveolar epithelial cells, Hermansky-Pudlak syndrome-associated interstitial pneumonia, apoptosis, lung fibrosis
hermansky-pudlak syndrome (HPS), a rare genetic disorder, is typically characterized by platelet dysfunction, occulocutaneous albinism, accumulation of ceroid lipofuscin in lysosomes, granulomatous colitis, and pulmonary fibrosis (16). Clinical diagnosis of HPS is established by hair and skin hypopigmentation, ocular abnormalities, and microscopic visualization of platelets revealing absence of dense bodies (16). Until now, HPS has been most frequently characterized among patients of Puerto Rican descent (2), but the disease is prevalent all over the world. Mutations in HPS genes are primarily known to affect the regulation and function of various lysosome-related organelles (LROs) such as platelet-dense granules, pigment cell melanosomes, and lung lamellar bodies (8, 12).
Several HPS mutations have been described previously, but pulmonary fibrosis known as HPS-associated interstitial pneumonia (HPSIP) seems to occur mostly in HPS1, HPS2, and HPS4, each encoding a different gene (1, 9, 17). HPSIP is a leading cause of death in these patients usually in the fourth or fifth decade of their life (9). Lungs of patients with HPSIP are typically characterized by swelling of alveolar epithelial type II cells (AECII) with an increase in size and number of lamellar bodies, termed “giant lamellar body degeneration” (34). Giant lamellar bodies are also observed within the AECII of HPS1/2 double-mutant mice (18), and we have previously reported that these mice spontaneously develop lung fibrosis associated with AECII-specific cellular stress and apoptosis (30). On the other hand, mice with mutations either in HPS1 or HPS2 genes show an increased predisposition to pulmonary fibrosis upon bleomycin challenge (56).
Whereas HPS2 encodes the β-subunit of the adaptor protein-3 (AP-3) that sorts proteins from endosomes to LROs (40), HPS1 and HPS4 proteins are subunits of the biogenesis of LRO complex-3 (BLOC-3), which plays a role in the distribution of lysosomes and LROs (15). Such effects of HPS gene products on lysosomes was also supported by a study in the lungs of HPS1/2 mice, which demonstrated severe defects in surfactant secretion from the lamellar bodies (18), which are the LROs of the AECII. In the same vein, we previously reported accumulation of surfactant proteins associated with severe lysosomal stress and apoptosis of AECII in HPS1/2 mice as well as in a patient with HPS1 with interstitial pneumonia (30). On the basis of these observations, we hypothesized that these lysosome-associated disturbances in HPSIP might be a result of altered autophagy, a basic homeostasis mechanism of a cell that degrades long-lived proteins and organelles via lysosomes (14) to promote cellular homeostasis. Both selective (4) and nonselective forms (45) of autophagy exist. Classically, autophagy is divided into macroautophagy, microautophagy, and chaperone-mediated autophagy (33). Autophagy basically involves formation of double-membrane structures called autophagosomes, which carry long-lived proteins or dysfunctional organelles in their lumen to fuse with lysosomes and degrade their contents (25). This is a dynamic process and requires the coordinated function of several autophagy-related (Atg) gene products (33). Microtubule-associated protein 1 light chain-3β (MAP1LC3B; referred to as LC3B hereafter) is one of the important autophagy-related proteins, and its lipidated form (LC3BII) is a marker for the autophagosomes (50, 51). It interacts with sequestosome 1 (SQSTM1)/p62, a substrate for autophagy that carries the cargo to the autophagosomes for degradation (38). Autophagy is tightly regulated in the cells at a basal level, and any functional defect in this degradation pathway results in the intracellular accumulation of toxic substrates, which has deleterious effects on various biological processes (44, 55). Dysfunctional autophagy has been indicated to play an important role in the development of several pathologies, including lysosomal storage diseases (LSDs) (10, 37, 46), neurodegenerative diseases (23), and several organ-specific diseases, including lung fibrosis (3, 39).
Because lysosomal stress has been reported in HPSIP, we hypothesized that autophagy pathway plays a key role in the development and progression of HPSIP. Hence, we aimed to study this pathway in HPS1/2 mice, HPS1 human lungs, as well as to study the in vitro effects of HPS1 knockdown on autophagy.
MATERIALS AND METHODS
Mice.
Breeding pairs of HPS1 and HPS2 monomutant mice were purchased from the Jackson Laboratory, whereas the HPS1/2 double-mutant mice breeding pairs were received as a gift from Richard Swank (Roswell Park, Buffalo, NY). Background strain for these mice was C57BL/6J. All mice were kept under pathogen-free conditions and were genotyped as described before (30). Both the University Animal Care Committee and the Federal Authorities for Animal Research of the Regierungspraesidium Giessen (Hessen, Germany) approved the study protocol.
Human lung sections, immunohistochemistry, and Western blot.
Written, informed consent was obtained, and subjects were enrolled in a protocol (04-HG-0211) approved by the Institutional Review Board of the National Human Genome Research Institute. For human subjects with HPS1, tissue was procured from a postmortem lung specimen and an explanted lung sample. Information about lungs from organ donors (used as controls) has been described before (24). All specimens from human and mice were fixed in 4% formaldehyde and embedded in formalin, and serial sections (3–6 μm) were performed. Immunostaining was performed using ZytoChem plus Broad Spectrum (AP-Fast red and HRP-DAB, Zytomed Systems) kit according to the manufacturer's instructions and as described before (30) for immunohistolocalization of LC3B (Abcam) and SQSTM1/p62 (Sigma-Aldrich). Stained sections were scanned with Hamamatsu scanner (Nanozoomer 2.0 RS). NDP.view2 software was used to make the images and analysis. Lung homogenates and cell lysates were denatured and subjected to Western blotting as described previously (30) to detect LC3B, green fluorescent protein (GFP), GAPDH, ATG5, myc (Abcam), p62 and ATG7 (Sigma-Aldrich), transcription factor EB (TFEB) (Proteintech), and lysosome-associated membrane protein 2 (LAMP2) (Santa Cruz Biotechnology).
Cloning, cell culture, and siRNA transfection.
Full-length LC3B (gene bank accession number: NM_026160) was cloned into pEGFP-C1 expression vector and pCMV-myc-Tag3 (Agilent Technologies). Standard protocols were used for cloning, and A549 cells were transfected with GFP-LC3B or myc-LC3B plasmids. Cells were harvested after the indicated time points to isolate protein or RNA for further analysis. A549 cells were transfected with 50 nM human HPS1 siRNA (Santa Cruz Biotechnology) or scrambled siRNA (Dharmacon) in serum-free medium and cultured for the indicated time in the same medium without antibiotics followed by harvesting them for protein or RNA isolation.
Immunofluorescence.
Immunofluorescence was performed according to the previously described protocols (29). Briefly, following plasmid or siRNA transfection for the indicated time points, cells were washed with ice-cold PBS and fixed in 4% paraformaldehyde. Permeabilization was performed with 0.1% Triton X-100, and nonspecific binding was blocked with 10% donkey serum followed by overnight incubation with primary antibodies against cleaved caspase-3 (Cell Signaling Technology), LC3B, LAMP1, caspase-8 (Abcam), and p62 (Sigma-Aldrich). Slides were then washed and incubated with respective Alexa Fluor probes (Alexa Fluor 488 or 555, Life Technologies) raised in donkey. Microscopy was performed using a Leica M205 FA fluorescent stereoscope (Leica Microsystems) equipped with a Leica DFC360 FX camera, and all images were captured using an ×63 objective lens. Immunofluorescence images were analyzed using Leica Application Suite Advanced Fluorescence (LAS AF) software, version 4.3. Quantification of the immunofluorescence images was performed using the JACOP plugin from ImageJ to calculate Pearson's coefficient (7). Briefly, for quantification, 15–30 random regions per well were imaged and subjected to the JACOP plugin. Image-acquisition parameters were kept constant for all images during the process of obtaining images. Pearson's coefficient of colocalization was calculated using Costes automatic threshold method for each of these pair of images having two different fluorophores.
PCR and RT-PCR.
Cells were harvested, and total RNA was isolated (RNeasy Plus Mini Kit, Qiagen). RNA was reverse transcribed to cDNA (Omniscript RT Kit, Qiagen) and then subjected to PCR (HotstarTaq Polymerase PCR Kit, Qiagen) using primers for human HPS1, β-actin, GAPDH, and p62 with gene bank accession numbers NM_000195.3, NM_001101.3, NM_002046.5, and NM_003900.4 respectively, using the following primers: HPS1: forward: 5'GGACTTCTTGCTGGTGAAGAG3′, reverse: 5′CATCTGGAGTTTGTACCCCATG3′; β-actin: forward: 5′ACCCTGAAGTACCCCATCG3′, reverse: 5′CAGCCTGGATAGCAACGTAC3′; p62: forward: 5′TGGACCCATCTGTCTTCAAA3′, reverse: 5′ TCTGGGAGAGGGACTCAATC3′; GAPDH: forward: 5′ACCCAGAAGACTGTGGATGG3′, reverse: 5′GTGTCGCTGTTGAAGTCAGAG3′.
Electron microscopy and distribution pattern of immunogold labeling for LC3B in control and HPS1/2 mice.
A perfusion fixation of mice lungs was performed using a multipurpose fixation solution (4% paraformaldehyde, 0.1% glutaraldehyde in 0.2 M HEPES buffer) according to recently published methods (53). Three lungs each [wild-type (WT) vs. HPS1/2] were subjected to a systematic uniform sampling (52) and embedded in lowicryl resin following a freeze substitution procedure. Immunogold labeling for LC3B (Abcam) was carried out as described before (29). Quantitative analysis of gold labeling was performed to determine cellular compartments in which a preferential labeling exists. As advocated by the current standards for quantitative evaluation of lung structures, we determined the relative labeling index (RLI) followed by χ2 analysis for statistical assessment of distribution pattern (19, 32). A systematic uniform area sampling was performed to sample AECII. Three lungs for each group (WT vs HPS1/2) were included, and two sections per lung were studied. The following compartments were defined, and volume fractions (Vv) and surface densities within AECII cells were determined by point and intersection counting: lumen of lamellar body, the limiting membrane (area 100 nm around the limiting membrane), mitochondria, nucleus, and cytosol. In addition, gold particles (Ngold observed) in these five compartments were counted. Supplemental Fig. S1 demonstrates examples of gold particles in different compartments of the cell (supplemental material for this article is available online at the American Journal of Physiology Lung Cellular and Molecular Physiology website). Gold particles were seen also in cytosol and counted accordingly. The cell size of AECII cells was much larger in HPS1/2 mice than in WT mice, and volume fractions of different compartments differed extensively (Table 1). For instance, the volume fraction of lamellar bodies in AECII was 61% in HPS1/2 and 13% in WT. Using volume fractions, the expected number of gold particles per subcellular compartment (Ngold expected) was calculated by multiplication of the sum of all counted gold particles in AECII cells and the volume fraction of the subcellular compartment. Ngold expected is the number of particles we would have counted in the different compartments if labeling was random. Ngold observed and Ngold expected were used in a further step to calculate the RLI as follows: RLI = Ngold observed/Ngold expected.
Table 1.
Stereological analysis of LC3B labeling
| Compartment | Ngold observed | Vv(comp/cell) | Ngold expected | RLI |
|---|---|---|---|---|
| WT | ||||
| LB | 246 | 0.13 | 54 | 4.55 |
| LM | 67 | 0.04 | 16 | 3.69 |
| Mitochondria | 15 | 0.16 | 66 | 0.22 |
| Nucleus | 4 | 0.19 | 79 | 0.05 |
| Cytosol | 84 | 0.49 | 203 | 0.41 |
| Total | 416 | 1 | 418 | |
| HPS1/2 | ||||
| LB | 1,830 | 0.61 | 1,158 | 1.58 |
| LM | 27 | 0.04 | 71 | 0.38 |
| Mitochondria | 0 | 0.01 | 18 | 0 |
| Nucleus | 6 | 0.05 | 100 | 0.06 |
| Cytosol | 34 | 0.30 | 566 | 0.06 |
| Total | 1,897 | 1 | 1,913 | |
In wild-type (WT) mice, the number of observed gold particles (Ngold observed) is larger than the expected number of gold particles (Ngold expected) in lamellar bodies (LB) and the limiting membrane (LM). Accordingly, the relative labeling index (RLI) is >1 in these compartments, suggesting a nonrandom distribution of gold particles and a preferable binding to LM and LB. To test the hypothesis that the distribution is nonrandom, a χ2 test was added. With a total χ2 of 997 and a degree of freedom (dF) of 4, the null hypothesis of random distribution must be rejected (P < 0.05). In HPS1/2 mice, Ngold observed > Ngold expected was found in LB but not in LM. Hence the RLI is > 1 in LB but < 1 in LM so that a preferable binding can be attributed to the LB but not to the LM. A total χ2 of 1013 and a dF of 4 results in a P < 0.05 so that the null hypothesis of random distribution of gold particles must be rejected. LC3B, light chain 3β; HPS, Hermansky-Pudlak syndrome.
Statistics.
For statistical comparison of the difference between groups, one-way ANOVA was applied followed by Bonferroni's multiple comparison as a posttest. GraphPad Prism 5.0 was employed. P values were as follows: *P < 0.05, **P < 0.01, and ***P < 0.001. Distribution patterns of immunogold labeling in different ultrastructural compartments of AECII were assessed using χ2 analysis and a degree of freedom of 4 (in view of 5 defined ultrastructural compartments).
RESULTS
Defective autophagy in HPSIP.
To study the regulation of autophagy under HPSIP conditions, we first analyzed the lungs of 3- and 9-mo-old HPS1/2 mice. Whole-lung homogenates from these mice were subjected to Western blotting of key autophagy marker proteins. Compared with age-matched WT mice lungs as well as HPS1 and HPS2 monomutant mice, we observed an increase in LC3BII, the lipidated form of LC3B, indicating the formation of autophagosomes in HPS1/2 mice lungs (Fig. 1A). Supporting this, HPS1/2 mice lung homogenates (both 3 and 9 mo old) showed an increase in the levels of autophagy proteins p62, Atg7, and Atg5 and the regulator of lysosomal biogenesis, TFEB. Accumulation of p62 together with an increase in LC3BII is already indicative of a defective autophagy pathway. Interestingly, an increase in p62 was observed in the older HPS1 mice (9 mo) but not in the younger HPS1 mice (3 mo). Correspondingly, LAMP2, a lysosomal marker protein, was increased in both the double-mutant as well as in the monomutant HPS mice compared with age-matched controls (Fig. 1A).
Fig. 1.

Defective autophagy in the alveolar epithelial type II cells (AECII) of Hermansky-Pudlak syndrome (HPS)1/2 mice. A: 3- and 9-mo-old HPS1/2, HPS1, HPS2, and wild-type (WT) mice lung homogenates were subjected to Western blotting to detect the indicated markers of autophagy. B: immunogold labeling for light chain 3β (LC3B) on lung sections of HPS1/2 and WT mice. Lamellar bodies (LBs) within AECII of WT or HPS1/2 mice lungs are depicted here. Arrow heads indicate LC3B gold labeling on the limiting membrane of LBs in WT mice. *LC3B gold labeling within the lumen of LBs in HPS1/2 mice lungs. LM, lamellar membrane of LBs. C and D: serial sections of HPS1/2 and WT mice lungs were stained for p62 and prosurfactant protein C (SP-C). Original magnification: ×200; scale bar = 100 μm (C). High-magnification images for the selected regions are depicted in D. Arrows indicate p62-positive AECII in HPS1/2 mice lung sections. Original magnification: ×400; scale bar = 50 μm (D). Representative images from 3 different experiments are shown with n = 3–5 mice. ATG, autophagy-related gene protein; TFEB, transcription factor EB; Lamp, lysosome-associated membrane protein.
Furthermore, we performed transmission electron microscopy on HPS1/2 and WT mice lungs. Immunogold labeling for LC3B was performed on a qualitative basis. Gold particles were found in lamellar bodies but occasionally also in other compartments, such as cytosol and mitochondria in both WT and HPS1/2 mice. However, whereas in WT mice gold particles were often observed in close proximity to the limiting membrane of lamellar bodies, this was not the case in HPS1/2 mice (Fig. 1B), in which the label was almost exclusively found inside the lamellar bodies. Hence, the distribution pattern of labeling for LC3B appeared to differ between WT and HPS1/2. To detect the preferential binding to AECII compartments in dependency of the genotype on a statistical basis, we determined the RLI, followed by the χ2 analyses to test the distribution of labeling for randomness (Table 1), as advocated by the ATS/ERS standards for quantitative evaluation of lung structures (19). On the basis of the RLI > 1 and χ2 analysis with a P < 0.05 (degree of freedom = 4), a preferential labeling for LC3B was found in the lamellar body as well as the limiting membrane in WT mice, whereas, in HPS1/2, a preferential binding of LC3B to the lamellar body, but not the limiting membrane, was encountered (RLI < 1). These data clearly demonstrate a different subcellular distribution of LC3B in WT vs. HPS1/2 mice (Fig. 1D, Table 1). Table 1 demonstrates a high number of observed gold particles in lamellar bodies, which is an increase of more than sevenfold compared with WT mice. The larger number of observed gold particles in lamellar bodies is above all a consequence of enlarged cells and enlarged lamellar bodies, which contribute 62% to the volume of AECII compared with WT mice, in which the lamellar bodies are smaller and contribute to only 13% of the volume of AECII (Table 1).
We next performed immunohistochemistry for p62 and surfactant protein C on serial lung sections of HPS1/2 and WT mice lung sections. Interestingly, we observed an intense staining for p62 within the AECII of HPS1/2 mice lung sections (Fig. 1, C and D), reflecting defective autophagy. Supporting these observations, AECII of one patient with HPS1 also displayed an increased staining for p62 (Fig. 2, A and B). Results until this point indicated an accumulation of p62 within the AECII in the lungs of HPS1/2 mice and patient with HPS1, pointing toward defective autophagy under conditions of HPS-associated lung fibrosis.
Fig. 2.

Defective autophagy in AECII of lung of a patient with HPS1. Serial lung sections from 1 patient with HPS-1 and healthy donor were immunostained either for p62 or pro-SP-C. A: original magnification: ×200; scale bar = 100 μm. B: high-magnification images for the selected regions are shown, and arrows indicate p62-positive AECII in the lung sections of a patient with HPS1. Scale bar = 50 μm. C: low- and high-magnification images for negative control in which primary antibody was omitted are shown. Representative images from 3 independent experiments are shown.
Knockdown of HPS1 in A549 cells leads to defective autophagy and induces upregulation of apoptosis-related proteins.
To further examine the effect of the loss of HPS1 gene on autophagy in vitro, we performed siRNA-mediated knockdown of HPS1 in the A549 cell line. After 24 h of transfection, knockdown of HPS1 led to vacuolization in A549 cells, a feature that was not so prominent in scrambled or in untreated cells (Fig. 3, A–C). Supporting this observation, Western blot analysis showed a prominent and significant increase in both LC3BII and p62 in A549 cells following HPS1 knockdown for 24 h (Fig. 3, D–F) and 48 h (not shown). It has been suggested that the transcriptional activation of p62 might counteract the degradation of p62 protein via autophagy, which might lead to either steady-state levels of p62 protein or, sometimes, an increase in p62 protein levels (41). In such cases, p62 may not be used as a marker for autophagy-mediated clearance. Hence, to identify whether the increase in p62 protein level was a mere reflection of its transcriptional regulation, we analyzed the transcript levels of p62 in untreated cells or cells transfected with scrambled siRNA or HPS1 siRNA. As indicated in Fig. 3, G and H, semiquantitative PCR showed that the transcription of p62 is not upregulated following HPS1 knockdown in A549 cells. Taken together, our results clearly indicate a defect in autophagy pathway, as evident by simultaneous LC3B and p62 accumulation attributable to loss of functional HPS1 protein in vitro.
Fig. 3.
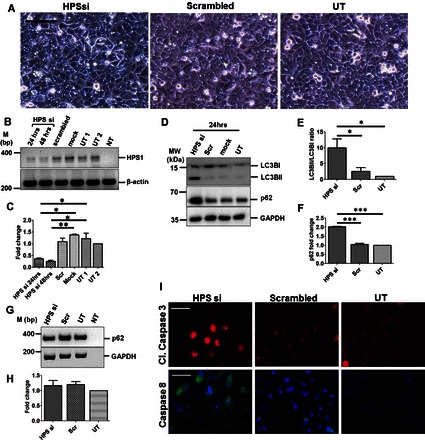
siRNA-mediated knockdown of HPS1 in A549 cells results in defective autophagy and increased apoptosis. A: phase-contrast images of A549 cells after 24 h of transfection with HPS1 or scrambled siRNA or untransfected cells (UT). Scale bar = 100 μm. B: representative agarose gel image of semiquantitative PCR performed for HPS1 and β-actin from cDNA prepared from the RNA isolated from untransfected or HPS1 or scrambled or mock-transfected A549 cells. β-Actin was used as a housekeeping gene. C: densitometric quantification of HPS1 transcript levels after HPS1 knockdown compared with scrambled, mock, and untreated cells. D: representative Western blot images for LC3B, p62, and GAPDH from cell lysates prepared from untransfected, mock, or scrambled (scr) siRNA or HPS1 siRNA-transfected A549 cells after 24 h. E and F: graphical representation of densitometry analysis from Western blots shown in D is shown as ratio of LC3BII/LC3BI (E) and fold change in p62 protein level (F) compared with untransfected controls. *P < 0.05, **P < 0.01, ***P < 0.001. G: representative agarose gel image from semiquantitative RT-PCR for p62 is shown. GAPDH was used as a housekeeping gene. H: quantification of p62 transcript levels is represented as fold change compared with untransfected controls. I: representative immunofluorescence images for cleaved caspase-3 (red) and caspase-8 (green) following HPS1 or scrambled siRNA transfection or untransfected cells. DAPI (blue) was used to stain cell nuclei. Scale bar = 10 μm. Western blot images and immunofluorescence images are represented here from n = 3 independent experiments. UT1, untransfected sample 1; UT2, untransfected sample 2; NT, no template sample.
In line with our previous observations (30), we observed severe cell death after 24 h of HPS1 knockdown that was already evident under the cell culture microscope. Autophagy has been recently shown to regulate the clearance of active caspases, especially caspase-8 (57). We therefore analyzed caspase-8 and caspase-3 levels following HPS1 knockdown. As evident from Fig. 3I, HPS1 knockdown led to an increased staining of the apoptosis markers, cleaved caspase-3 and caspase 8, compared with scrambled siRNA transfected or untransfected cells.
Overexpression of LC3B before HPS1 knockdown protects cells against p62 accumulation.
On the basis of the previous results, we hypothesized that autophagy flux might be decreased following HPS1 knockdown. To test this, we performed coimmunofluorescence for the autophagosome marker LC3B and the lysosomal marker LAMP1 following knockdown of HPS1 gene alongside with scrambled siRNA-transfected or untreated cells to study whether the fusion event between autophagosomes and lysosomes occurs following HPS1 knockdown. Interestingly, we observed primarily nuclear as well as cytosolic localization of LC3B, and this LC3B signal did not colocalize with LAMP1 (Fig. 4A). Quantification of the fluorescence signals also revealed a significant decrease in colocalization between LC3B and LAMP1 (Fig. 4B), again implicating defective autophagic degradation in cells with HPS1 knockdown, whereas a diffuse staining for LC3B and normal LAMP1 distribution were observed in untreated cells or scrambled siRNA-transfected cells. Generally, use of autophagy inhibitors like bafilomycin A1 or chloroquine is recommended to study autophagy flux on a biochemical level. However, according to the guidelines of autophagy (21), prolonged exposure (>8 h) of cells to these inhibitors is not recommended because of lysosomal leakage. Hence, we aimed to assess the GFP-LC3 cleavage, which was also suggested to study the flux. In this approach, the LC3 part of the protein degrades more rapidly than the GFP, and detection of the GFP fragment alone indicates an increased autophagy flux (21). We therefore transfected A549 cells with GFP-LC3B for 24 h followed by the transfection of scrambled siRNA or HPS1 siRNA for another 24 h. Following HPS1 knockdown, the GFP-LC3B colocalized with LAMP1 (Fig. 4, C and D), and, using anti GFP antibody, we observed free GFP fragment in cells transfected with GFP-LC3B and HPS1 siRNA, whereas bands corresponding to GFP-LC3B were observed in all samples transfected with the plasmid (Fig. 5A). The detection of cleaved GFP fragment in GFP-LC3B and HPS1 siRNA-transfected cells indicated an increase in autophagy flux. However, one striking observation that we noted from these experiments is that cells transfected with GFP-LC3B and HPS1 siRNA did not display the kind of severe vacuolization compared with HPS1 siRNA transfection alone (Fig. 5B). To understand the mechanism behind this observation, we transfected cells with GFP-LC3B, followed by HPS1 siRNA or scrambled siRNA or left them untreated. As indicated in Fig. 5C, cells transfected with GFP-LC3B showed a decrease in HPS1 knockdown-induced LC3B (endogenous) as well as p62 accumulation (Fig. 5, C and D). This to us indicated that the GFP cleavage as observed in Fig. 5A was a result of the activation of autophagy because of GFP-LC3B transfection per se, a feature that is not observed upon HPS1 knockdown alone. To further confirm that this induction of autophagy was due to overexpression of LC3B and not due to GFP, we transfected cells with myc-LC3B plasmid, followed by transfection with HPS1 siRNA or scrambled siRNA or left them untreated with or without myc-LC3B transfection. We observed that cells transfected with myc-LC3B also showed a decrease in accumulation of p62 (Fig. 5, E and F), indicating that the observed effects are LC3B specific.
Fig. 4.

Endogenous LC3B and LAMP1 do not colocalize following HPS1 knockdown. A: A549 cells were either left untreated or were transfected with HPS1 siRNA or scrambled siRNA for 24 h followed by immunofluorescence for endogenous LC3B (red) and LAMP1 (green). B: quantification of the LC3B-LAMP1 colocalization signal is depicted as Pearson's coefficient. ***P < 0.001. C: A549 cells were transfected with GFP-LC3B plasmid for 24 h and were left untreated or transfected with HPS1 siRNA or scrambled siRNA for another 24 h followed by immunofluorescence for LAMP1. DAPI was used to stain the nuclei. Scale bar = 10 μm. D: quantification of the colocalization signals of LC3B with LAMP1 is depicted as Pearson's coefficient. ns, not significant. Representative images from n = 3 independent experiments are shown.
Fig. 5.

Overexpression of LC3B reduces HPS1 knockdown-induced p62 accumulation. A: A549 cells transfected with green fluorescent protein (GFP)-LC3B were left either untransfected or transfected with mock, HPS1, or scrambled siRNA for another 24 h. Cell lysates were subjected to Western blotting using anti-GFP antibody. Empty GFP plasmid-transfected cells were taken as controls to detect free GFP. *Bands corresponding to free GFP. B: phase-contrast images of cells indicating vacuolar structures in HPS1 siRNA transfection that are not detectable in GFP-LC3 and HPS1 siRNA-transfected cells. C–F: A549 cells were transfected with GFP-LC3B (C and D) or myc-LC3B (E and F) followed by transfection with scrambled or HPS1 siRNA for 24 h. Western blots and densitometry quantification for the indicated proteins show fold change compared with untransfected controls (D and F). G and H: representative Western blot images depicting p62 and GAPDH (G) and quantification of p62 Western blot (H) from cells transfected with HPS1 or scrambled siRNA, which were treated with 100 mM rapamycin. *P < 0.05. Representative images and analysis from n = 3 independent experiments are shown.
It is well documented that rapamycin is an inhibitor of mammalian target of rapamycin, which leads to the activation of autophagy (6). We hence asked whether activating the autophagy pathway with rapamycin in HPS1 knockdown cells would also exert similar effects like LC3B overexpression. We treated the cells with 100 nM rapamycin followed by HPS1 siRNA or scrambled siRNA transfection or left them untransfected. Cells treated with rapamycin did show an increase in LC3BII levels (not shown), but there was no decrease in HPS1 knockdown-induced p62 accumulation following rapamycin treatment (Fig. 5, G and H). This readout indicated that rapamycin treatment did not reverse or attenuate the p62 accumulation induced by HPS1 knockdown.
DISCUSSION
In the present study, we document that the autophagy pathway is defective in HPS1/2 mice as well as in human patients with HPS1. In addition, we report that in vitro knockdown of HPS1 gene in A549 cells results in apoptosis that is associated with defective autophagy. Overexpression of GFP-LC3B or myc-LC3B rescued HPS1 knockdown-mediated defective autophagy by promoting autophagosome-lysosome fusion, thereby normalizing p62 levels (Fig. 6).
Fig. 6.

Proposed model for defective autophagy following HPS1 knockdown. A: under healthy conditions in which autophagy is functional, p62 is recruited into the autophagosomes, which fuse with lysosomes to form autophagolysosomes, in which p62 and other contents are degraded. B: under conditions of HPS1 knockdown in A549 cells, fusion between autophagosomes and lysosomes is impaired, thereby resulting in defective autophagy (LC3B and p62 accumulation). C: overexpressing LC3B (GFP-LC3B is shown as example) before knocking down HPS1 leads to the fusion of GFP-LC3B-labeled autophagosomes and lysosomes, resulting in functional autophagolysosomes and eventually p62 degradation.
HPS gene products are known to affect LROs of the body, causing impaired lysosomal trafficking and secretion and therefore cell- and organ-specific disturbances. HPS1 and HPS4 proteins are two subunits of a large protein called BLOC-3 (35). Functions of BLOC-3 complex remain obscure, but few studies have demonstrated that this protein complex might regulate microtubule-dependent movement and distribution of LROs (15). Although direct studies reporting autophagy regulation in the absence of HPS1 are scarce, there is one study that showed that tyrosine-related proteins, LAMP1, and LAMP3 were sequestered in large membranous structures resembling macroautophagosomes in melanocytes derived from patients with HPS1 (5). In contrast, AP-3, affected in patients with HPS2, is the only HPS protein of which functions are well characterized. HPS2 encodes mutations in the β3A subunit of the AP-3 complex, and this complex has been indicated to sort membrane proteins from endosomes to lysosomes and tissue-specific LROs (40). Of note, it has been reported that AP-3-null cells selectively mistarget LAMP1 and LAMP2 proteins (47). LAMP2 is a receptor for autophagosome-lysosome fusion, and its mistargeting by AP-3-deficient fibroblasts indicates dysregulated autophagy. Likewise, our present study reveals defective autophagy in the absence of functional HPS1 protein in humans and in the absence of both HPS1 and AP-3 proteins in mice. Besides species-specific differences, loss of both HPS1 and AP-3 proteins in mice might collectively lead to accumulation of autophagosomes, thereby resulting in the early lysosomal stress events as reported for HPS1/2 mice (30).
We observed a total increase in LC3B and p62 in HPS1/2 mice as well as in vitro following HPS1 knockdown in A549 cells. According to the classical autophagy pathway, an increase in LC3B with a parallel increase in p62 indicates deficient autophagy (41). Supporting this, we also did not observe a colocalization of LC3B with the lysosomal marker LAMP1. In line with this observation, a study published by Zhen et al., (58) while this manuscript was in revision showed p62 accumulation and impaired autophagosome-lysosome fusion in brain tissue of buff mutant mice, an HPS mouse model that also exhibits hypopigmentation and platelet storage pool deficiency. Furthermore, as an attempt to study autophagy flux following knockdown of HPS1 in vitro, we performed GFP cleavage assay by transfecting cells with GFP-tagged LC3B. Interestingly, in such cells, we observed a decrease in cellular vacuolization compared with cells in which HPS1 knockdown alone was performed, indicating that exogenous LC3B was protective, in part through induction of autophagy. This observation is in agreement with previously reported studies, in which overexpression of autophagy-related genes leading to antiapoptotic effects were shown (31, 42).
Altered autophagy has been indicated in the pathophysiology of several LSDs. In Niemann-Pick type C (NPC), a complex sphingolipidosis disorder, accumulation of autophagosomes was reported in skin fibroblasts from patients with NPC (37) and in brains of NPC mice (26). It has been suggested that induction of autophagy with impairment of autophagy flux leads to the severe aspects like generation of reactive oxygen species and accumulation of ubiquitinated proteins in the NPC neuropathology (28). Similarly, in other sphingolipidoses like Gaucher disease and Fabry disease, induction of autophagy and accumulation of autophagy substrates including p62 were reported in several tissues of patients as well as in mouse models of these diseases (11, 27, 36, 48). Such impaired autophagy flux was observed also in other LSDs like mucolipidosis (54), Pompe disease (43), Dannon disease (49), and neuronal ceroid lipofuscinoses (22). Similarly, we now report impaired autophagy resulting in accumulation of autophagy substrate p62 in vitro in the absence of HPS1 protein. Supporting this, we observed that LC3B immunogold labeling could preferentially be located in the lumen of lamellar bodies but not on the limiting membrane of lamellar bodies of the HPS1/2 mice compared with control mice. Whether this impaired autophagy contributes to the giant lamellar body degeneration that is characteristic to the HPSIP needs to be further studied. Another intriguing observation that stems from our present work is the nuclear localization of LC3B in cells in which the HPS1 gene was knocked down. Both LC3BI and LC3BII were shown to localize to nucleus (13), and it is known that nuclear LC3B is deacetylated to drive autophagy under starvation conditions (20); however, the function of nuclear LC3B under other settings is not yet clear. Why HPS1 gene knockdown should drive nuclear localization of endogenous LC3B remains an open question.
Altered autophagy has been reported in lung fibrosis recently. In a mouse model of amiodarone-induced lung fibrosis, we showed an increase in autophagy flux and autophagy-dependent apoptosis of alveolar epithelial cells (29). In patients with idiopathic pulmonary fibrosis, it was reported that autophagy was not induced despite the activation of the upstream pathways that induce autophagy (39). In the present study, we report that autophagy is impaired in HPS-associated lung fibrosis. On the basis of this information, it seems that autophagy is regulated differently depending on the kind of insult. Considering the complex regulation of autophagy and the integrated functions of the autophagosomes, lysosomes, and LROs, this observation may not be surprising. In any case, one of the important features shared by these different types of lung fibrosis is lysosomal stress in the alveolar epithelial cells that results from altered autophagy, either too much or too little.
We conclude that defective autophagy may be an important mechanism behind lysosomal stress events and epithelial cell apoptosis in the development of HPS-associated lung fibrosis.
GRANTS
This work was funded in part by the Excellence Cluster Cardio-Pulmonary System (ECCPS), Giessen, Germany and in part by the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: S.A., L.K., S.C., I.H., C.R., M.K., B.R.G., S.B., M.O., and P.M. performed experiments; S.A., L.K., A.G., and P.M. analyzed data; S.A., L.K., M.O., A.G., and P.M. interpreted results of experiments; S.A. and P.M. prepared figures; S.A. and P.M. drafted manuscript; S.A., L.K., S.C., I.H., C.R., M.K., B.R.G., S.B., W.S., M.O., A.G., and P.M. approved final version of manuscript; S.C., B.R.G., A.G., and P.M. edited and revised manuscript; W.S., A.G., and P.M. conception and design of research.
REFERENCES
- 1.Anderson PD, Huizing M, Claassen DA, White J, Gahl WA. Hermansky-Pudlak syndrome type 4 (HPS-4): clinical and molecular characteristics. Hum Genet 113: 10–17, 2003. [DOI] [PubMed] [Google Scholar]
- 2.Anikster Y, Huizing M, White J, Shevchenko YO, Fitzpatrick DL, Touchman JW, Compton JG, Bale SJ, Swank RT, Gahl WA, Toro JR. Mutation of a new gene causes a unique form of Hermansky-Pudlak syndrome in a genetic isolate of central Puerto Rico. Nat Genet 28: 376–380, 2001. [DOI] [PubMed] [Google Scholar]
- 3.Araya J, Kojima J, Takasaka N, Ito S, Fujii S, Hara H, Yanagisawa H, Kobayashi K, Tsurushige C, Kawaishi M, Kamiya N, Hirano J, Odaka M, Morikawa T, Nishimura SL, Kawabata Y, Hano H, Nakayama K, Kuwano K. Insufficient autophagy in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 304: L56–L69, 2013. [DOI] [PubMed] [Google Scholar]
- 4.Blommaart EF, Luiken JJ, Meijer AJ. Autophagic proteolysis: control and specificity. Histochem J 29: 365–385, 1997. [DOI] [PubMed] [Google Scholar]
- 5.Boissy RE, Zhao Y, Gahl WA. Altered protein localization in melanocytes from Hermansky-Pudlak syndrome: support for the role of the HPS gene product in intracellular trafficking. Lab Invest 78: 1037–1048, 1998. [PubMed] [Google Scholar]
- 6.Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, Nixon RA. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer's disease. J Neurosci 28: 6926–6937, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc 224: 213–232, 2006. [DOI] [PubMed] [Google Scholar]
- 8.Bonifacino JS. Insights into the biogenesis of lysosome-related organelles from the study of the Hermansky-Pudlak syndrome. Ann NY Acad Sci 1038: 103–114, 2004. [DOI] [PubMed] [Google Scholar]
- 9.Brantly M, Avila NA, Shotelersuk V, Lucero C, Huizing M, Gahl WA. Pulmonary function and high-resolution CT findings in patients with an inherited form of pulmonary fibrosis, Hermansky-Pudlak syndrome, due to mutations in HPS-1. Chest 117: 129–136, 2000. [DOI] [PubMed] [Google Scholar]
- 10.Cao Y, Espinola JA, Fossale E, Massey AC, Cuervo AM, MacDonald ME, Cotman SL. Autophagy is disrupted in a knock-in mouse model of juvenile neuronal ceroid lipofuscinosis. J Biol Chem 281: 20483–20493, 2006. [DOI] [PubMed] [Google Scholar]
- 11.Chevrier M, Brakch N, Celine L, Genty D, Ramdani Y, Moll S, Djavaheri-Mergny M, Brasse-Lagnel C, Annie Laquerriere AL, Barbey F, Bekri S. Autophagosome maturation is impaired in Fabry disease. Autophagy 6: 589–599, 2010. [DOI] [PubMed] [Google Scholar]
- 12.Dell'Angelica EC, Mullins C, Caplan S, Bonifacino JS. Lysosome-related organelles. FASEB J 14: 1265–1278, 2000. [DOI] [PubMed] [Google Scholar]
- 13.Drake KR, Kang M, Kenworthy AK. Nucleocytoplasmic distribution and dynamics of the autophagosome marker EGFP-LC3. PLoS One 5: e9806, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eskelinen EL. New insights into the mechanisms of macroautophagy in mammalian cells. Int Rev Cell Mol Biol 266: 207–247, 2008. [DOI] [PubMed] [Google Scholar]
- 15.Falcon-Perez JM, Nazarian R, Sabatti C, Dell'Angelica EC. Distribution and dynamics of Lamp1-containing endocytic organelles in fibroblasts deficient in BLOC-3. J Cell Sci 118: 5243–5255, 2005. [DOI] [PubMed] [Google Scholar]
- 16.Gahl WA, Brantly M, Kaiser-Kupfer MI, Iwata F, Hazelwood S, Shotelersuk V, Duffy LF, Kuehl EM, Troendle J, Bernardini I. Genetic defects and clinical characteristics of patients with a form of oculocutaneous albinism (Hermansky-Pudlak syndrome). N Engl J Med 338: 1258–1264, 1998. [DOI] [PubMed] [Google Scholar]
- 17.Gochuico BR, Huizing M, Golas GA, Scher CD, Tsokos M, Denver SD, Frei-Jones MJ, Gahl WA. Interstitial lung disease and pulmonary fibrosis in Hermansky-Pudlak syndrome type 2, an adaptor protein-3 complex disease. Mol Med 18: 56–64, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guttentag SH, Akhtar A, Tao JQ, Atochina E, Rusiniak ME, Swank RT, Bates SR. Defective surfactant secretion in a mouse model of Hermansky-Pudlak syndrome. Am J Respir Cell Mol Biol 33: 14–21, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hsia CC, Hyde DM, Ochs M, Weibel ER. An official research policy statement of the American Thoracic Society/European Respiratory Society: standards for quantitative assessment of lung structure. Am J Respir Crit Care Med 181: 394–418, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang R, Xu Y, Wan W, Shou X, Qian J, You Z, Liu B, Chang C, Zhou T, Lippincott-Schwartz J, Liu W. Deacetylation of nuclear LC3 drives autophagy initiation under starvation. Mol Cell 57: 456–466, 2015. [DOI] [PubMed] [Google Scholar]
- 21.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, Ahn HJ, Ait-Mohamed O, Ait-Si-Ali S, Akematsu T, Akira S, Al-Younes HM, Al-Zeer MA, Albert ML, Albin RL, Alegre-Abarrategui J, Aleo MF, Alirezaei M, Almasan A, Almonte-Becerril M, Amano A, Amaravadi R, Amarnath S, Amer AO, Andrieu-Abadie N, Anantharam V, Ann DK, Anoopkumar-Dukie S, Aoki H, Apostolova N, Arancia G, Aris JP, Asanuma K, Asare NY, Ashida H, Askanas V, Askew DS, Auberger P, Baba M, Backues SK, Baehrecke EH, Bahr BA, Bai XY, Bailly Y, Baiocchi R, Baldini G, Balduini W, Ballabio A, Bamber BA, Bampton ET, Banhegyi G, Bartholomew CR, Bassham DC, Bast RC Jr, Batoko H, Bay BH, Beau I, Bechet DM, Begley TJ, Behl C, Behrends C, Bekri S, Bellaire B, Bendall LJ, Benetti L, Berliocchi L, Bernardi H, Bernassola F, Besteiro S, Bhatia-Kissova I, Bi X, Biard-Piechaczyk M, Blum JS, Boise LH, Bonaldo P, Boone DL, Bornhauser BC, Bortoluci KR, Bossis I, Bost F, Bourquin JP, Boya P, Boyer-Guittaut M, Bozhkov PV, Brady NR, Brancolini C, Brech A, Brenman JE, Brennand A, Bresnick EH, Brest P, Bridges D, Bristol ML, Brookes PS, Brown EJ, Brumell JH, Brunetti-Pierri N, Brunk UT, Bulman DE, Bultman SJ, Bultynck G, Burbulla LF, Bursch W, Butchar JP, Buzgariu W, Bydlowski SP, Cadwell K, Cahova M, Cai D, Cai J, Cai Q, Calabretta B, Calvo-Garrido J, Camougrand N, Campanella M, Campos-Salinas J, Candi E, Cao L, Caplan AB, Carding SR, Cardoso SM, Carew JS, Carlin CR, Carmignac V, Carneiro LA, Carra S, Caruso RA, Casari G, Casas C, Castino R, Cebollero E, Cecconi F, Celli J, Chaachouay H, Chae HJ, Chai CY, Chan DC, Chan EY, Chang RC, Che CM, Chen CC, Chen GC, Chen GQ, Chen M, Chen Q, Chen SS, Chen W, Chen X, Chen YG, Chen Y, Chen YJ, Chen Z, Cheng A, Cheng CH, Cheng Y, Cheong H, Cheong JH, Cherry S, Chess-Williams R, Cheung ZH, Chevet E, Chiang HL, Chiarelli R, Chiba T, Chin LS, Chiou SH, Chisari FV, Cho CH, Cho DH, Choi AM, Choi D, Choi KS, Choi ME, Chouaib S, Choubey D, Choubey V, Chu CT, Chuang TH, Chueh SH, Chun T, Chwae YJ, Chye ML, Ciarcia R, Ciriolo MR, Clague MJ, Clark RS, Clarke PG, Clarke R, Codogno P, Coller HA, Colombo MI, Comincini S, Condello M, Condorelli F, Cookson MR, Coombs GH, Coppens I, Corbalan R, Cossart P, Costelli P, Costes S, Coto-Montes A, Couve E, Coxon FP, Cregg JM, Crespo JL, Cronje MJ, Cuervo AM, Cullen JJ, Czaja MJ, D'Amelio M, Darfeuille-Michaud A, Davids LM, Davies FE, De Felici M, de Groot JF, de Haan CA, De Martino L, De Milito A, De Tata V, Debnath J, Degterev A, Dehay B, Delbridge LM, Demarchi F, Deng YZ, Dengjel J, Dent P, Denton D, Deretic V, Desai SD, Devenish RJ, Di Gioacchino M, Di Paolo G, Di Pietro C, Diaz-Araya G, Diaz-Laviada I, Diaz-Meco MT, Diaz-Nido J, Dikic I, Dinesh-Kumar SP, Ding WX, Distelhorst CW, Diwan A, Djavaheri-Mergny M, Dokudovskaya S, Dong Z, Dorsey FC, Dosenko V, Dowling JJ, Doxsey S, Dreux M, Drew ME, Duan Q, Duchosal MA, Duff K, Dugail I, Durbeej M, Duszenko M, Edelstein CL, Edinger AL, Egea G, Eichinger L, Eissa NT, Ekmekcioglu S, El-Deiry WS, Elazar Z, Elgendy M, Ellerby LM, Eng KE, Engelbrecht AM, Engelender S, Erenpreisa J, Escalante R, Esclatine A, Eskelinen EL, Espert L, Espina V, Fan H, Fan J, Fan QW, Fan Z, Fang S, Fang Y, Fanto M, Fanzani A, Farkas T, Farre JC, Faure M, Fechheimer M, Feng CG, Feng J, Feng Q, Feng Y, Fesus L, Feuer R, Figueiredo-Pereira ME, Fimia GM, Fingar DC, Finkbeiner S, Finkel T, Finley KD, Fiorito F, Fisher EA, Fisher PB, Flajolet M, Florez-McClure ML, Florio S, Fon EA, Fornai F, Fortunato F, Fotedar R, Fowler DH, Fox HS, Franco R, Frankel LB, Fransen M, Fuentes JM, Fueyo J, Fujii J, Fujisaki K, Fujita E, Fukuda M, Furukawa RH, Gaestel M, Gailly P, Gajewska M, Galliot B, Galy V, Ganesh S, Ganetzky B, Ganley IG, Gao FB, Gao GF, Gao J, Garcia L, Garcia-Manero G, Garcia-Marcos M, Garmyn M, Gartel AL, Gatti E, Gautel M, Gawriluk TR, Gegg ME, Geng J, Germain M, Gestwicki JE, Gewirtz DA, Ghavami S, Ghosh P, Giammarioli AM, Giatromanolaki AN, Gibson SB, Gilkerson RW, Ginger ML, Ginsberg HN, Golab J, Goligorsky MS, Golstein P, Gomez-Manzano C, Goncu E, Gongora C, Gonzalez CD, Gonzalez R, Gonzalez-Estevez C, Gonzalez-Polo RA, Gonzalez-Rey E, Gorbunov NV, Gorski S, Goruppi S, Gottlieb RA, Gozuacik D, Granato GE, Grant GD, Green KN, Gregorc A, Gros F, Grose C, Grunt TW, Gual P, Guan JL, Guan KL, Guichard SM, Gukovskaya AS, Gukovsky I, Gunst J, Gustafsson AB, Halayko AJ, Hale AN, Halonen SK, Hamasaki M, Han F, Han T, Hancock MK, Hansen M, Harada H, Harada M, Hardt SE, Harper JW, Harris AL, Harris J, Harris SD, Hashimoto M, Haspel JA, Hayashi S, Hazelhurst LA, He C, He YW, Hebert MJ, Heidenreich KA, Helfrich MH, Helgason GV, Henske EP, Herman B, Herman PK, Hetz C, Hilfiker S, Hill JA, Hocking LJ, Hofman P, Hofmann TG, Hohfeld J, Holyoake TL, Hong MH, Hood DA, Hotamisligil GS, Houwerzijl EJ, Hoyer-Hansen M, Hu B, Hu CA, Hu HM, Hua Y, Huang C, Huang J, Huang S, Huang WP, Huber TB, Huh WK, Hung TH, Hupp TR, Hur GM, Hurley JB, Hussain SN, Hussey PJ, Hwang JJ, Hwang S, Ichihara A, Ilkhanizadeh S, Inoki K, Into T, Iovane V, Iovanna JL, Ip NY, Isaka Y, Ishida H, Isidoro C, Isobe K, Iwasaki A, Izquierdo M, Izumi Y, Jaakkola PM, Jaattela M, Jackson GR, Jackson WT, Janji B, Jendrach M, Jeon JH, Jeung EB, Jiang H, Jiang JX, Jiang M, Jiang Q, Jiang X, Jimenez A, Jin M, Jin S, Joe CO, Johansen T, Johnson DE, Johnson GV, Jones NL, Joseph B, Joseph SK, Joubert AM, Juhasz G, Juillerat-Jeanneret L, Jung CH, Jung YK, Kaarniranta K, Kaasik A, Kabuta T, Kadowaki M, Kagedal K, Kamada Y, Kaminskyy VO, Kampinga HH, Kanamori H, Kang C, Kang KB, Kang KI, Kang R, Kang YA, Kanki T, Kanneganti TD, Kanno H, Kanthasamy AG, Kanthasamy A, Karantza V, Kaushal GP, Kaushik S, Kawazoe Y, Ke PY, Kehrl JH, Kelekar A, Kerkhoff C, Kessel DH, Khalil H, Kiel JA, Kiger AA, Kihara A, Kim DR, Kim DH, Kim EK, Kim HR, Kim JS, Kim JH, Kim JC, Kim JK, Kim PK, Kim SW, Kim YS, Kim Y, Kimchi A, Kimmelman AC, King JS, Kinsella TJ, Kirkin V, Kirshenbaum LA, Kitamoto K, Kitazato K, Klein L, Klimecki WT, Klucken J, Knecht E, Ko BC, Koch JC, Koga H, Koh JY, Koh YH, Koike M, Komatsu M, Kominami E, Kong HJ, Kong WJ, Korolchuk VI, Kotake Y, Koukourakis MI, Kouri Flores JB, Kovacs AL, Kraft C, Krainc D, Kramer H, Kretz-Remy C, Krichevsky AM, Kroemer G, Kruger R, Krut O, Ktistakis NT, Kuan CY, Kucharczyk R, Kumar A, Kumar R, Kumar S, Kundu M, Kung HJ, Kurz T, Kwon HJ, La Spada AR, Lafont F, Lamark T, Landry J, Lane JD, Lapaquette P, Laporte JF, Laszlo L, Lavandero S, Lavoie JN, Layfield R, Lazo PA, Le W, Le Cam L, Ledbetter DJ, Lee AJ, Lee BW, Lee GM, Lee J, Lee JH, Lee M, Lee MS, Lee SH, Leeuwenburgh C, Legembre P, Legouis R, Lehmann M, Lei HY, Lei QY, Leib DA, Leiro J, Lemasters JJ, Lemoine A, Lesniak MS, Lev D, Levenson VV, Levine B, Levy E, Li F, Li JL, Li L, Li S, Li W, Li XJ, Li YB, Li YP, Liang C, Liang Q, Liao YF, Liberski PP, Lieberman A, Lim HJ, Lim KL, Lim K, Lin CF, Lin FC, Lin J, Lin JD, Lin K, Lin WW, Lin WC, Lin YL, Linden R, Lingor P, Lippincott-Schwartz J, Lisanti MP, Liton PB, Liu B, Liu CF, Liu K, Liu L, Liu QA, Liu W, Liu YC, Liu Y, Lockshin RA, Lok CN, Lonial S, Loos B, Lopez-Berestein G, Lopez-Otin C, Lossi L, Lotze MT, Low P, Lu B, Lu Z, Luciano F, Lukacs NW, Lund AH, Lynch-Day MA, Ma Y, Macian F, MacKeigan JP, Macleod KF, Madeo F, Maiuri L, Maiuri MC, Malagoli D, Malicdan MC, Malorni W, Man N, Mandelkow EM, Manon S, Manov I, Mao K, Mao X, Mao Z, Marambaud P, Marazziti D, Marcel YL, Marchbank K, Marchetti P, Marciniak SJ, Marcondes M, Mardi M, Marfe G, Marino G, Markaki M, Marten MR, Martin SJ, Martinand-Mari C, Martinet W, Martinez-Vicente M, Masini M, Matarrese P, Matsuo S, Matteoni R, Mayer A, Mazure NM, McConkey DJ, McConnell MJ, McDermott C, McDonald C, McInerney GM, McKenna SL, McLaughlin B, McLean PJ, McMaster CR, McQuibban GA, Meijer AJ, Meisler MH, Melendez A, Melia TJ, Melino G, Mena MA, Menendez JA, Menna-Barreto RF, Menon MB, Menzies FM, Mercer CA, Merighi A, Merry DE, Meschini S, Meyer CG, Meyer TF, Miao CY, Miao JY, Michels PA, Michiels C, Mijaljica D, Milojkovic A, Minucci S, Miracco C, Miranti CK, Mitroulis I, Miyazawa K, Mizushima N, Mograbi B, Mohseni S, Molero X, Mollereau B, Mollinedo F, Momoi T, Monastyrska I, Monick MM, Monteiro MJ, Moore MN, Mora R, Moreau K, Moreira PI, Moriyasu Y, Moscat J, Mostowy S, Mottram JC, Motyl T, Moussa CE, Muller S, Munger K, Munz C, Murphy LO, Murphy ME, Musaro A, Mysorekar I, Nagata E, Nagata K, Nahimana A, Nair U, Nakagawa T, Nakahira K, Nakano H, Nakatogawa H, Nanjundan M, Naqvi NI, Narendra DP, Narita M, Navarro M, Nawrocki ST, Nazarko TY, Nemchenko A, Netea MG, Neufeld TP, Ney PA, Nezis IP, Nguyen HP, Nie D, Nishino I, Nislow C, Nixon RA, Noda T, Noegel AA, Nogalska A, Noguchi S, Notterpek L, Novak I, Nozaki T, Nukina N, Nurnberger T, Nyfeler B, Obara K, Oberley TD, Oddo S, Ogawa M, Ohashi T, Okamoto K, Oleinick NL, Oliver FJ, Olsen LJ, Olsson S, Opota O, Osborne TF, Ostrander GK, Otsu K, Ou JH, Ouimet M, Overholtzer M, Ozpolat B, Paganetti P, Pagnini U, Pallet N, Palmer GE, Palumbo C, Pan T, Panaretakis T, Pandey UB, Papackova Z, Papassideri I, Paris I, Park J, Park OK, Parys JB, Parzych KR, Patschan S, Patterson C, Pattingre S, Pawelek JM, Peng J, Perlmutter DH, Perrotta I, Perry G, Pervaiz S, Peter M, Peters GJ, Petersen M, Petrovski G, Phang JM, Piacentini M, Pierre P, Pierrefite-Carle V, Pierron G, Pinkas-Kramarski R, Piras A, Piri N, Platanias LC, Poggeler S, Poirot M, Poletti A, Pous C, Pozuelo-Rubio M, Praetorius-Ibba M, Prasad A, Prescott M, Priault M, Produit-Zengaffinen N, Progulske-Fox A, Proikas-Cezanne T, Przedborski S, Przyklenk K, Puertollano R, Puyal J, Qian SB, Qin L, Qin ZH, Quaggin SE, Raben N, Rabinowich H, Rabkin SW, Rahman I, Rami A, Ramm G, Randall G, Randow F, Rao VA, Rathmell JC, Ravikumar B, Ray SK, Reed BH, Reed JC, Reggiori F, Regnier-Vigouroux A, Reichert AS, Reiners JJ Jr, Reiter RJ, Ren J, Revuelta JL, Rhodes CJ, Ritis K, Rizzo E, Robbins J, Roberge M, Roca H, Roccheri MC, Rocchi S, Rodemann HP, Rodriguez de Cordoba S, Rohrer B, Roninson IB, Rosen K, Rost-Roszkowska MM, Rouis M, Rouschop KM, Rovetta F, Rubin BP, Rubinsztein DC, Ruckdeschel K, Rucker EB 3rd, Rudich A, Rudolf E, Ruiz-Opazo N, Russo R, Rusten TE, Ryan KM, Ryter SW, Sabatini DM, Sadoshima J, Saha T, Saitoh T, Sakagami H, Sakai Y, Salekdeh GH, Salomoni P, Salvaterra PM, Salvesen G, Salvioli R, Sanchez AM, Sanchez-Alcazar JA, Sanchez-Prieto R, Sandri M, Sankar U, Sansanwal P, Santambrogio L, Saran S, Sarkar S, Sarwal M, Sasakawa C, Sasnauskiene A, Sass M, Sato K, Sato M, Schapira AH, Scharl M, Schatzl HM, Scheper W, Schiaffino S, Schneider C, Schneider ME, Schneider-Stock R, Schoenlein PV, Schorderet DF, Schuller C, Schwartz GK, Scorrano L, Sealy L, Seglen PO, Segura-Aguilar J, Seiliez I, Seleverstov O, Sell C, Seo JB, Separovic D, Setaluri V, Setoguchi T, Settembre C, Shacka JJ, Shanmugam M, Shapiro IM, Shaulian E, Shaw RJ, Shelhamer JH, Shen HM, Shen WC, Sheng ZH, Shi Y, Shibuya K, Shidoji Y, Shieh JJ, Shih CM, Shimada Y, Shimizu S, Shintani T, Shirihai OS, Shore GC, Sibirny AA, Sidhu SB, Sikorska B, Silva-Zacarin EC, Simmons A, Simon AK, Simon HU, Simone C, Simonsen A, Sinclair DA, Singh R, Sinha D, Sinicrope FA, Sirko A, Siu PM, Sivridis E, Skop V, Skulachev VP, Slack RS, Smaili SS, Smith DR, Soengas MS, Soldati T, Song X, Sood AK, Soong TW, Sotgia F, Spector SA, Spies CD, Springer W, Srinivasula SM, Stefanis L, Steffan JS, Stendel R, Stenmark H, Stephanou A, Stern ST, Sternberg C, Stork B, Stralfors P, Subauste CS, Sui X, Sulzer D, Sun J, Sun SY, Sun ZJ, Sung JJ, Suzuki K, Suzuki T, Swanson MS, Swanton C, Sweeney ST, Sy LK, Szabadkai G, Tabas I, Taegtmeyer H, Tafani M, Takacs-Vellai K, Takano Y, Takegawa K, Takemura G, Takeshita F, Talbot NJ, Tan KS, Tanaka K, Tang D, Tanida I, Tannous BA, Tavernarakis N, Taylor GS, Taylor GA, Taylor JP, Terada LS, Terman A, Tettamanti G, Thevissen K, Thompson CB, Thorburn A, Thumm M, Tian F, Tian Y, Tocchini-Valentini G, Tolkovsky AM, Tomino Y, Tonges L, Tooze SA, Tournier C, Tower J, Towns R, Trajkovic V, Travassos LH, Tsai TF, Tschan MP, Tsubata T, Tsung A, Turk B, Turner LS, Tyagi SC, Uchiyama Y, Ueno T, Umekawa M, Umemiya-Shirafuji R, Unni VK, Vaccaro MI, Valente EM, Van den Berghe G, van der Klei IJ, van Doorn W, van Dyk LF, van Egmond M, van Grunsven LA, Vandenabeele P, Vandenberghe WP, Vanhorebeek I, Vaquero EC, Velasco G, Vellai T, Vicencio JM, Vierstra RD, Vila M, Vindis C, Viola G, Viscomi MT, Voitsekhovskaja OV, von Haefen C, Votruba M, Wada K, Wade-Martins R, Walker CL, Walsh CM, Walter J, Wan XB, Wang A, Wang C, Wang D, Wang F, Wang G, Wang H, Wang HG, Wang HD, Wang J, Wang K, Wang M, Wang RC, Wang X, Wang YJ, Wang Y, Wang Z, Wang ZC, Wansink DG, Ward DM, Watada H, Waters SL, Webster P, Wei L, Weihl CC, Weiss WA, Welford SM, Wen LP, Whitehouse CA, Whitton JL, Whitworth AJ, Wileman T, Wiley JW, Wilkinson S, Willbold D, Williams RL, Williamson PR, Wouters BG, Wu C, Wu DC, Wu WK, Wyttenbach A, Xavier RJ, Xi Z, Xia P, Xiao G, Xie Z, Xu DZ, Xu J, Xu L, Xu X, Yamamoto A, Yamashina S, Yamashita M, Yan X, Yanagida M, Yang DS, Yang E, Yang JM, Yang SY, Yang W, Yang WY, Yang Z, Yao MC, Yao TP, Yeganeh B, Yen WL, Yin JJ, Yin XM, Yoo OJ, Yoon G, Yoon SY, Yorimitsu T, Yoshikawa Y, Yoshimori T, Yoshimoto K, You HJ, Youle RJ, Younes A, Yu L, Yu SW, Yu WH, Yuan ZM, Yue Z, Yun CH, Yuzaki M, Zabirnyk O, Silva-Zacarin E, Zacks D, Zacksenhaus E, Zaffaroni N, Zakeri Z, Zeh HJ 3rd, Zeitlin SO, Zhang H, Zhang HL, Zhang J, Zhang JP, Zhang L, Zhang MY, Zhang XD, Zhao M, Zhao YF, Zhao Y, Zhao ZJ, Zheng X, Zhivotovsky B, Zhong Q, Zhou CZ, Zhu C, Zhu WG, Zhu XF, Zhu X, Zhu Y, Zoladek T, Zong WX, Zorzano A, Zschocke J, Zuckerbraun B. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8: 445–544, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koike M, Shibata M, Waguri S, Yoshimura K, Tanida I, Kominami E, Gotow T, Peters C, von Figura K, Mizushima N, Saftig P, Uchiyama Y. Participation of autophagy in storage of lysosomes in neurons from mouse models of neuronal ceroid-lipofuscinoses (Batten disease). Am J Pathol 167: 1713–1728, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441: 880–884, 2006. [DOI] [PubMed] [Google Scholar]
- 24.Korfei M, von der Beck D, Henneke I, Markart P, Ruppert C, Mahavadi P, Ghanim B, Klepetko W, Fink L, Meiners S, Kramer OH, Seeger W, Vancheri C, Guenther A. Comparative proteome analysis of lung tissue from patients with idiopathic pulmonary fibrosis (IPF), non-specific interstitial pneumonia (NSIP) and organ donors. J Proteomics 85: 109–128, 2013. [DOI] [PubMed] [Google Scholar]
- 25.Lawrence BP, Brown WJ. Autophagic vacuoles rapidly fuse with pre-existing lysosomes in cultured hepatocytes. J Cell Sci 102: 515–526, 1992. [DOI] [PubMed] [Google Scholar]
- 26.Liao G, Yao Y, Liu J, Yu Z, Cheung S, Xie A, Liang X, Bi X. Cholesterol accumulation is associated with lysosomal dysfunction and autophagic stress in Npc1 −/− mouse brain. Am J Pathol 171: 962–975, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liebau MC, Braun F, Hopker K, Weitbrecht C, Bartels V, Muller RU, Brodesser S, Saleem MA, Benzing T, Schermer B, Cybulla M, Kurschat CE. Dysregulated autophagy contributes to podocyte damage in Fabry's disease. PLoS One 8: e63506, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lieberman AP, Puertollano R, Raben N, Slaugenhaupt S, Walkley SU, Ballabio A. Autophagy in lysosomal storage disorders. Autophagy 8: 719–730, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mahavadi P, Knudsen L, Venkatesan S, Henneke I, Hegermann J, Wrede C, Ochs M, Ahuja S, Chillappagari S, Ruppert C, Seeger W, Korfei M, Guenther A. Regulation of macroautophagy in amiodarone induced pulmonary fibrosis. J Pathol Clin Res 1: 252–263, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mahavadi P, Korfei M, Henneke I, Liebisch G, Schmitz G, Gochuico BR, Markart P, Bellusci S, Seeger W, Ruppert C, Guenther A. Epithelial stress and apoptosis underlie Hermansky-Pudlak syndrome-associated interstitial pneumonia. Am J Respir Crit Care Med 182: 207–219, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mai S, Muster B, Bereiter-Hahn J, Jendrach M. Autophagy proteins LC3B, ATG5 and ATG12 participate in quality control after mitochondrial damage and influence lifespan. Autophagy 8: 47–62, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mayhew TM, Lucocq JM. Quantifying immunogold labelling patterns of cellular compartments when they comprise mixtures of membranes (surface-occupying) and organelles (volume-occupying). Histochem Cell Biol 129: 367–378, 2008. [DOI] [PubMed] [Google Scholar]
- 33.Mizushima N. Autophagy: process and function. Genes Dev 21: 2861–2873, 2007. [DOI] [PubMed] [Google Scholar]
- 34.Nakatani Y, Nakamura N, Sano J, Inayama Y, Kawano N, Yamanaka S, Miyagi Y, Nagashima Y, Ohbayashi C, Mizushima M, Manabe T, Kuroda M, Yokoi T, Matsubara O. Interstitial pneumonia in Hermansky-Pudlak syndrome: significance of florid foamy swelling/degeneration (giant lamellar body degeneration) of type-2 pneumocytes. Virchows Arch 437: 304–313, 2000. [DOI] [PubMed] [Google Scholar]
- 35.Nazarian R, Falcon-Perez JM, Dell'Angelica EC. Biogenesis of lysosome-related organelles complex 3 (BLOC-3): a complex containing the Hermansky-Pudlak syndrome (HPS) proteins HPS1 and HPS4. Proc Natl Acad Sci USA 100: 8770–8775, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Osellame LD, Duchen MR. Defective quality control mechanisms and accumulation of damaged mitochondria link Gaucher and Parkinson diseases. Autophagy 9: 1633–1635, 2013. [DOI] [PubMed] [Google Scholar]
- 37.Pacheco CD, Kunkel R, Lieberman AP. Autophagy in Niemann-Pick C disease is dependent upon Beclin-1 and responsive to lipid trafficking defects. Hum Mol Genet 16: 1495–1503, 2007. [DOI] [PubMed] [Google Scholar]
- 38.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282: 24131–24145, 2007. [DOI] [PubMed] [Google Scholar]
- 39.Patel AS, Lin L, Geyer A, Haspel JA, An CH, Cao J, Rosas IO, Morse D. Autophagy in idiopathic pulmonary fibrosis. PLoS One 7: e41394, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peden AA, Oorschot V, Hesser BA, Austin CD, Scheller RH, Klumperman J. Localization of the AP-3 adaptor complex defines a novel endosomal exit site for lysosomal membrane proteins. J Cell Biol 164: 1065–1076, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Puissant A, Fenouille N, Auberger P. When autophagy meets cancer through p62/SQSTM1. Am J Cancer Res 2: 397–413, 2012. [PMC free article] [PubMed] [Google Scholar]
- 42.Pyo JO, Yoo SM, Ahn HH, Nah J, Hong SH, Kam TI, Jung S, Jung YK. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat Commun 4: 2300, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Raben N, Baum R, Schreiner C, Takikita S, Mizushima N, Ralston E, Plotz P. When more is less: excess and deficiency of autophagy coexist in skeletal muscle in Pompe disease. Autophagy 5: 111–113, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schultz ML, Tecedor L, Chang M, Davidson BL. Clarifying lysosomal storage diseases. Trends Neurosci 34: 401–410, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seglen PO, Gordon PB, Holen I. Non-selective autophagy. Semin Cell Biol 1: 441–448, 1990. [PubMed] [Google Scholar]
- 46.Settembre C, Fraldi A, Jahreiss L, Spampanato C, Venturi C, Medina D, de Pablo R, Tacchetti C, Rubinsztein DC, Ballabio A. A block of autophagy in lysosomal storage disorders. Hum Mol Genet 17: 119–129, 2008. [DOI] [PubMed] [Google Scholar]
- 47.Styers ML, Salazar G, Love R, Peden AA, Kowalczyk AP, Faundez V. The endo-lysosomal sorting machinery interacts with the intermediate filament cytoskeleton. Mol Biol Cell 15: 5369–5382, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sun Y, Liou B, Ran H, Skelton MR, Williams MT, Vorhees CV, Kitatani K, Hannun YA, Witte DP, Xu YH, Grabowski GA. Neuronopathic Gaucher disease in the mouse: viable combined selective saposin C deficiency and mutant glucocerebrosidase (V394L) mice with glucosylsphingosine and glucosylceramide accumulation and progressive neurological deficits. Hum Mol Genet 19: 1088–1097, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tanaka Y, Guhde G, Suter A, Eskelinen EL, Hartmann D, Lullmann-Rauch R, Janssen PM, Blanz J, von Figura K, Saftig P. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature 406: 902–906, 2000. [DOI] [PubMed] [Google Scholar]
- 50.Tanida I, Ueno T, Kominami E. Human light chain 3/MAP1LC3B is cleaved at its carboxyl-terminal Met121 to expose Gly120 for lipidation and targeting to autophagosomal membranes. J Biol Chem 279: 47704–47710, 2004. [DOI] [PubMed] [Google Scholar]
- 51.Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol 36: 2503–2518, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tschanz S, Schneider JP, Knudsen L. Design-based stereology: Planning, volumetry and sampling are crucial steps for a successful study. Ann Anat 196: 3–11, 2014. [DOI] [PubMed] [Google Scholar]
- 53.Vasilescu DM, Gao Z, Saha PK, Yin L, Wang G, Haefeli-Bleuer B, Ochs M, Weibel ER, Hoffman EA. Assessment of morphometry of pulmonary acini in mouse lungs by nondestructive imaging using multiscale microcomputed tomography. Proc Natl Acad Sci USA 109: 17105–17110, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vergarajauregui S, Connelly PS, Daniels MP, Puertollano R. Autophagic dysfunction in mucolipidosis type IV patients. Hum Mol Genet 17: 2723–2737, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vitner EB, Platt FM, Futerman AH. Common and uncommon pathogenic cascades in lysosomal storage diseases. J Biol Chem 285: 20423–20427, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Young LR, Pasula R, Gulleman PM, Deutsch GH, McCormack FX. Susceptibility of Hermansky-Pudlak mice to bleomycin-induced type II cell apoptosis and fibrosis. Am J Respir Cell Mol Biol 37: 67–74, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang L, Huang X, Qin C, Brinkman K, Gong Y, Wang S, Huang K. First spectroscopic identification of pyrocarbonate for high CO2 flux membranes containing highly interconnected three dimensional ionic channels. Phys Chem Chem Phys 15: 13147–13152, 2013. [DOI] [PubMed] [Google Scholar]
- 58.Zhen Y, Li W. Impairment of autophagosome-lysosome fusion in the buff mutant mice with the VPS33A(D251E) mutation. Autophagy 11: 1608–1622, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]